
Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Cancer: Current Use and Future Prospects



Abstract
1. Introduction
1.1. Cancer
1.2. Treatment
1.2.1. Non-TKI Agents
1.2.2. TKI Agents
2. EGFRs
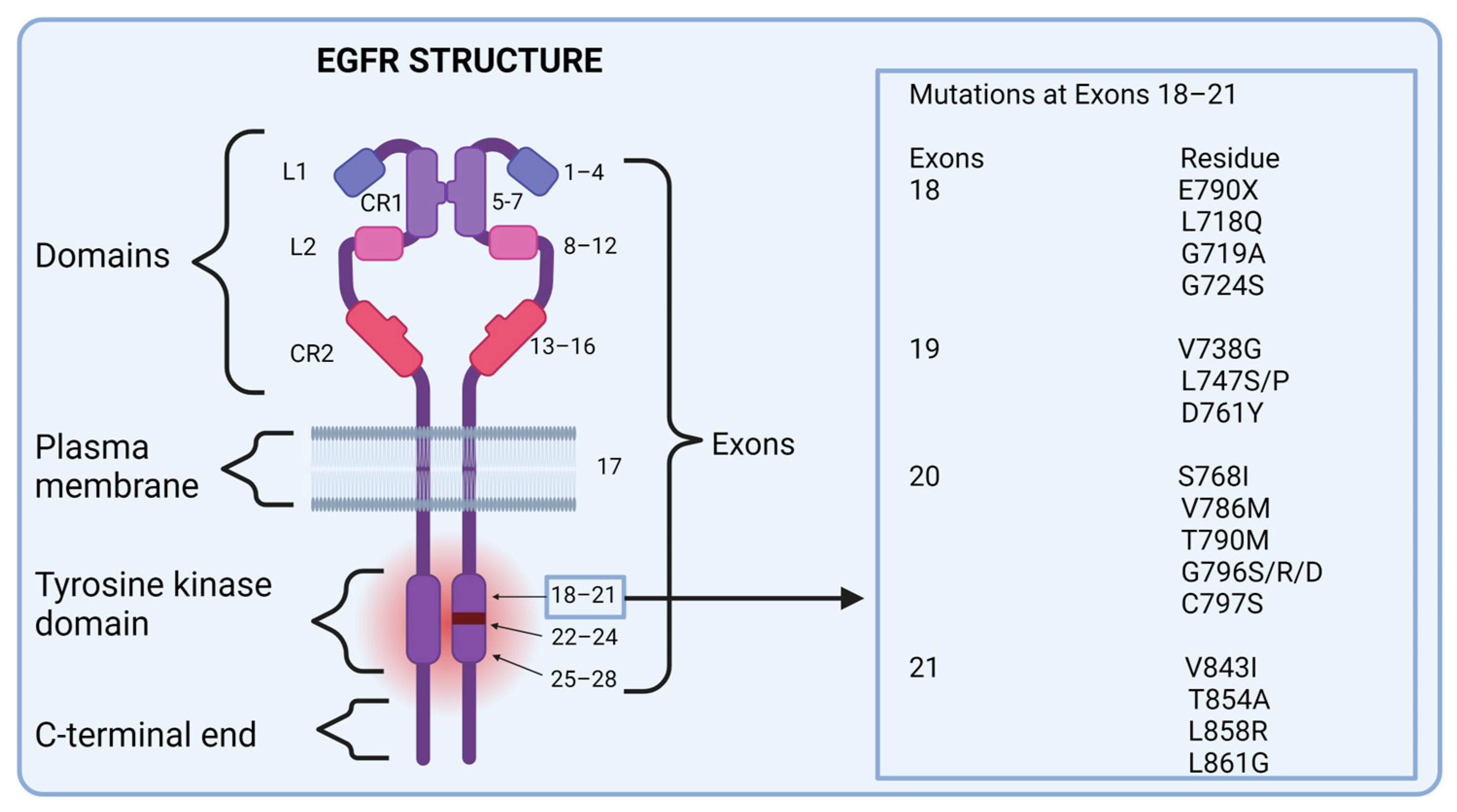
2.1. Structure and Function
2.2. Physiological Activation
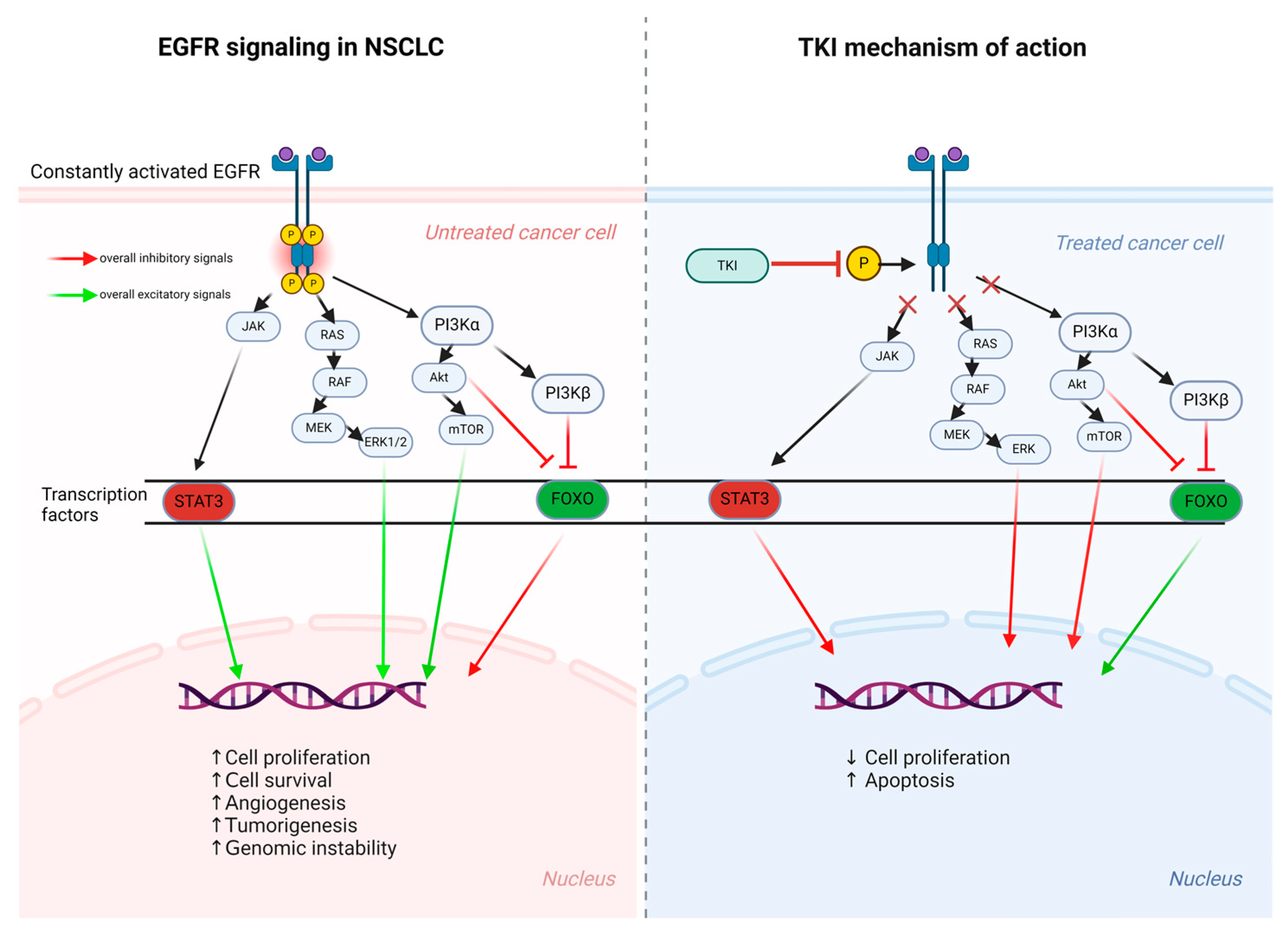
2.3. Signaling Pathways
2.4. The Forkhead Box FOXO Regulation
2.5. Additional Oncogenic Signaling
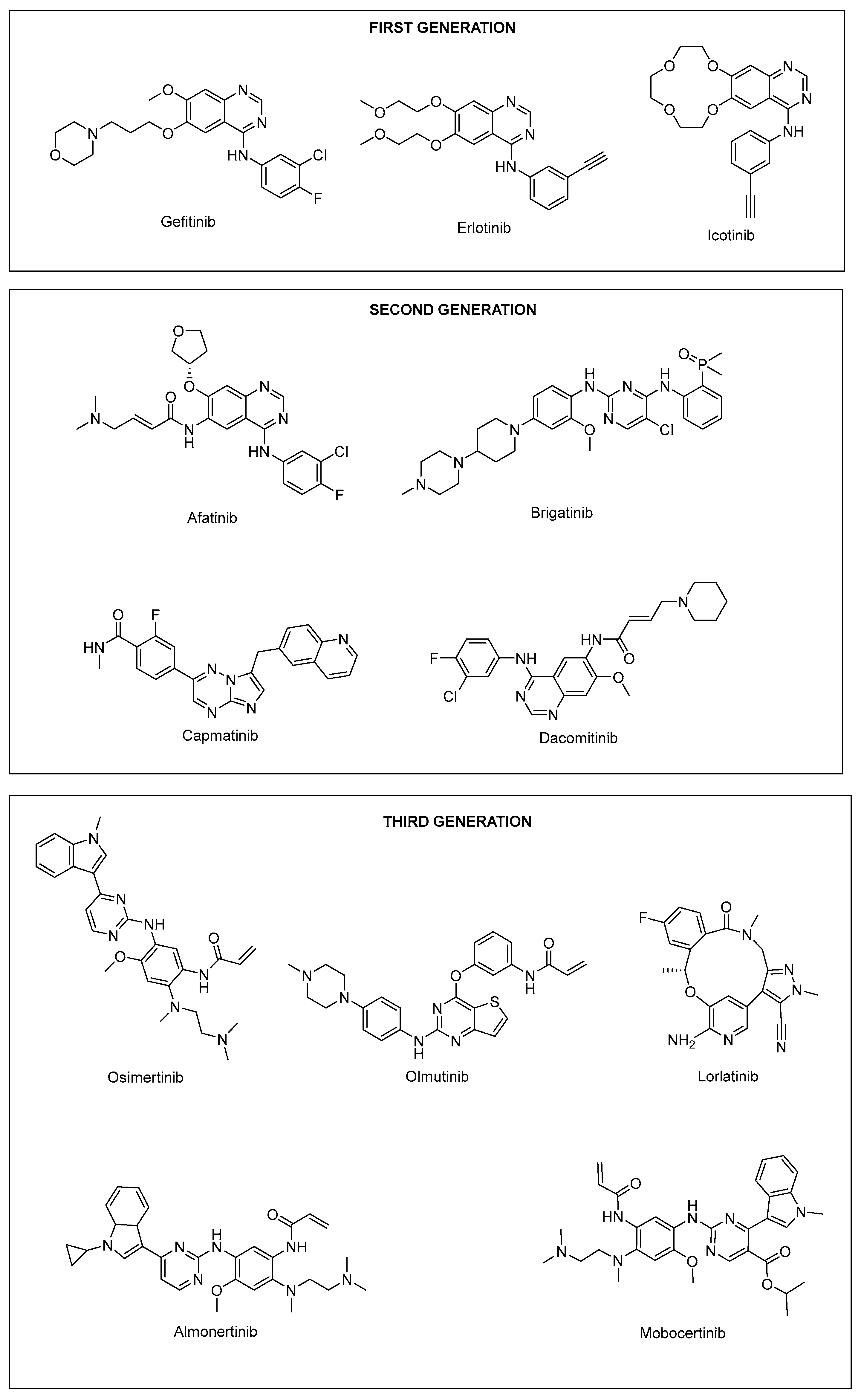
3. TKI Development
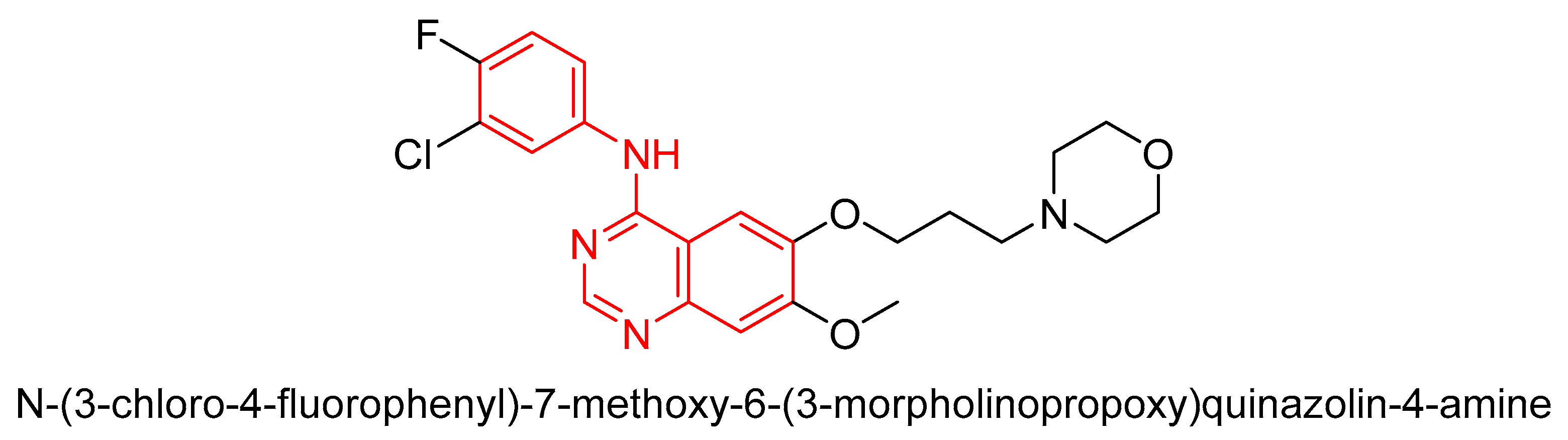
3.1. First-Generation TKI
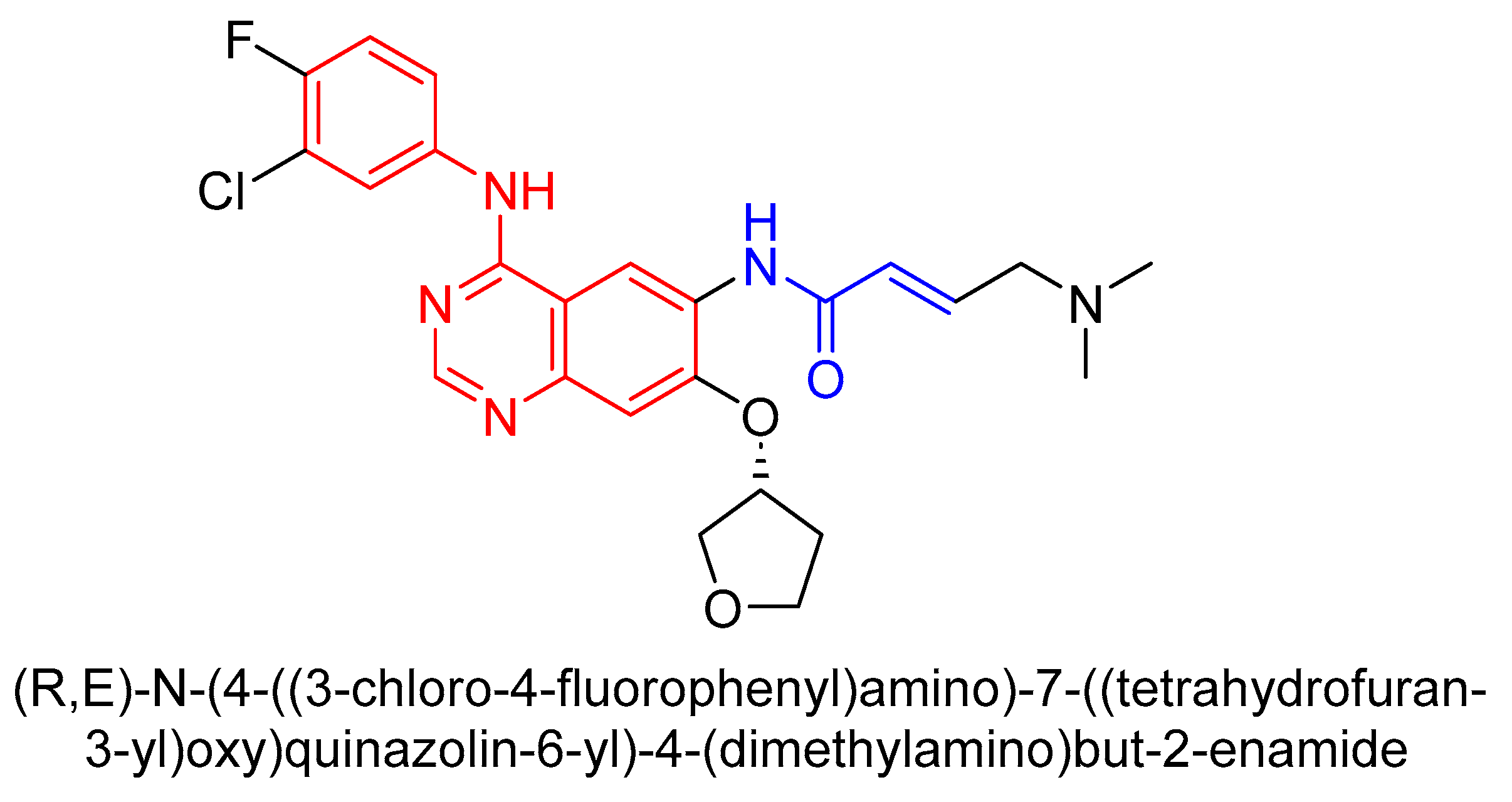
