
NR5A1/SF-1 Collaborates with Inhibin α and the Androgen Receptor

 , , and
, , and
Abstract
:1. Introduction
2. Results
2.1. Phenotypic Characterization
2.2. Genotypic Characterization
2.3. Characterization of the Identified Variants in NR5A1/SF-1, NR1H2, and INHA
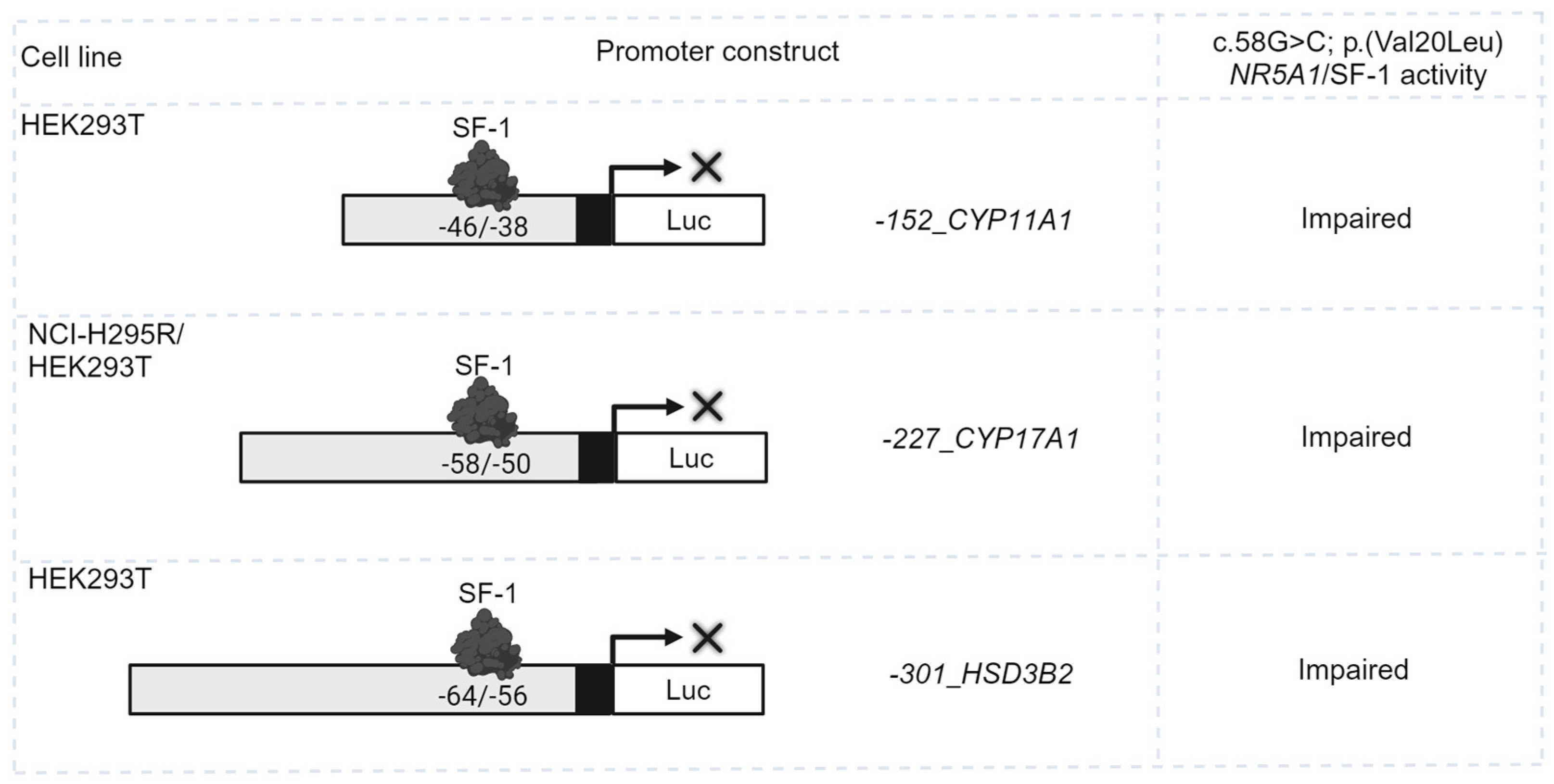
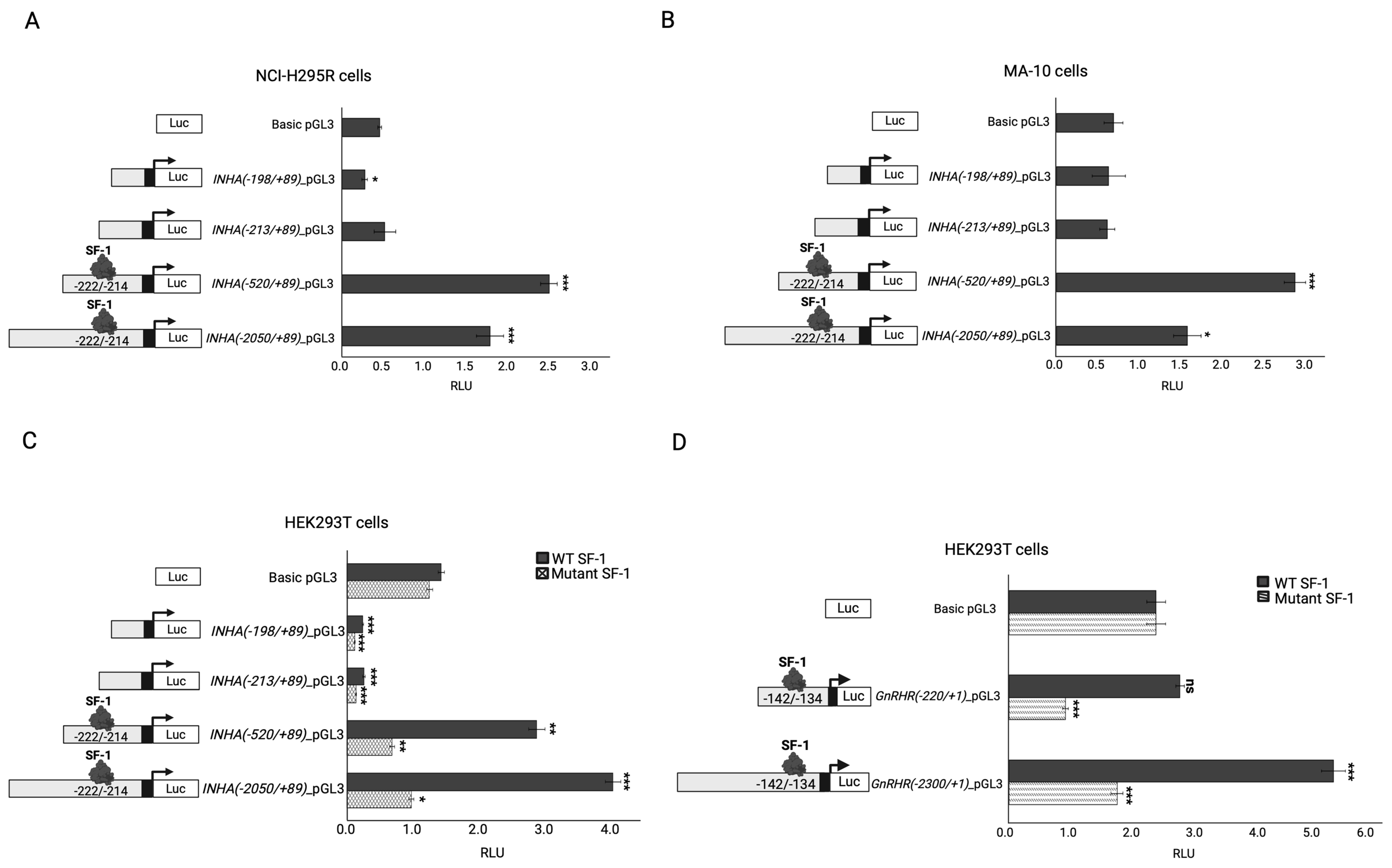
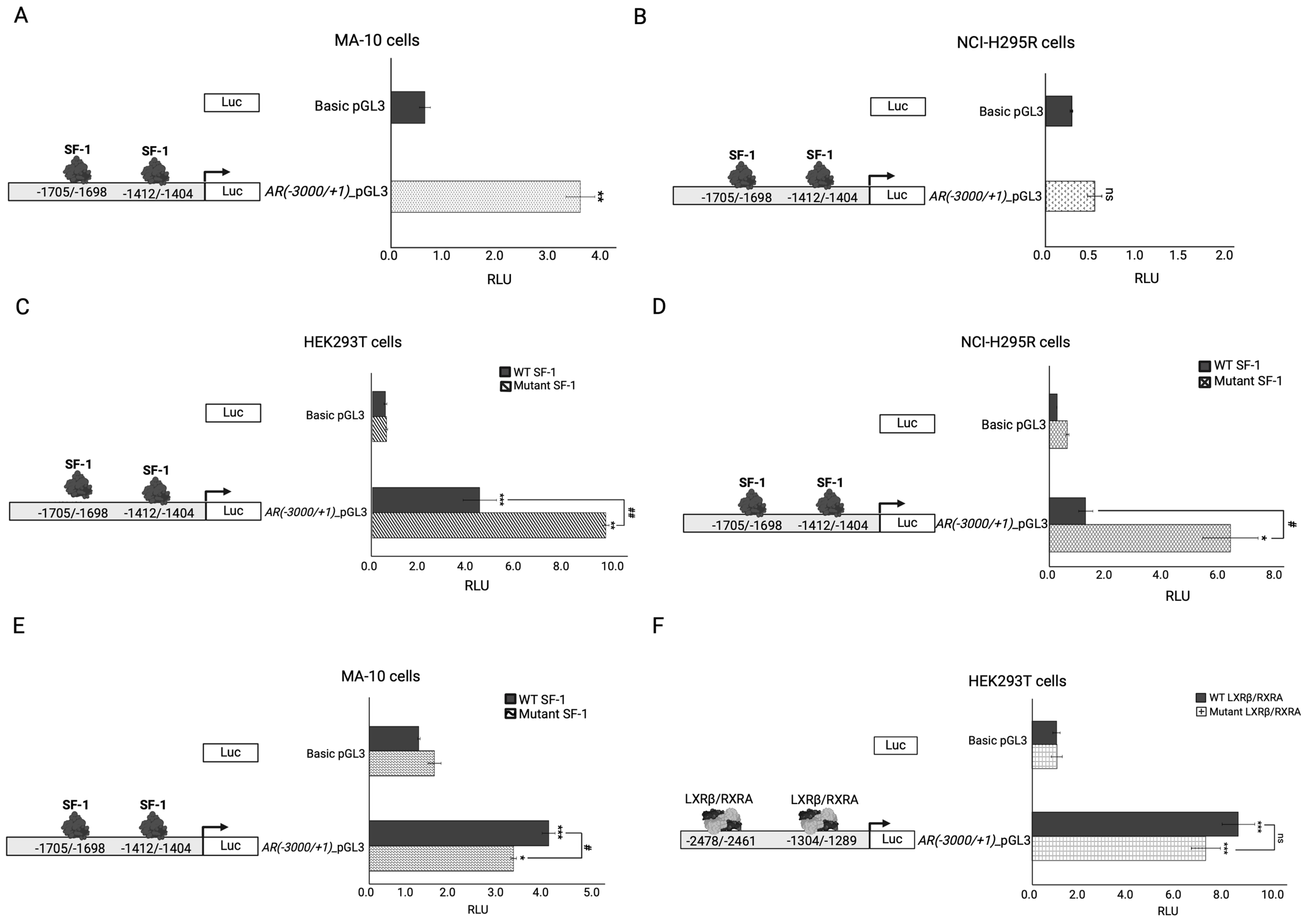
2.4. In Vitro Functional Testing of Selected Variants
3. Discussion
4. Materials and Methods
4.1. Participants
4.2. In Silico Analyses and Variant Classification
4.3. Plasmids
4.4. Cloning
4.5. In Vitro Testing of Transactivation Activity by Dual Luciferase Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schimmer, B.P.; White, P.C. Minireview: Steroidogenic factor 1: Its roles in differentiation, development, and disease. Mol. Endocrinol. 2010, 24, 1322–1337. [Google Scholar] [PubMed]
