
Understanding the Role of the SMN Complex Component GEMIN5 and Its Functional Relationship with Demethylase KDM6B in the Flunarizine-Mediated Neuroprotection of Motor Neuron Disease Spinal Muscular Atrophy
 , , , , and
, , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
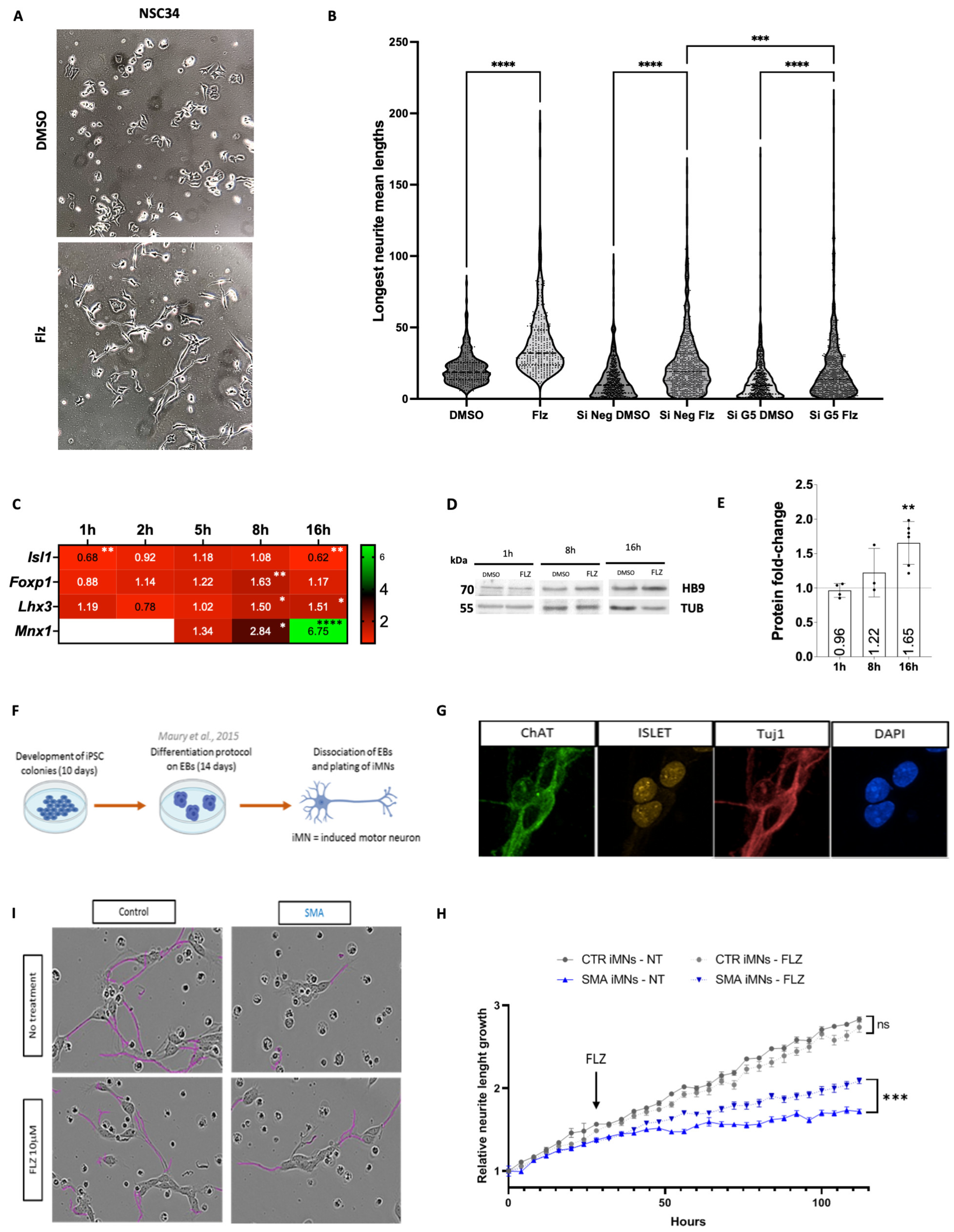
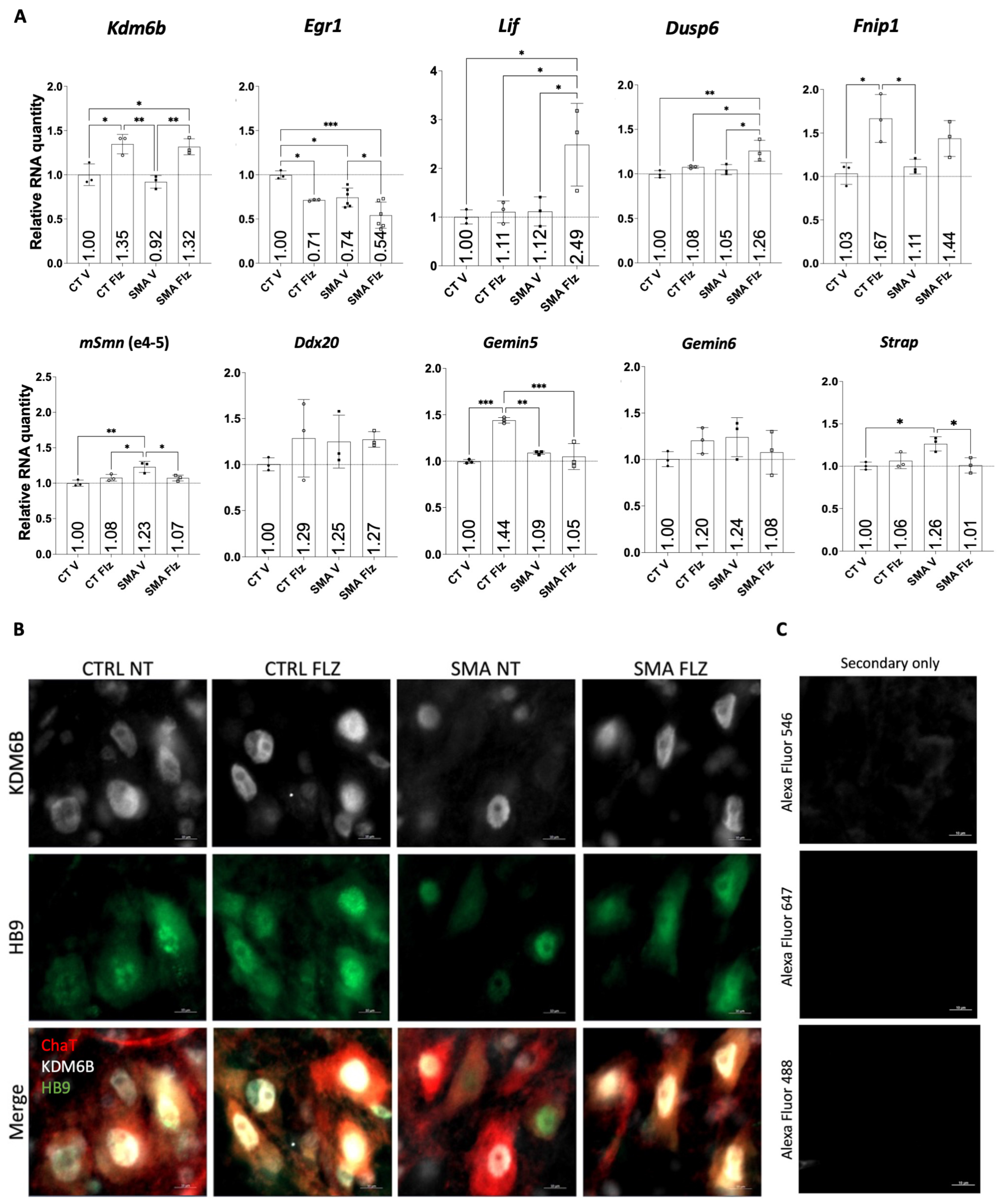
2.1. SMN-Complex Component Gemin5 Expression Levels Are Modulated by Flunarizine
2.2. Expression of Key Motor Neuron Genes Is Modulated by Flunarizine
2.3. Flunarizine-Modulated Transcripts Are Immunoprecipitated by GEMIN5
2.4. Gemin5 Depletion Mimics the Early Time Point of Flunarizine Treatment
2.5. Neurite Outgrowth Is Promoted in Cell Cultures by Flunarizine
2.6. Flunarizine Stimulates KDM6B and HB9 Expression in Motor Neurons of Smn-Deficient Mice
3. Discussion
4. Materials and Methods
4.1. Cell Cultures, Flunarizine Treatment, and Immunodetection
4.2. Neurite Tracking with IncuCyte®
4.3. Transfection Assays
4.4. Immunoblotting Analysis
4.5. RNA Extraction and Expression Analysis
4.6. RNA Immunoprecipitation
4.7. Animal Procedure and Spinal Cord Tissue Experiment
4.8. Immunofluorescence Microscopy, Image Acquisition, Processing, and Analysis
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lefebvre, S.; Bürglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M. Identification and Characterization of a Spinal Muscular Atrophy-Determining Gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Kolb, S.J.; Kissel, J.T. Spinal Muscular Atrophy. Arch. Neurol. 2011, 68, 979–984. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, S.; Burlet, P.; Liu, Q.; Bertrandy, S.; Clermont, O.; Munnich, A.; Dreyfuss, G.; Melki, J. Correlation between Severity and SMN Protein Level in Spinal Muscular Atrophy. Nat. Genet. 1997, 16, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Lorson, C.L.; Hahnen, E.; Androphy, E.J.; Wirth, B. A Single Nucleotide in the SMN Gene Regulates Splicing and Is Responsible for Spinal Muscular Atrophy. Proc. Natl. Acad. Sci. USA 1999, 96, 6307–6311. [Google Scholar] [CrossRef]
- Monani, U.R.; Lorson, C.L.; Parsons, D.W.; Prior, T.W.; Androphy, E.J.; Burghes, A.H.; McPherson, J.D. A Single Nucleotide Difference That Alters Splicing Patterns Distinguishes the SMA Gene SMN1 from the Copy Gene SMN2. Hum. Mol. Genet. 1999, 8, 1177–1183. [Google Scholar] [CrossRef]
- Calucho, M.; Bernal, S.; Alías, L.; March, F.; Venceslá, A.; Rodríguez-Álvarez, F.J.; Aller, E.; Fernández, R.M.; Borrego, S.; Millán, J.M.; et al. Correlation between SMA Type and SMN2 Copy Number Revisited: An Analysis of 625 Unrelated Spanish Patients and a Compilation of 2834 Reported Cases. Neuromuscul. Disord. NMD 2018, 28, 208–215. [Google Scholar] [CrossRef]
- Bowerman, M. Recent Advances and Future Perspectives in the Development of Therapeutic Approaches for Neurodegenerative Diseases. Brain Sci. 2020, 10, 633. [Google Scholar] [CrossRef]
- Finkel, R.S.; Fischbeck, K.H. Maybe Too Much of a Good Thing in Gene Therapy. Nat. Neurosci. 2021, 24, 901–902. [Google Scholar] [CrossRef] [PubMed]
- Chaytow, H.; Faller, K.M.E.; Huang, Y.-T.; Gillingwater, T.H. Spinal Muscular Atrophy: From Approved Therapies to Future Therapeutic Targets for Personalized Medicine. Cell Rep. Med. 2021, 2, 100346. [Google Scholar] [CrossRef]
- Mendell, J.R.; Al-Zaidy, S.; Shell, R.; Arnold, W.D.; Rodino-Klapac, L.R.; Prior, T.W.; Lowes, L.; Alfano, L.; Berry, K.; Church, K.; et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1713–1722. [Google Scholar] [CrossRef]