3.2. Second-Generation TKI
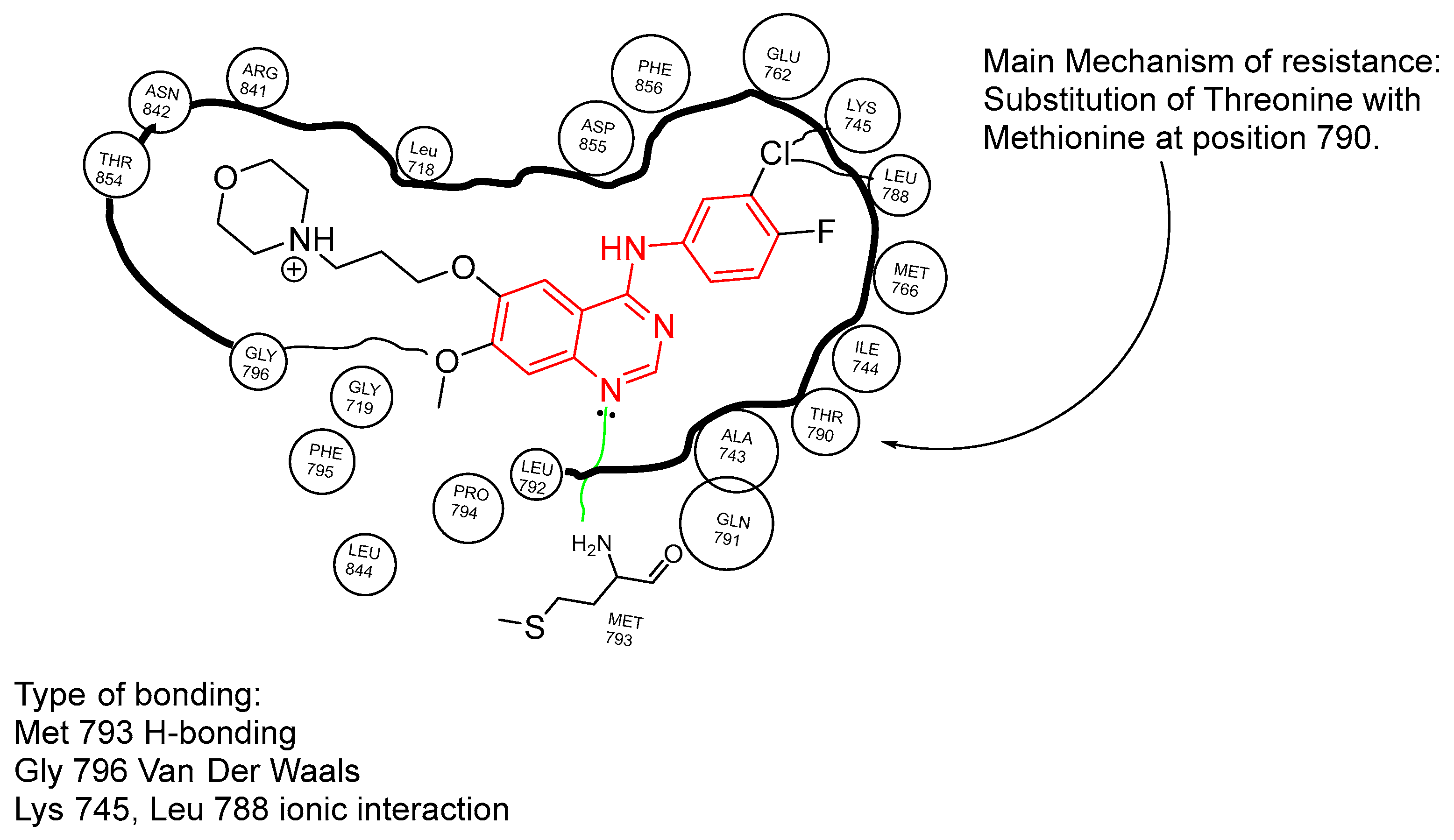
3.2.1. Structure and Mechanism of Second-Generation TKIs
3.2.2. Clinical Trials and Efficacy
3.2.3. Trial Comparison with Erlotinib (Table 3)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Afatinib Group | Erlotinib Group |
|---|---|---|
| PFS | 2.6 months | 1.9 months |
| Overall survival | 7.9 months | 6.9 months |
| Objective response rate (%) | 5.5 | 2.8 |
| Disease control rate (%) | 50.5 | 39.5 |
| Grade 3 adverse effects (%) | 57.1 | 57.5 |
3.2.4. Trial Comparison with Cisplatin (Table 4)