- Wong, M.; Ikeda, Y.; Luo, X.; Caron, K.M.; Weber, T.J.; Swain, A.; Schimmer, B.P.; Parker, K.L. Steroidogenic factor 1 plays multiple roles in endocrine development and function. Recent. Prog. Horm. Res. 1997, 52, 167–182, discussion 182–184. [Google Scholar]
- Luo, X.; Ikeda, Y.; Parker, K.L. A cell-specific nuclear receptor is essential for adrenal and gonadal development and sexual differentiation. Cell 1994, 77, 481–490. [Google Scholar]
- Bland, M.L.; Jamieson, C.A.; Akana, S.F.; Bornstein, S.R.; Eisenhofer, G.; Dallman, M.F.; Ingraham, H.A. Haploinsufficiency of steroidogenic factor-1 in mice disrupts adrenal development leading to an impaired stress response. Proc. Natl. Acad. Sci. USA 2000, 97, 14488–14493. [Google Scholar] [PubMed]
- Kouri, C.; Sommer, G.; Martinez de Lapiscina, I.; Elzenaty, R.N.; Tack, L.J.W.; Cools, M.; Ahmed, S.F.; Fluck, C.E.; SF1next Study Group. Clinical and genetic characteristics of a large international cohort of individuals with rare NR5A1/SF-1 variants of sex development. EBioMedicine 2024, 99, 104941. [Google Scholar]
- Naamneh Elzenaty, R.; Martinez de Lapiscina, I.; Kouri, C.; Sauter, K.S.; Sommer, G.; Castano, L.; Fluck, C.E.; SF1next Study Group. Characterization of 35 novel NR5A1/SF-1 variants identified in individuals with atypical sexual development: The SF1next study. J. Clin. Endocrinol. Metab. 2024. [Google Scholar] [CrossRef]
- Fabbri-Scallet, H.; de Sousa, L.M.; Maciel-Guerra, A.T.; Guerra-Junior, G.; de Mello, M.P. Mutation update for the NR5A1 gene involved in DSD and infertility. Hum. Mutat. 2020, 41, 58–68. [Google Scholar]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar]