- Mercuri, E.; Darras, B.T.; Chiriboga, C.A.; Day, J.W.; Campbell, C.; Connolly, A.M.; Iannaccone, S.T.; Kirschner, J.; Kuntz, N.L.; Saito, K.; et al. Nusinersen versus Sham Control in Later-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2018, 378, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Poirier, A.; Weetall, M.; Heinig, K.; Bucheli, F.; Schoenlein, K.; Alsenz, J.; Bassett, S.; Ullah, M.; Senn, C.; Ratni, H.; et al. Risdiplam Distributes and Increases SMN Protein in Both the Central Nervous System and Peripheral Organs. Pharmacol. Res. Perspect. 2018, 6, e00447. [Google Scholar] [CrossRef] [PubMed]
- Schrank, B.; Götz, R.; Gunnersen, J.M.; Ure, J.M.; Toyka, K.V.; Smith, A.G.; Sendtner, M. Inactivation of the Survival Motor Neuron Gene, a Candidate Gene for Human Spinal Muscular Atrophy, Leads to Massive Cell Death in Early Mouse Embryos. Proc. Natl. Acad. Sci. USA 1997, 94, 9920–9925. [Google Scholar] [CrossRef] [PubMed]
- Hsieh-Li, H.M.; Chang, J.G.; Jong, Y.J.; Wu, M.H.; Wang, N.M.; Tsai, C.H.; Li, H. A Mouse Model for Spinal Muscular Atrophy. Nat. Genet. 2000, 24, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Pellizzoni, L. Chaperoning Ribonucleoprotein Biogenesis in Health and Disease. EMBO Rep. 2007, 8, 340–345. [Google Scholar] [CrossRef]
- Massenet, S.; Pellizzoni, L.; Paushkin, S.; Mattaj, I.W.; Dreyfuss, G. The SMN Complex Is Associated with snRNPs throughout Their Cytoplasmic Assembly Pathway. Mol. Cell. Biol. 2002, 22, 6533–6541. [Google Scholar] [CrossRef]
- Meister, G.; Eggert, C.; Fischer, U. SMN-Mediated Assembly of RNPs: A Complex Story. Trends Cell Biol. 2002, 12, 472–478. [Google Scholar] [CrossRef]
- Donlin-Asp, P.G.; Bassell, G.J.; Rossoll, W. A Role for the Survival of Motor Neuron Protein in mRNP Assembly and Transport. Curr. Opin. Neurobiol. 2016, 39, 53–61. [Google Scholar] [CrossRef]
- Liu, Q.; Dreyfuss, G. A Novel Nuclear Structure Containing the Survival of Motor Neurons Protein. EMBO J. 1996, 15, 3555–3565. [Google Scholar] [CrossRef]
- Gall, J.G. Cajal Bodies: The First 100 Years. Annu. Rev. Cell Dev. Biol. 2000, 16, 273–300. [Google Scholar] [CrossRef]
- Schilling, M.; Prusty, A.B.; Boysen, B.; Oppermann, F.S.; Riedel, Y.L.; Husedzinovic, A.; Rasouli, H.; König, A.; Ramanathan, P.; Reymann, J.; et al. TOR Signaling Regulates Liquid Phase Separation of the SMN Complex Governing snRNP Biogenesis. Cell Rep. 2021, 35, 109277. [Google Scholar] [CrossRef] [PubMed]
- Nussbacher, J.K.; Tabet, R.; Yeo, G.W.; Lagier-Tourenne, C. Disruption of RNA Metabolism in Neurological Diseases and Emerging Therapeutic Interventions. Neuron 2019, 102, 294–320. [Google Scholar] [CrossRef]
- Sapaly, D.; Dos Santos, M.; Delers, P.; Biondi, O.; Quérol, G.; Houdebine, L.; Khoobarry, K.; Girardet, F.; Burlet, P.; Armand, A.-S.; et al. Small-Molecule Flunarizine Increases SMN Protein in Nuclear Cajal Bodies and Motor Function in a Mouse Model of Spinal Muscular Atrophy. Sci. Rep. 2018, 8, 2075. [Google Scholar] [CrossRef]
- Younis, I.; Berg, M.; Kaida, D.; Dittmar, K.; Wang, C.; Dreyfuss, G. Rapid-Response Splicing Reporter Screens Identify Differential Regulators of Constitutive and Alternative Splicing. Mol. Cell. Biol. 2010, 30, 1718–1728. [Google Scholar] [CrossRef] [PubMed]
- von Lewinski, F.; Keller, B.U. Ca2+, Mitochondria and Selective Motoneuron Vulnerability: Implications for ALS. Trends Neurosci. 2005, 28, 494–500. [Google Scholar] [CrossRef]