| Characteristic | Afatinib Group | Cisplatin Group |
|---|---|---|
| PFS | 11.1 months | 6.9 months |
| Overall survival | 33.3 months | 21.1 months |
| Tumor size change (%) | −56.0 | −23.0 |
| Adverse effects discontinuation (%) | 8.0 | 12.0 |
3.2.5. Key Findings
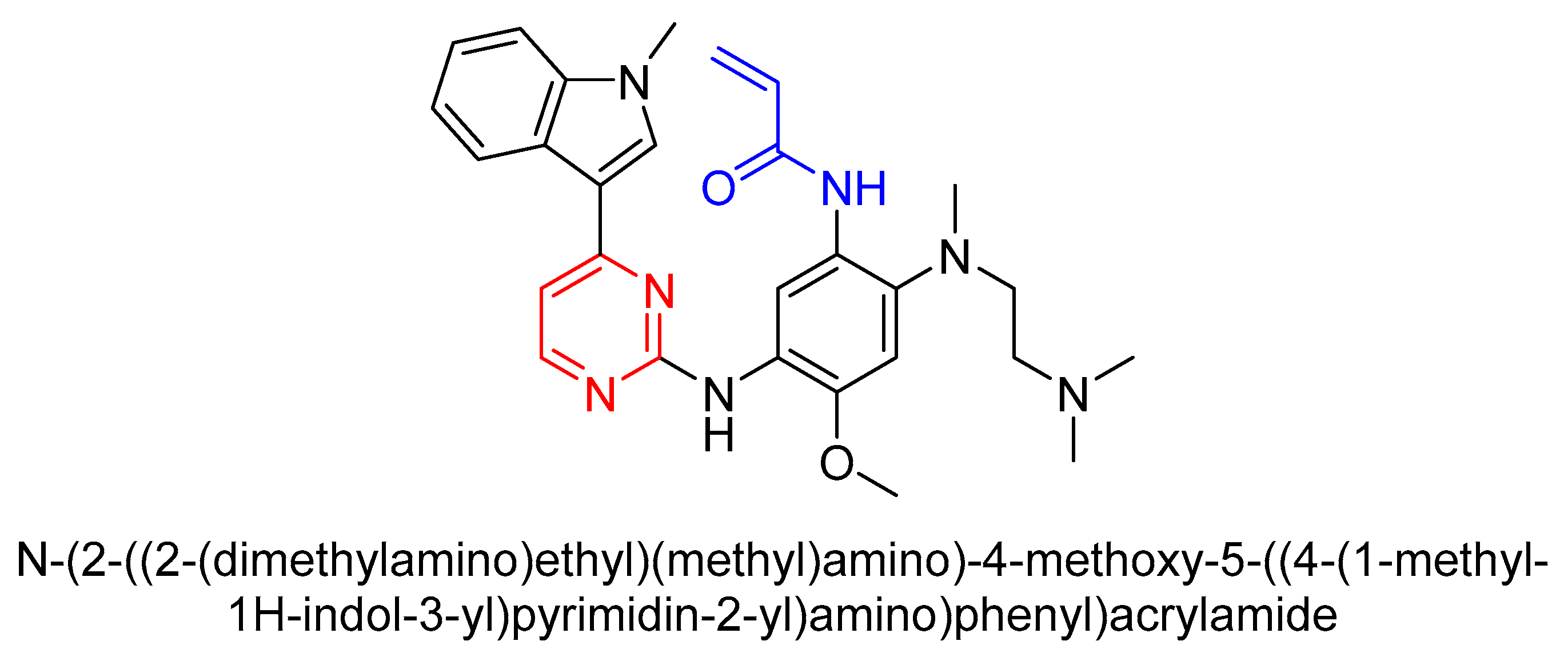
3.3. Third-Generation TKI
4. Future for TKIs
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

References
- National Health Sevice (NHS). Overveiw—Cancer. Available online: https://www.england.nhs.uk/statistics/statistical-work-areas/cancer-patient-experience-survey/ (accessed on 11 September 2024).