- Zhang, Z.; Lotti, F.; Dittmar, K.; Younis, I.; Wan, L.; Kasim, M.; Dreyfuss, G. SMN Deficiency Causes Tissue-Specific Perturbations in the Repertoire of snRNAs and Widespread Defects in Splicing. Cell 2008, 133, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Doktor, T.K.; Hua, Y.; Andersen, H.S.; Brøner, S.; Liu, Y.H.; Wieckowska, A.; Dembic, M.; Bruun, G.H.; Krainer, A.R.; Andresen, B.S. RNA-Sequencing of a Mouse-Model of Spinal Muscular Atrophy Reveals Tissue-Wide Changes in Splicing of U12-Dependent Introns. Nucleic Acids Res. 2017, 45, 395–416. [Google Scholar] [CrossRef]
- Sapaly, D.; Delers, P.; Coridon, J.; Salman, B.; Letourneur, F.; Dumont, F.; Lefebvre, S. The Small-Molecule Flunarizine in Spinal Muscular Atrophy Patient Fibroblasts Impacts on the Gemin Components of the SMN Complex and TDP43, an RNA-Binding Protein Relevant to Motor Neuron Diseases. Front. Mol. Biosci. 2020, 7, 55. [Google Scholar] [CrossRef]
- Black, J.C.; Van Rechem, C.; Whetstine, J.R. Histone Lysine Methylation Dynamics: Establishment, Regulation, and Biological Impact. Mol. Cell 2012, 48, 491–507. [Google Scholar] [CrossRef]
- d’Ydewalle, C.; Ramos, D.M.; Pyles, N.J.; Ng, S.-Y.; Gorz, M.; Pilato, C.M.; Ling, K.; Kong, L.; Ward, A.J.; Rubin, L.L.; et al. The Antisense Transcript SMN-AS1 Regulates SMN Expression and Is a Novel Therapeutic Target for Spinal Muscular Atrophy. Neuron 2017, 93, 66–79. [Google Scholar] [CrossRef]
- Yang, L.; Zha, Y.; Ding, J.; Ye, B.; Liu, M.; Yan, C.; Dong, Z.; Cui, H.; Ding, H.-F. Histone Demethylase KDM6B Has an Anti-Tumorigenic Function in Neuroblastoma by Promoting Differentiation. Oncogenesis 2019, 8, 3. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Cho, H.; Lee, J.W.; Lee, S.-K. The Histone Demethylase Kdm6b Regulates Subtype Diversification of Mouse Spinal Motor Neurons during Development. Nat. Commun. 2022, 13, 958. [Google Scholar] [CrossRef] [PubMed]
- Arber, S.; Han, B.; Mendelsohn, M.; Smith, M.; Jessell, T.M.; Sockanathan, S. Requirement for the Homeobox Gene Hb9 in the Consolidation of Motor Neuron Identity. Neuron 1999, 23, 659–674. [Google Scholar] [CrossRef]
- Sager, R.A.; Woodford, M.R.; Backe, S.J.; Makedon, A.M.; Baker-Williams, A.J.; DiGregorio, B.T.; Loiselle, D.R.; Haystead, T.A.; Zachara, N.E.; Prodromou, C.; et al. Post-Translational Regulation of FNIP1 Creates a Rheostat for the Molecular Chaperone Hsp90. Cell Rep. 2019, 26, 1344–1356.e5. [Google Scholar] [CrossRef]
- Bizarro, J.; Dodré, M.; Huttin, A.; Charpentier, B.; Schlotter, F.; Branlant, C.; Verheggen, C.; Massenet, S.; Bertrand, E. NUFIP and the HSP90/R2TP Chaperone Bind the SMN Complex and Facilitate Assembly of U4-Specific Proteins. Nucleic Acids Res. 2015, 43, 8973–8989. [Google Scholar] [CrossRef]
- Battle, D.J.; Lau, C.-K.; Wan, L.; Deng, H.; Lotti, F.; Dreyfuss, G. The Gemin5 Protein of the SMN Complex Identifies snRNAs. Mol. Cell 2006, 23, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Yong, J.; Kasim, M.; Bachorik, J.L.; Wan, L.; Dreyfuss, G. Gemin5 Delivers snRNA Precursors to the SMN Complex for snRNP Biogenesis. Mol. Cell 2010, 38, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, C.E.; Cunningham, D.; Venkataramany, A.S.; Chandler, D.S. Heat Increases Full-Length SMN Splicing: Promise for Splice-Augmenting Therapies for SMA. Hum. Genet. 2022, 141, 239–256. [Google Scholar] [CrossRef]
- Manford, A.G.; Mena, E.L.; Shih, K.Y.; Gee, C.L.; McMinimy, R.; Martínez-González, B.; Sherriff, R.; Lew, B.; Zoltek, M.; Rodríguez-Pérez, F.; et al. Structural Basis and Regulation of the Reductive Stress Response. Cell 2021, 184, 5375–5390.e16. [Google Scholar] [CrossRef]