- NIH, National Cancer Institiute. How Cancer Is Diagnosed. 17 January 2023. Available online: https://www.cancer.gov/about-cancer/understanding/statistics (accessed on 11 September 2024).
- Roy Chowdhury, S.; Bohara, A.K. Measuring the societal burden of cancer: A case of lost productivity in Nepal. Public Health 2020, 185, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Landeiro, F.; Harris, C.; Groves, D.; O’Neill, S.; Jandu, K.S.; Tacconi, E.M.C.; Field, S.; Patel, N.; Göpfert, A.; Hagson, H.; et al. The economic burden of cancer, coronary heart disease, dementia, and stroke in England in 2018, with projection to 2050: An evaluation of two cohort studies. Lancet Healthy Longev. 2024, 5, e514–e523. [Google Scholar] [CrossRef]
- Chen, S.; Cao, Z.; Prettner, K.; Kuhn, M.; Yang, J.; Jiao, L.; Wang, Z.; Li, W.; Geldsetzer, P.; Bärnighausen, T.; et al. Estimates and Projections of the Global Economic Cost of 29 Cancers in 204 Countries and Territories From 2020 to 2050. JAMA Oncol. 2023, 9, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Macmillan Cancer Support, Research, Cancer Statistics in the UK. Available online: https://www.macmillan.org.uk/about-us/what-we-do/research/cancer-statistics-fact-sheet (accessed on 16 September 2024).
- Karati, D.; Mahadik, K.R.; Trivedi, P.; Kumar, D. Alkylating Agents, the Road Less Traversed, Changing Anticancer Therapy. Anticancer Agents Med. Chem. 2022, 22, 1478–1495. [Google Scholar] [CrossRef]
- Peters, G.J. Novel Developments in the Use of Antimetabolites. Nucleosides Nucleotides Nucleic Acids 2014, 33, 358–374. [Google Scholar] [CrossRef]
- Kümler, I.; Knoop, A.S.; Jessing, C.A.R.; Ejlertsen, B.; Nielsen, D.L. Review of hormone-based treatments in postmenopausal patients with advanced breast cancer focusing on aromatase inhibitors and fulvestrant. ESMO Open 2016, 1, e000062. [Google Scholar] [CrossRef]
- Stanton, R.A.; Gernert, K.M.; Nettles, J.H.; Aneja, R. Drugs that target dynamic microtubules: A new molecular perspective. Med. Res. Rev. 2011, 31, 443–481. [Google Scholar] [CrossRef] [PubMed]
- Yip, H.Y.K.; Papa, A. Signaling Pathways in Cancer: Therapeutic Targets, Combinatorial Treatments, and New Developments. Cells 2021, 10, 659. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, G.; Rakshit, S.; Sarkar, K. HDAC inhibitors: Targets for tumor therapy, immune modulation and lung diseases. Transl. Oncol. 2022, 16, 101312. [Google Scholar] [CrossRef]
- Bharti, A.C.; Vishnoi, K.; Singh, S.M.; Aggarwal, B.B. Chapter 1—Pathways Linked to Cancer Chemoresistance and Their Targeting by Nutraceuticals. In Role of Nutraceuticals in Cancer Chemosensitization; Bharti, A.C., Aggarwal, B.B., Eds.; Academic Press: Cambridge, MA, USA, 2018; Volume 2, pp. 1–30. [Google Scholar]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 5460033, Cisplatin; National Library of Medicine (US): Bethesda, MD, USA, 2004. [Google Scholar]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef]
- Zhang, C.; Xu, C.; Gao, X.; Yao, Q. Platinum-based drugs for cancer therapy and anti-tumor strategies. Theranostics 2022, 12, 2115–2132. [Google Scholar] [CrossRef] [PubMed]
- Cazeau, N. Mobile Health Interventions: Examining Medication Adherence Outcomes Among Patients with Cancer. Clin. J. Oncol. Nurs. 2021, 25, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Al Musaimi, O. Peptide Therapeutics: Unveiling the Potential against Cancer—A Journey through 1989. Cancers 2024, 16, 1032. Available online: https://www.mdpi.com/2072-6694/16/5/1032 (accessed on 16 September 2024). [CrossRef]
- Ogitani, Y.; Abe, Y.; Iguchi, T.; Yamaguchi, J.; Terauchi, T.; Kitamura, M.; Goto, K.; Goto, M.; Oitate, M.; Yukinaga, H.; et al. Wide application of a novel topoisomerase I inhibitor-based drug conjugation technology. Bioorganic Med. Chem. Lett. 2016, 26, 5069–5072. [Google Scholar] [CrossRef]
- Lahiry, P.; Torkamani, A.; Schork, N.J.; Hegele, R.A. Kinase mutations in human disease: Interpreting genotype-phenotype relationships. Nat. Rev. Genet. 2010, 11, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Longo, P.A.; Tarrant, M.K.; Kim, K.; Head, S.; Leahy, D.J.; Cole, P.A. Mechanistic insights into the activation of oncogenic forms of EGF receptor. Nat. Struct. Mol. Biol. 2011, 18, 1388–1393. [Google Scholar] [CrossRef]
- van den Bent, M.J.; Brandes, A.A.; Rampling, R.; Kouwenhoven, M.C.; Kros, J.M.; Carpentier, A.F.; Clement, P.M.; Frenay, M.; Campone, M.; Baurain, J.F.; et al. Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: EORTC brain tumor group study 26034. J. Clin. Oncol. 2009, 27, 1268–1274. [Google Scholar] [CrossRef]
- Fiala, O.; Pesek, M.; Finek, J.; Benesova, L.; Bortlicek, Z.; Minarik, M. Comparison of EGFR-TKI and chemotherapy in the first-line treatment of advanced EGFR mutation-positive NSCLC. Neoplasma 2013, 60, 425–431. [Google Scholar] [CrossRef]
- Eek, D.; Krohe, M.; Mazar, I.; Horsfield, A.; Pompilus, F.; Friebe, R.; Shields, A.L. Patient-reported preferences for oral versus intravenous administration for the treatment of cancer: A review of the literature. Patient Prefer. Adherence 2016, 10, 1609–1621. [Google Scholar] [CrossRef]
- Bergonzini, C.; Leonetti, A.; Tiseo, M.; Giovannetti, E.; Peters, G.J. Is there a role for dacomitinib, a second-generation irreversible inhibitor of the epidermal-growth factor receptor tyrosine kinase, in advanced non-small cell lung cancer? Expert Opin. Pharmacother. 2020, 21, 1287–1298. [Google Scholar] [CrossRef]
- MRC Protein Phosphoryltion and Ubiquitylation Unit, University of Dundee, Inhibitors Approved for Clinical Use.
- Culy, C.R.; Faulds, D. Gefitinib. Drugs 2002, 62, 2237–2248. [Google Scholar] [CrossRef] [PubMed]
- Gridelli, C.; Bareschino, M.A.; Schettino, C.; Rossi, A.; Maione, P.; Ciardiello, F. Erlotinib in Non-Small Cell Lung Cancer Treatment: Current Status and Future Development. Oncologist 2007, 12, 840–849. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Fu, Y.; Wang, D.; Wang, J. Icotinib enhances lung cancer cell radiosensitivity in vitro and in vivo by inhibiting MAPK/ERK and AKT activation. Clin. Exp. Pharmacol. Physiol. 2018, 45, 969–977. [Google Scholar] [CrossRef]
- Wang, M.; Yuang-Chi Chang, A. Molecular mechanism of action and potential biomarkers of growth inhibition of synergistic combination of afatinib and dasatinib against gefitinib-resistant non-small cell lung cancer cells. Oncotarget 2018, 9, 16533–16546. [Google Scholar] [CrossRef]
- Markham, A. Brigatinib: First Global Approval. Drugs 2017, 77, 1131–1135. [Google Scholar] [CrossRef]
- Vansteenkiste, J.F.; Van De Kerkhove, C.; Wauters, E.; Van Mol, P. Capmatinib for the treatment of non-small cell lung cancer. Expert Rev. Anticancer Ther. 2019, 19, 659–671. [Google Scholar] [CrossRef]
- Santarpia, M.; Liguori, A.; Karachaliou, N.; Gonzalez-Cao, M.; Daffinà, M.G.; D’Aveni, A.; Marabello, G.; Altavilla, G.; Rosell, R. Osimertinib in the treatment of non-small-cell lung cancer: Design, development and place in therapy. Lung Cancer Targets Ther. 2017, 8, 109–125. [Google Scholar] [CrossRef]
- Kou, S.-B.; Lin, Z.-Y.; Wang, B.-L.; Shi, J.-H.; Liu, Y.-X. Evaluation of the binding behavior of olmutinib (HM61713) with model transport protein: Insights from spectroscopic and molecular docking studies. J. Mol. Struct. 2021, 1224, 129024. [Google Scholar] [CrossRef]
- Yang, J.; Gong, W. Lorlatinib for the treatment of anaplastic lymphoma kinase-positive non-small cell lung cancer. Expert Rev. Clin. Pharmacol. 2019, 12, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-P.; Hung, T.-H.; Lusvarghi, S.; Chu, Y.-H.; Hsiao, S.-H.; Huang, Y.-H.; Chang, Y.-T.; Ambudkar, S.V. The third-generation EGFR inhibitor almonertinib (HS-10296) resensitizes ABCB1-overexpressing multidrug-resistant cancer cells to chemotherapeutic drugs. Biochem. Pharmacol. 2021, 188, 114516. [Google Scholar] [CrossRef]
- Duke, E.S.; Stapleford, L.; Drezner, N.; Amatya, A.K.; Mishra-Kalyani, P.S.; Shen, Y.-L.; Maxfield, K.; Zirkelbach, J.F.; Bi, Y.; Liu, J.; et al. FDA Approval Summary: Mobocertinib for Metastatic Non–Small Cell Lung Cancer with EGFR Exon 20 Insertion Mutations. Clin. Cancer Res. 2023, 29, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, N.; Iqbal, N. Human Epidermal Growth Factor Receptor 2 (HER2) in Cancers: Overexpression and Therapeutic Implications. Mol. Biol. Int. 2014, 2014, 852748. [Google Scholar] [CrossRef]
- Singh, S.; Sadhukhan, S.; Sonawane, A. 20 years since the approval of first EGFR-TKI, gefitinib: Insight and foresight. Biochim. Et Biophys. Acta (BBA)—Rev. Cancer 2023, 1878, 188967. [Google Scholar] [CrossRef]
- Burgess, A.W. EGFR family: Structure physiology signalling and therapeutic targets†. Growth Factors 2008, 26, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Pretelli, G.; Spagnolo, C.C.; Ciappina, G.; Santarpia, M.; Pasello, G. Overview on Therapeutic Options in Uncommon EGFR Mutant Non-Small Cell Lung Cancer (NSCLC): New Lights for an Unmet Medical Need. Int. J. Mol. Sci. 2023, 24, 8878. [Google Scholar] [CrossRef] [PubMed]
- Verma, N.; Rai, A.K.; Kaushik, V.; Brünnert, D.; Chahar, K.R.; Pandey, J.; Goyal, P. Identification of gefitinib off-targets using a structure-based systems biology approach; their validation with reverse docking and retrospective data mining. Sci. Rep. 2016, 6, 33949. [Google Scholar]
- da Cunha Santos, G.; Shepherd, F.A.; Tsao, M.S. EGFR mutations and lung cancer. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 49–69. [Google Scholar]
- Kumar, R. John Mendelsohn’s journey in cancer biology and therapy. Cancer Biol. Ther. 2020, 21, 389–390. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 123631, Gefitinib; National Library of Medicine (US): Bethesda, MD, USA, 2004. [Google Scholar]
- Koizumi, F.; Kanzawa, F.; Ueda, Y.; Koh, Y.; Tsukiyama, S.; Taguchi, F.; Tamura, T.; Saijo, N.; Nishio, K. Synergistic interaction between the EGFR tyrosine kinase inhibitor gefitinib (“Iressa”) and the DNA topoisomerase I inhibitor CPT-11 (irinotecan) in human colorectal cancer cells. Int. J. Cancer 2004, 108, 464–472. [Google Scholar] [CrossRef]
- Rawluk, J.; Waller, C.F. Gefitinib. In Small Molecules in Oncology; Martens, U.M., Ed.; Springer International Publishing: Cham, Switzerland, 2018; pp. 235–246. [Google Scholar] [CrossRef]
- Maemondo, M.; Inoue, A.; Kobayashi, K.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Harada, M.; Yoshizawa, H.; Kinoshita, I.; et al. Gefitinib or Chemotherapy for Non–Small-Cell Lung Cancer with Mutated EGFR. N. Engl. J. Med. 2010, 362, 2380–2388. [Google Scholar] [CrossRef]
- American Cancer Society. Cancer Facts and Figures; American Cancer Society: Atlanta, GA, USA, 2023. [Google Scholar]
- Walser, T.; Cui, X.; Yanagawa, J.; Lee, J.M.; Heinrich, E.; Lee, G.; Sharma, S.; Dubinett, S.M. Smoking and Lung Cancer. Proc. Am. Thorac. Soc. 2008, 5, 811–815. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Li, J. Medicine is not health care, food is health care: Plant metabolic engineering, diet and human health. New Phytol. 2017, 216, 699–719. [Google Scholar] [CrossRef] [PubMed]
- Gefitinib Drug Approval Package (fda.gov). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2003/021399_iressa.cfm (accessed on 11 September 2024).
- Onitsuka, T.; Uramoto, H.; Nose, N.; Takenoyama, M.; Hanagiri, T.; Sugio, K.; Yasumoto, K. Acquired resistance to gefitinib: The contribution of mechanisms other than the T790M, MET, and HGF status. Lung Cancer 2010, 68, 198–203. [Google Scholar] [CrossRef]
- Wecker, H.; Waller, C.F. Afatinib. In Small Molecules in Oncology; Martens, U.M., Ed.; Springer International Publishing: Cham, Switzerland, 2018; pp. 199–215. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 10184653, Afatinib; National Library of Medicine (US): Bethesda, MD, USA, 2004. [Google Scholar]
- Cohen, P.; Cross, D.; Jänne, P.A. Kinase drug discovery 20 years after imatinib: Progress and future directions. Nat. Rev. Drug Discov. 2021, 20, 551–569. [Google Scholar] [CrossRef]
- 2015 Boehringer Ingelheim International GmbH. GIOTRIF ® (AFATINIB *) 3. DATA OVERVIEW. Available online: https://www.boehringer-ingelheim.com/sites/default/files/Infographics/afatinib_backgrounder.pdf (accessed on 11 September 2024).