- Yin, Y.; Xu, D.; Mao, Y.; Xiao, L.; Sun, Z.; Liu, J.; Zhou, D.; Xu, Z.; Liu, L.; Fu, T.; et al. FNIP1 Regulates Adipocyte Browning and Systemic Glucose Homeostasis in Mice by Shaping Intracellular Calcium Dynamics. J. Exp. Med. 2022, 219, e20212491. [Google Scholar] [CrossRef]
- Reyes, N.L.; Banks, G.B.; Tsang, M.; Margineantu, D.; Gu, H.; Djukovic, D.; Chan, J.; Torres, M.; Liggitt, H.D.; Hirenallur-S, D.K.; et al. Fnip1 Regulates Skeletal Muscle Fiber Type Specification, Fatigue Resistance, and Susceptibility to Muscular Dystrophy. Proc. Natl. Acad. Sci. USA 2015, 112, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Cashman, N.R.; Durham, H.D.; Blusztajn, J.K.; Oda, K.; Tabira, T.; Shaw, I.T.; Dahrouge, S.; Antel, J.P. Neuroblastoma × Spinal Cord (NSC) Hybrid Cell Lines Resemble Developing Motor Neurons. Dev. Dyn. 1992, 194, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Sims, T.J.; Vaughn, J.E. The Generation of Neurons Involved in an Early Reflex Pathway of Embryonic Mouse Spinal Cord. J. Comp. Neurol. 1979, 183, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Klingauf, M.; Stanĕk, D.; Neugebauer, K.M. Enhancement of U4/U6 Small Nuclear Ribonucleoprotein Particle Association in Cajal Bodies Predicted by Mathematical Modeling. Mol. Biol. Cell 2006, 17, 4972–4981. [Google Scholar] [CrossRef]
- Sawyer, I.A.; Sturgill, D.; Sung, M.-H.; Hager, G.L.; Dundr, M. Cajal Body Function in Genome Organization and Transcriptome Diversity. BioEssays News Rev. Mol. Cell. Dev. Biol. 2016, 38, 1197–1208. [Google Scholar] [CrossRef]
- Carvalho, T.; Almeida, F.; Calapez, A.; Lafarga, M.; Berciano, M.T.; Carmo-Fonseca, M. The Spinal Muscular Atrophy Disease Gene Product, SMN: A Link between snRNP Biogenesis and the Cajal (Coiled) Body. J. Cell Biol. 1999, 147, 715–728. [Google Scholar] [CrossRef]
- Renvoisé, B.; Quérol, G.; Verrier, E.R.; Burlet, P.; Lefebvre, S. A Role for Protein Phosphatase PP1γ in SMN Complex Formation and Subnuclear Localization to Cajal Bodies. J. Cell Sci. 2012, 125 Pt 12, 2862–2874. [Google Scholar] [CrossRef]
- Šimčíková, D.; Gelles-Watnick, S.; Neugebauer, K.M. Tudor–Dimethylarginine Interactions: The Condensed Version. Trends Biochem. Sci. 2023, 48, 689–698. [Google Scholar] [CrossRef]
- Grimmler, M.; Otter, S.; Peter, C.; Müller, F.; Chari, A.; Fischer, U. Unrip, a Factor Implicated in Cap-Independent Translation, Associates with the Cytosolic SMN Complex and Influences Its Intracellular Localization. Hum. Mol. Genet. 2005, 14, 3099–3111. [Google Scholar] [CrossRef]
- Lee, S.; Shang, Y.; Redmond, S.A.; Urisman, A.; Tang, A.A.; Li, K.H.; Burlingame, A.L.; Pak, R.A.; Jovičić, A.; Gitler, A.D.; et al. Activation of HIPK2 Promotes ER Stress-Mediated Neurodegeneration in Amyotrophic Lateral Sclerosis. Neuron 2016, 91, 41–55. [Google Scholar] [CrossRef]
- Weydt, P.; Yuen, E.C.; Ransom, B.R.; Möller, T. Increased Cytotoxic Potential of Microglia from ALS-Transgenic Mice. Glia 2004, 48, 179–182. [Google Scholar] [CrossRef] [PubMed]
- De Conti, L.; Akinyi, M.V.; Mendoza-Maldonado, R.; Romano, M.; Baralle, M.; Buratti, E. TDP-43 Affects Splicing Profiles and Isoform Production of Genes Involved in the Apoptotic and Mitotic Cellular Pathways. Nucleic Acids Res. 2015, 43, 8990–9005. [Google Scholar] [CrossRef] [PubMed]
- Curtis, R.; Scherer, S.S.; Somogyi, R.; Adryan, K.M.; Ip, N.Y.; Zhu, Y.; Lindsay, R.M.; DiStefano, P.S. Retrograde Axonal Transport of LIF Is Increased by Peripheral Nerve Injury: Correlation with Increased LIF Expression in Distal Nerve. Neuron 1994, 12, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Giess, R.; Beck, M.; Goetz, R.; Nitsch, R.M.; Toyka, K.V.; Sendtner, M. Potential Role of LIF as a Modifier Gene in the Pathogenesis of Amyotrophic Lateral Sclerosis. Neurology 2000, 54, 1003–1005. [Google Scholar] [CrossRef]
- Henderson, C.E.; Phillips, H.S.; Pollock, R.A.; Davies, A.M.; Lemeulle, C.; Armanini, M.; Simmons, L.; Moffet, B.; Vandlen, R.A.; Koliatsos, V.E.; et al. GDNF: A Potent Survival Factor for Motoneurons Present in Peripheral Nerve and Muscle. Science 1994, 266, 1062–1064. [Google Scholar] [CrossRef]
- Machado, V.; Haas, S.J.-P.; von Bohlen Und Halbach, O.; Wree, A.; Krieglstein, K.; Unsicker, K.; Spittau, B. Growth/Differentiation Factor-15 Deficiency Compromises Dopaminergic Neuron Survival and Microglial Response in the 6-Hydroxydopamine Mouse Model of Parkinson’s Disease. Neurobiol. Dis. 2016, 88, 1–15. [Google Scholar] [CrossRef]
- Nichterwitz, S.; Nijssen, J.; Storvall, H.; Schweingruber, C.; Comley, L.H.; Allodi, I.; van der Lee, M.; Deng, Q.; Sandberg, R.; Hedlund, E. LCM-Seq Reveals Unique Transcriptional Adaptation Mechanisms of Resistant Neurons and Identifies Protective Pathways in Spinal Muscular Atrophy. Genome Res. 2020, 30, 1083–1096. [Google Scholar] [CrossRef]
- Burgess, R.W.; Nguyen, Q.T.; Son, Y.J.; Lichtman, J.W.; Sanes, J.R. Alternatively Spliced Isoforms of Nerve- and Muscle-Derived Agrin: Their Roles at the Neuromuscular Junction. Neuron 1999, 23, 33–44. [Google Scholar] [CrossRef]
- Solomon, E.R.; Caldwell, K.K.; Allan, A.M. A Novel Method for the Normalization of ChIP-qPCR Data. MethodsX 2021, 8, 101504. [Google Scholar] [CrossRef]
- Workman, E.; Kalda, C.; Patel, A.; Battle, D.J. Gemin5 Binds to the Survival Motor Neuron mRNA to Regulate SMN Expression. J. Biol. Chem. 2015, 290, 15662–15669. [Google Scholar] [CrossRef]
- Francisco-Velilla, R.; Fernandez-Chamorro, J.; Dotu, I.; Martinez-Salas, E. The Landscape of the Non-Canonical RNA-Binding Site of Gemin5 Unveils a Feedback Loop Counteracting the Negative Effect on Translation. Nucleic Acids Res. 2018, 46, 7339–7353. [Google Scholar] [CrossRef] [PubMed]
- Lampe, S.; Kunze, M.; Scholz, A.; Brauß, T.F.; Winslow, S.; Simm, S.; Keller, M.; Heidler, J.; Wittig, I.; Brüne, B.; et al. Identification of the TXNIP IRES and Characterization of the Impact of Regulatory IRES Trans-Acting Factors. Biochim. Biophys. Acta Gene Regul. Mech. 2018, 1861, 147–157. [Google Scholar] [CrossRef] [PubMed]
- McWhorter, M.L.; Monani, U.R.; Burghes, A.H.M.; Beattie, C.E. Knockdown of the Survival Motor Neuron (Smn) Protein in Zebrafish Causes Defects in Motor Axon Outgrowth and Pathfinding. J. Cell Biol. 2003, 162, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Patel, T.; Hammelman, J.; Aziz, S.; Jang, S.; Closser, M.; Michaels, T.L.; Blum, J.A.; Gifford, D.K.; Wichterle, H. Transcriptional Dynamics of Murine Motor Neuron Maturation in Vivo and in Vitro. Nat. Commun. 2022, 13, 5427. [Google Scholar] [CrossRef] [PubMed]
- Maury, Y.; Côme, J.; Piskorowski, R.A.; Salah-Mohellibi, N.; Chevaleyre, V.; Peschanski, M.; Martinat, C.; Nedelec, S. Combinatorial analysis of developmental cues efficiently converts human pluripotent stem cells into multiple neuronal subtypes. Nat. Biotechnol. 2015, 33, 89–96. [Google Scholar] [CrossRef]