- Park, K.; Tan, E.H.; O’Byrne, K.; Zhang, L.; Boyer, M.; Mok, T.; Hirsh, V.; Yang, J.C.; Lee, K.H.; Lu, S.; et al. Afatinib versus gefitinib as first-line treatment of patients with EGFR mutation-positive non-small-cell lung cancer (LUX-Lung 7): A phase 2B, open-label, randomised controlled trial. Lancet Oncol. 2016, 17, 577–589. [Google Scholar] [CrossRef]
- Afatinib Drug Approval Package (fda.gov). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/201292Orig1s000TOC.cfm (accessed on 11 September 2024).
- Dungo, R.T.; Keating, G.M. Afatinib: First global approval. Drugs 2013, 73, 1503–1515. [Google Scholar] [CrossRef]
- Masuda, T.; Miura, S.; Sato, Y.; Tachihara, M.; Bessho, A.; Nakamura, A.; Miyawaki, T.; Yoshimine, K.; Mori, M.; Shiraishi, H.; et al. Significance of micro-EGFR T790M mutations on EGFR-tyrosine kinase inhibitor efficacy in non-small cell lung cancer. Sci. Rep. 2023, 13, 19729. [Google Scholar] [CrossRef]
- Sakuma, Y.; Yamazaki, Y.; Nakamura, Y.; Yoshihara, M.; Matsukuma, S.; Nakayama, H.; Yokose, T.; Kameda, Y.; Koizume, S.; Miyagi, Y. WZ4002, a third-generation EGFR inhibitor, can overcome anoikis resistance in EGFR-mutant lung adenocarcinomas more efficiently than Src inhibitors. Lab. Investig. 2012, 92, 371–383. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 71496458, Osimertinib; National Library of Medicine (US): Bethesda, MD, USA, 2004. [Google Scholar]
- Schmid, S.; Li, J.J.N.; Leighl, N.B. Mechanisms of osimertinib resistance and emerging treatment options. Lung Cancer 2020, 147, 123–129. [Google Scholar] [CrossRef]
- Abourehab, M.A.S.; Alqahtani, A.M.; Youssif, B.G.M.; Gouda, A.M. Globally Approved EGFR Inhibitors: Insights into Their Syntheses, Target Kinases, Biological Activities, Receptor Interactions, and Metabolism. Molecules 2021, 26, 6677. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Vansteenkiste, J.; Planchard, D.; Cho, B.C.; Gray, J.E.; Ohe, Y.; Zhou, C.; Reungwetwattana, T.; Cheng, Y.; Chewaskulyong, B.; et al. Overall Survival with Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC. N. Engl. J. Med. 2019, 382, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2024/208065s030lbl.pdf (accessed on 11 September 2024).
- Greig, S.L. Osimertinib: First Global Approval. Drugs 2016, 76, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.M.; Fujino, T.; Kim, C.; Lee, G.; Lee, Y.H.; Kim, D.W.; Ahn, J.S.; Mitsudomi, T.; Jin, T.; Lee, S.Y. BBT-176, a Novel Fourth-Generation Tyrosine Kinase Inhibitor for Osimertinib-Resistant EGFR Mutations in Non-Small Cell Lung Cancer. Clin. Cancer Res. 2023, 29, 3004–3016. [Google Scholar] [CrossRef]
- Mansour, M.A.; AboulMagd, A.M.; Abbas, S.H.; Abdel-Rahman, H.M.; Abdel-Aziz, M. Insights into fourth generation selective inhibitors of (C797S) EGFR mutation combating non-small cell lung cancer resistance: A critical review. RSC Adv. 2023, 13, 18825–18853. [Google Scholar] [CrossRef]
- Lim, S.M.; Ahn, J.S.; Hong, M.H.; Kim, T.M.; Jung, H.A.; Jung, H.A.; Ou, S.H.I.; Jeong, S.; Lee, Y.H.; Yim, E.; et al. MA07.09 BBT-176, a 4th generation EGFR TKI, for Progressed NSCLC after EGFR TKI Therapy: PK, Safety and Efficacy from Phase 1 Study. J. Thorac. Oncol. 2022, 17, S70–S71. [Google Scholar] [CrossRef]
- Zhang, H.; Xie, R.; Ai-furas, H.; Li, Y.; Wu, Q.; Li, J.; Xu, F.; Xu, T. Design, Synthesis, and Biological Evaluation of Novel EGFR PROTACs Targeting Del19/T790M/C797S Mutation. ACS Med. Chem. Lett. 2022, 13, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Ye, X.; Wu, Y.; Shen, H.; Cai, Z.; Xia, F.; Min, W.; Hou, Y.; Wang, L.; Wang, X.; et al. Design, Synthesis, and Biological Evaluation of Novel EGFR PROTACs Targeting C797S Mutation. J. Med. Chem. 2024, 67, 7283–7300. [Google Scholar] [CrossRef]
- Wang, X.; Qin, Z.; Qiu, W.; Xu, K.; Bai, Y.; Zeng, B.; Ma, Y.; Yang, S.; Shi, Y.; Fan, Y. Novel EGFR inhibitors against resistant L858R/T790M/C797S mutant for intervention of non-small cell lung cancer. Eur. J. Med. Chem. 2024, 277, 116711. [Google Scholar] [CrossRef]
- Kageji, H.; Momose, T.; Nagamoto, Y.; Togashi, N.; Yasumatsu, I.; Nishikawa, Y.; Kihara, K.; Hiramoto, K.; Minami, M.; Kasanuki, N.; et al. Synthesis, activity, and their relationships of 2,4-diaminonicotinamide derivatives as EGFR inhibitors targeting C797S mutation. Bioorganic Med. Chem. Lett. 2024, 98, 129575. [Google Scholar] [CrossRef]
- Lee, E.J.; Oh, S.Y.; Lee, Y.W.; Kim, J.Y.; Kim, M.-J.; Kim, T.H.; Lee, J.B.; Hong, M.H.; Lim, S.M.; Baum, A.; et al. Discovery of a Novel Potent EGFR Inhibitor Against EGFR Activating Mutations and On-Target Resistance in NSCLC. Clin. Cancer Res. 2024, 30, 1582–1594. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Ding, X.; Leng, Z. Immunotherapy-based therapy as a promising treatment for EGFR-mutant advanced non-small cell lung cancer patients after EGFR-TKI resistance. Expert. Rev. Anticancer. Ther. 2023, 23, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Huang, H.; Sun, Y.; Jiang, Q.; Yu, Y. Immunotherapy strategies for EGFR-mutated advanced NSCLC after EGFR tyrosine-kinase inhibitors failure. Front. Oncol. 2023, 13, 1265236. [Google Scholar] [CrossRef]
- Yu, L.; Hu, Y.; Xu, J.; Qiao, R.; Zhong, H.; Han, B.; Xia, J.; Zhong, R. Multi-target angiogenesis inhibitor combined with PD-1 inhibitors may benefit advanced non-small cell lung cancer patients in late line after failure of EGFR-TKI therapy. Int. J. Cancer 2023, 153, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Wang, Z.; Fu, C.; Tao, H.; Liu, C. The efficacy and safety of PD-1 inhibitors for EGFR-mutant non-small cell lung cancer after tyrosine kinase inhibitor failure: A retrospective real-world cohort study. Ann. Transl. Med. 2023, 11, 157. [Google Scholar] [CrossRef]
- Amir, M.; Javed, S. A Review on the Therapeutic Role of TKIs in Case of CML in Combination With Epigenetic Drugs. Front. Genet. 2021, 12, 742802. [Google Scholar] [CrossRef]
- Sahafnejad, Z.; Ramazi, S.; Allahverdi, A. An Update of Epigenetic Drugs for the Treatment of Cancers and Brain Diseases: A Comprehensive Review. Genes 2023, 14, 873. [Google Scholar] [CrossRef]
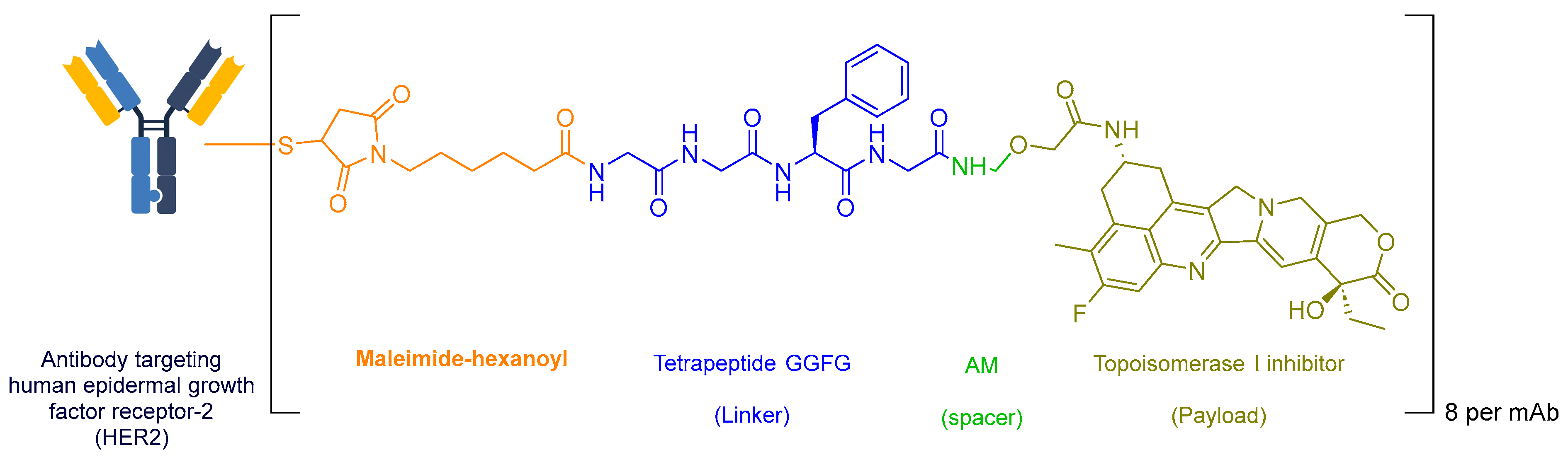
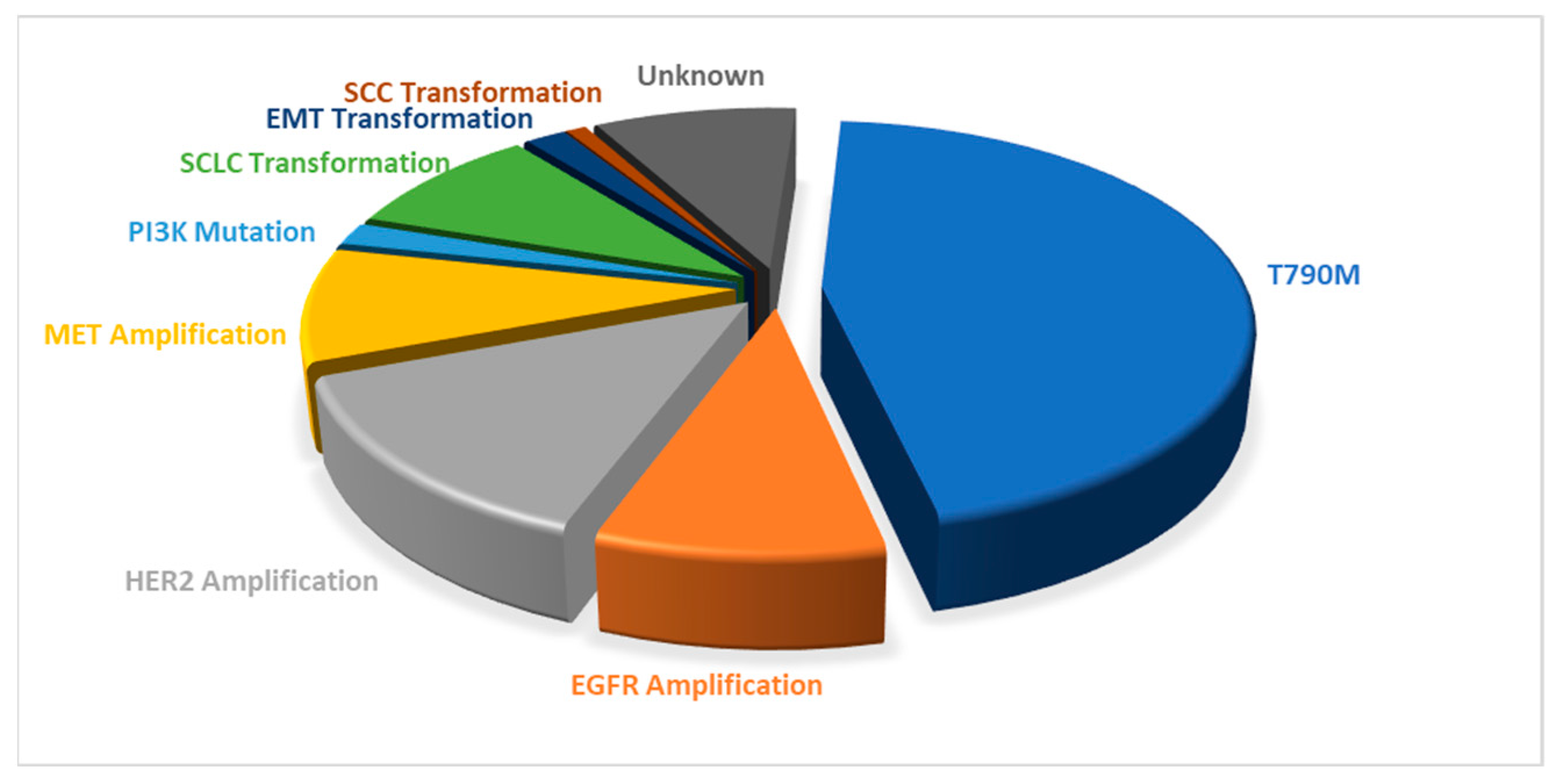
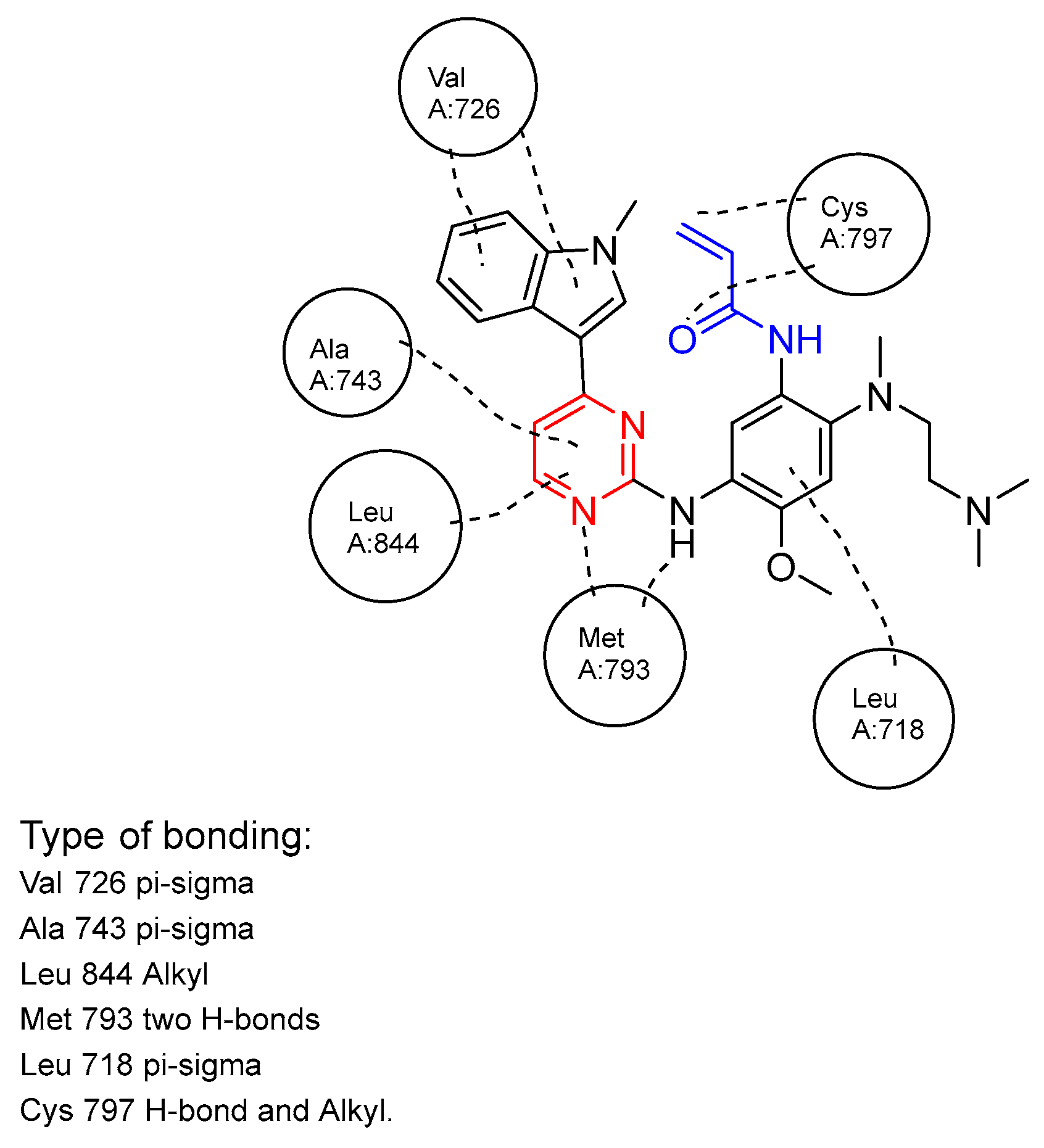
| Name | Mode of Action | Therapeutic Target | Administration in Adults | Approval Year | Reference Number |
|---|---|---|---|---|---|
| First-generation TKI | |||||
| Gefitinib [27] | Reversible inhibitor for EGFR | Advanced/ Metastatic NSCLC with exon 19 and 21 mutations | Oral Tablets | 2002 Japan 2003 FDA | NDA 206995 |
| Erlotinib [28] | Reversible inhibitor for EGFR | Advanced/ Metastatic NSCLC | Oral Tablets | 2004 FDA | NDA 021743 |
| Icotinib [29] | Inhibits MAPK/ERK and AKT in EGFR | Advanced/ Metastatic NSCLC, even exon 19 and 21 mutations | Oral Tablets | 2012 China | 220224-91-5 |
| Second-generation TKI | |||||
| Afatinib [30] | Irreversible inhibitors for EGFRs HER2 and HER4 | Exon 19 or exon L8585 in NSCLC | Oral Tablets | 2013 FDA 2013 EMA | NDA 201292 EMEA/H/C/002280 |
| Brigatinib [31] | Selective inhibitor of ALK, including T790M | ALK-positive metastatic NSCLC | Oral Tablets | 2017 FDA 2018 EMA | NDA 208772 EMEA/H/C/004257 |
| Dacomitinib [25] | Irreversible inhibitor of EGFR | Metastatic NSCLC, with exon 19 or 21 mutations | Oral Tablets | 2018 FDA 2019 EMA | NDA 211288 EMEA/H/C/004094 |