- Chang, T.; Zheng, W.; Tsark, W.; Bates, S.; Huang, H.; Lin, R.-J.; Yee, J.-K. Brief Report: Phenotypic Rescue of Induced Pluripotent Stem Cell-Derived Motoneurons of a Spinal Muscular Atrophy Patient. Stem Cells 2011, 29, 2090–2093. [Google Scholar] [CrossRef]
- Boza-Morán, M.G.; Martínez-Hernández, R.; Bernal, S.; Wanisch, K.; Also-Rallo, E.; Le Heron, A.; Alías, L.; Denis, C.; Girard, M.; Yee, J.-K.; et al. Decay in Survival Motor Neuron and Plastin 3 Levels during Differentiation of iPSC-Derived Human Motor Neurons. Sci. Rep. 2015, 5, 11696. [Google Scholar] [CrossRef]
- Mentis, G.Z.; Blivis, D.; Liu, W.; Drobac, E.; Crowder, M.E.; Kong, L.; Alvarez, F.J.; Sumner, C.J.; O’Donovan, M.J. Early Functional Impairment of Sensory-Motor Connectivity in a Mouse Model of Spinal Muscular Atrophy. Neuron 2011, 69, 453–467. [Google Scholar] [CrossRef]
- Branchu, J.; Biondi, O.; Chali, F.; Collin, T.; Leroy, F.; Mamchaoui, K.; Makoukji, J.; Pariset, C.; Lopes, P.; Massaad, C.; et al. Shift from Extracellular Signal-Regulated Kinase to AKT/cAMP Response Element-Binding Protein Pathway Increases Survival-Motor-Neuron Expression in Spinal-Muscular-Atrophy-Like Mice and Patient Cells. J. Neurosci. 2013, 33, 4280–4294. [Google Scholar] [CrossRef]
- Yoo, D.H.; Im, Y.S.; Oh, J.Y.; Gil, D.; Kim, Y.-O. DUSP6 Is a Memory Retention Feedback Regulator of ERK Signaling for Cellular Resilience of Human Pluripotent Stem Cells in Response to Dissociation. Sci. Rep. 2023, 13, 5683. [Google Scholar] [CrossRef]
- Polymenidou, M.; Lagier-Tourenne, C.; Hutt, K.R.; Huelga, S.C.; Moran, J.; Liang, T.Y.; Ling, S.-C.; Sun, E.; Wancewicz, E.; Mazur, C.; et al. Long Pre-mRNA Depletion and RNA Missplicing Contribute to Neuronal Vulnerability from Loss of TDP-43. Nat. Neurosci. 2011, 14, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Kwong, L.K.; Neumann, M.; Sampathu, D.M.; Lee, V.M.-Y.; Trojanowski, J.Q. TDP-43 Proteinopathy: The Neuropathology Underlying Major Forms of Sporadic and Familial Frontotemporal Lobar Degeneration and Motor Neuron Disease. Acta Neuropathol. 2007, 114, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Corbet, G.A.; Wheeler, J.R.; Parker, R.; Weskamp, K. TDP43 Ribonucleoprotein Granules: Physiologic Function to Pathologic Aggregates. RNA Biol. 2021, 18, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Faravelli, I.; Riboldi, G.M.; Rinchetti, P.; Lotti, F. The SMN Complex at the Crossroad between RNA Metabolism and Neurodegeneration. Int. J. Mol. Sci. 2023, 24, 2247. [Google Scholar] [CrossRef]
- Francisco-Velilla, R. The RNA-Binding Protein Gemin5 Binds Directly to the Ribosome and Regulates Global Translation. Nucleic Acids Res. 2016, 44, 8335–8351. [Google Scholar] [CrossRef]
- Garcia-Moreno, M.; Noerenberg, M.; Ni, S.; Järvelin, A.I.; González-Almela, E.; Lenz, C.E.; Bach-Pages, M.; Cox, V.; Avolio, R.; Davis, T.; et al. System-Wide Profiling of RNA-Binding Proteins Uncovers Key Regulators of Virus Infection. Mol. Cell 2019, 74, 196–211.e11. [Google Scholar] [CrossRef]
- Pacheco, A.; de Quinto, S.L.; Ramajo, J.; Fernández, N.; Martínez-Salas, E. A Novel Role for Gemin5 in mRNA Translation. Nucleic Acids Res. 2009, 37, 582. [Google Scholar] [CrossRef]
- Kour, S.; Rajan, D.S.; Fortuna, T.R.; Anderson, E.N.; Ward, C.; Lee, Y.; Lee, S.; Shin, Y.B.; Chae, J.-H.; Choi, M.; et al. Loss of Function Mutations in GEMIN5 Cause a Neurodevelopmental Disorder. Nat. Commun. 2021, 12, 2558. [Google Scholar] [CrossRef]