| Capmatinib [32] | Selective inhibitor of MET | Metastatic NSCLC with exon 14 mutation | Oral Tablets | 2020 FDA | NDA 213383 |
| Third-generation TKI | |||||
| Osimertinib [33] | Inhibits EGFR with exon 19 and T790M mutations | Resistant NSCLC from T790M mutation | Oral Tablets | 2015 FDA 2016 EMA | NDA 208065 EMEA/H/C/003905 |
| Olmutinib [34] | Inhibits EGFR with T790M mutation | Resistant NSCLC from T790M mutation | Oral Tablets | 2016 South Korea | HM61713 |
| Lorlatinib [35] | Inhibits ALK and ROS1 | ALK-positive metastatic NSCLC | Oral Tablets | 2018 FDA 2019 EMA | NDA 210868 EMEA/HC/004664 |
| Almonertinib [36] | Inhibits EGFR, including T790M mutations | Advanced/Metastatic NSCLC, with T790M mutation | Oral Tablet | 2020 China | HS-10296 |
| Mobocertinib [37] | Inhibits EGFR with exon 20 mutations | Resistant NSCLC from exon 20 mutation | Oral Tablets | 2021 FDA | NDA 214164 |
| Characteristic | Gefitinib Group | Carboplatin Group | |
|---|---|---|---|
| No. of Patients (% of Patients) | |||
| Response | Complete | 5 (4.4) | 0 |
| Partial | 79 (69.3) | 35 (30.7) | |
| Stabilized NSCLC | 18 (15.8) | 56 (49.1) | |
| Progressive disease | 11 (9.6) | 16 (14.0) | |
| Grade 1 side effect | Diarrhea | 32 | 7 |
| Appetite Loss | 7 | 39 | |
| Rash | 38 | 8 | |
| Neuropathy (sensory) | 0 | 28 | |
| ALT elevation | 20 | 31 | |
| Anemia | 19 | 35 | |
| Thrombocytopenia | 8 | 25 | |
| Progression free | Date | Survival (PFS) | |
| May 2009 | 10.4 months | 5.5 months | |
| December 2009 | 10.8 months | 5.4 months | |
| Characteristic | Osimertinib Group (N = 279) | Gefitinib Group (N = 277) | |
|---|---|---|---|
| No. of Patients (% of Patients) | |||
| Overall survival | At 12 months | 89 | 83 |
| At 24 months | 74 | 59 | |
| At 36 months | 54 | 44 | |
| Any grade of adverse effects | Diarrhea | 167 (60) | 162 (58) |
| Rash/Acne | 164 (59) | 219 (79) | |
| Nail Effects | 108 (39) | 95 (34) | |
| Dry Skin | 106 (38) | 102 (37) | |
| Stomatitis | 82 (29) | 60 (22) | |
| Decreased Appetite | 66 (24) | 58 (21) | |
| Cough | 60 (22) | 50 (18) | |
| Nausea | 55 (20) | 55 (20) | |
| Constipation | 51 (18) | 39 (14) | |
| Anemia | 44 (16) | 27 (10) | |
| Subgroup | No. of Patients | |
|---|---|---|
| Sex | Male | 206 |
| Female | 350 | |
| Age | Under 65 | 298 |
| Over 65 | 258 | |
| Race | Asian | 347 |
| Non-Asian | 209 | |
| Smoking history | Yes | 199 |
| No | 357 | |
| Dose (mg) | Age | Gender | Race | Tumor Size Change (%) |
|---|---|---|---|---|
| 160 to 320 | 43 | Female | Asian | −30.3 |
| 320 | 39 | Male | Asian | 0.0 |
| 320 | 52 | Female | Asian | −12.1 |
| 480 | 67 | Female | Asian | −11.8 |
| 480 | 54 | Male | Asian | −26.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dickerson, H.; Diab, A.; Al Musaimi, O. Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Cancer: Current Use and Future Prospects. Int. J. Mol. Sci. 2024, 25, 10008. https://doi.org/10.3390/ijms251810008
Dickerson H, Diab A, Al Musaimi O. Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Cancer: Current Use and Future Prospects. International Journal of Molecular Sciences. 2024; 25(18):10008. https://doi.org/10.3390/ijms251810008
Chicago/Turabian StyleDickerson, Henry, Ahmad Diab, and Othman Al Musaimi. 2024. "Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Cancer: Current Use and Future Prospects" International Journal of Molecular Sciences 25, no. 18: 10008. https://doi.org/10.3390/ijms251810008
APA StyleDickerson, H., Diab, A., & Al Musaimi, O. (2024). Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Cancer: Current Use and Future Prospects. International Journal of Molecular Sciences, 25(18), 10008. https://doi.org/10.3390/ijms251810008