- Rajan, D.S.; Kour, S.; Fortuna, T.R.; Cousin, M.A.; Barnett, S.S.; Niu, Z.; Babovic-Vuksanovic, D.; Klee, E.W.; Kirmse, B.; Innes, M.; et al. Autosomal Recessive Cerebellar Atrophy and Spastic Ataxia in Patients With Pathogenic Biallelic Variants in GEMIN5. Front. Cell Dev. Biol. 2022, 10, 783762. [Google Scholar] [CrossRef]
- Francisco-Velilla, R.; Embarc-Buh, A.; del Caño-Ochoa, F.; Abellan, S.; Vilar, M.; Alvarez, S.; Fernandez-Jaen, A.; Kour, S.; Rajan, D.S.; Pandey, U.B.; et al. Functional and Structural Deficiencies of Gemin5 Variants Associated with Neurological Disorders. Life Sci. Alliance 2022, 5, e202201403. [Google Scholar] [CrossRef]
- Cascajo-Almenara, M.V.; Juliá-Palacios, N.; Urreizti, R.; Sánchez-Cuesta, A.; Fernández-Ayala, D.M.; García-Díaz, E.; Oliva, C.; O Callaghan, M.D.M.; Paredes-Fuentes, A.J.; Moreno-Lozano, P.J.; et al. Mutations of GEMIN5 Are Associated with Coenzyme Q10 Deficiency: Long-Term Follow-up after Treatment. Eur. J. Hum. Genet. EJHG 2024, 32, 426–434. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, X.; Zhu, G.; Wan, L.; Liang, Y.; Li, N.; Huang, M.; Yang, G. Expanding the Clinical Phenotype and Genetic Spectrum of GEMIN5 Disorders: Early-Infantile Developmental and Epileptic Encephalopathies. Brain Behav. 2024, 14, e3535. [Google Scholar] [CrossRef]
- Rots, D.; Jakub, T.E.; Keung, C.; Jackson, A.; Banka, S.; Pfundt, R.; de Vries, B.B.A.; van Jaarsveld, R.H.; Hopman, S.M.J.; van Binsbergen, E.; et al. The Clinical and Molecular Spectrum of the KDM6B-Related Neurodevelopmental Disorder. Am. J. Hum. Genet. 2023, 110, 963–978. [Google Scholar] [CrossRef] [PubMed]
- Saida, K.; Tamaoki, J.; Sasaki, M.; Haniffa, M.; Koshimizu, E.; Sengoku, T.; Maeda, H.; Kikuchi, M.; Yokoyama, H.; Sakamoto, M.; et al. Pathogenic Variants in the Survival of Motor Neurons Complex Gene GEMIN5 Cause Cerebellar Atrophy. Clin. Genet. 2021, 100, 722–730. [Google Scholar] [CrossRef] [PubMed]
- Embarc-Buh, A.; Francisco-Velilla, R.; Garcia-Martin, J.A.; Abellan, S.; Ramajo, J.; Martinez-Salas, E. Gemin5-Dependent RNA Association with Polysomes Enables Selective Translation of Ribosomal and Histone mRNAs. Cell. Mol. Life Sci. 2022, 79, 490. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Cuvillier, J.M.; Lee, B.; Shen, R.; Lee, J.W.; Lee, S.-K. Fusion Protein Isl1–Lhx3 Specifies Motor Neuron Fate by Inducing Motor Neuron Genes and Concomitantly Suppressing the Interneuron Programs. Proc. Natl. Acad. Sci. USA 2012, 109, 3383–3388. [Google Scholar] [CrossRef]
- Mazzoni, E.O.; Mahony, S.; Closser, M.; Morrison, C.A.; Nedelec, S.; Williams, D.J.; An, D.; Gifford, D.K.; Wichterle, H. Synergistic Binding of Transcription Factors to Cell-Specific Enhancers Programs Motor Neuron Identity. Nat. Neurosci. 2013, 16, 1219–1227. [Google Scholar] [CrossRef]
- Cho, H.-H.; Cargnin, F.; Kim, Y.; Lee, B.; Kwon, R.-J.; Nam, H.; Shen, R.; Barnes, A.P.; Lee, J.W.; Lee, S.; et al. Isl1 Directly Controls a Cholinergic Neuronal Identity in the Developing Forebrain and Spinal Cord by Forming Cell Type-Specific Complexes. PLoS Genet. 2014, 10, e1004280. [Google Scholar] [CrossRef]
- Erb, M.; Lee, B.; Yeon Seo, S.; Lee, J.W.; Lee, S.; Lee, S.-K. The Isl1-Lhx3 Complex Promotes Motor Neuron Specification by Activating Transcriptional Pathways That Enhance Its Own Expression and Formation. eNeuro 2017, 4, ENEURO.0349-16.2017. [Google Scholar] [CrossRef]
- Thaler, J.; Harrison, K.; Sharma, K.; Lettieri, K.; Kehrl, J.; Pfaff, S.L. Active Suppression of Interneuron Programs within Developing Motor Neurons Revealed by Analysis of Homeodomain Factor HB9. Neuron 1999, 23, 675–687. [Google Scholar] [CrossRef]
- Lee, S.; Shen, R.; Cho, H.-H.; Kwon, R.-J.; Seo, S.Y.; Lee, J.W.; Lee, S.-K. STAT3 Promotes Motor Neuron Differentiation by Collaborating with Motor Neuron-Specific LIM Complex. Proc. Natl. Acad. Sci. USA 2013, 110, 11445–11450. [Google Scholar] [CrossRef] [PubMed]
- Liau, E.S.; Jin, S.; Chen, Y.-C.; Liu, W.-S.; Calon, M.; Nedelec, S.; Nie, Q.; Chen, J.-A. Single-Cell Transcriptomic Analysis Reveals Diversity within Mammalian Spinal Motor Neurons. Nat. Commun. 2023, 14, 46. [Google Scholar] [CrossRef] [PubMed]
- Kanning, K.C.; Kaplan, A.; Henderson, C.E. Motor Neuron Diversity in Development and Disease. Annu. Rev. Neurosci. 2010, 33, 409–440. [Google Scholar] [CrossRef] [PubMed]
- Simon, C.M.; Dai, Y.; Van Alstyne, M.; Koutsioumpa, C.; Pagiazitis, J.G.; Chalif, J.I.; Wang, X.; Rabinowitz, J.E.; Henderson, C.E.; Pellizzoni, L.; et al. Converging Mechanisms of P53 Activation Drive Motor Neuron Degeneration in Spinal Muscular Atrophy. Cell Rep. 2017, 21, 3767–3780. [Google Scholar] [CrossRef]
- Delers, P.; Sapaly, D.; Salman, B.; Waard, S.D.; Waard, M.D.; Lefebvre, S. A Link between Agrin Signalling and Cav3.2 at the Neuromuscular Junction in Spinal Muscular Atrophy. Sci. Rep. 2022, 12, 18960. [Google Scholar] [CrossRef]
- Labbadia, J.; Morimoto, R.I. Repression of the Heat Shock Response Is a Programmed Event at the Onset of Reproduction. Mol. Cell 2015, 59, 639–650. [Google Scholar] [CrossRef]
- Merkwirth, C.; Jovaisaite, V.; Durieux, J.; Matilainen, O.; Jordan, S.D.; Quiros, P.M.; Steffen, K.K.; Williams, E.G.; Mouchiroud, L.; Tronnes, S.U.; et al. Two Conserved Histone Demethylases Regulate Mitochondrial Stress-Induced Longevity. Cell 2016, 165, 1209–1223. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salman, B.; Bon, E.; Delers, P.; Cottin, S.; Pasho, E.; Ciura, S.; Sapaly, D.; Lefebvre, S. Understanding the Role of the SMN Complex Component GEMIN5 and Its Functional Relationship with Demethylase KDM6B in the Flunarizine-Mediated Neuroprotection of Motor Neuron Disease Spinal Muscular Atrophy. Int. J. Mol. Sci. 2024, 25, 10039. https://doi.org/10.3390/ijms251810039
Salman B, Bon E, Delers P, Cottin S, Pasho E, Ciura S, Sapaly D, Lefebvre S. Understanding the Role of the SMN Complex Component GEMIN5 and Its Functional Relationship with Demethylase KDM6B in the Flunarizine-Mediated Neuroprotection of Motor Neuron Disease Spinal Muscular Atrophy. International Journal of Molecular Sciences. 2024; 25(18):10039. https://doi.org/10.3390/ijms251810039
Chicago/Turabian StyleSalman, Badih, Emeline Bon, Perrine Delers, Steve Cottin, Elena Pasho, Sorana Ciura, Delphine Sapaly, and Suzie Lefebvre. 2024. "Understanding the Role of the SMN Complex Component GEMIN5 and Its Functional Relationship with Demethylase KDM6B in the Flunarizine-Mediated Neuroprotection of Motor Neuron Disease Spinal Muscular Atrophy" International Journal of Molecular Sciences 25, no. 18: 10039. https://doi.org/10.3390/ijms251810039
APA StyleSalman, B., Bon, E., Delers, P., Cottin, S., Pasho, E., Ciura, S., Sapaly, D., & Lefebvre, S. (2024). Understanding the Role of the SMN Complex Component GEMIN5 and Its Functional Relationship with Demethylase KDM6B in the Flunarizine-Mediated Neuroprotection of Motor Neuron Disease Spinal Muscular Atrophy. International Journal of Molecular Sciences, 25(18), 10039. https://doi.org/10.3390/ijms251810039