- Camats, N.; Fernandez-Cancio, M.; Audi, L.; Schaller, A.; Fluck, C.E. Broad phenotypes in heterozygous NR5A1 46,XY patients with a disorder of sex development: An oligogenic origin? Eur. J. Hum. Genet. 2018, 26, 1329–1338. [Google Scholar]
- Camats, N.; Fluck, C.E.; Audi, L. Oligogenic Origin of Differences of Sex Development in Humans. Int. J. Mol. Sci. 2020, 21, 1809. [Google Scholar] [CrossRef]
- Martinez de Lapiscina, I.; Kouri, C.; Aurrekoetxea, J.; Sanchez, M.; Naamneh Elzenaty, R.; Sauter, K.S.; Camats, N.; Grau, G.; Rica, I.; Rodriguez, A.; et al. Genetic reanalysis of patients with a difference of sex development carrying the NR5A1/SF-1 variant p.Gly146Ala has discovered other likely disease-causing variations. PLoS ONE 2023, 18, e0287515. [Google Scholar]
- Martinez de LaPiscina, I.; Mahmoud, R.A.; Sauter, K.S.; Esteva, I.; Alonso, M.; Costa, I.; Rial-Rodriguez, J.M.; Rodriguez-Estevez, A.; Vela, A.; Castano, L.; et al. Variants of STAR, AMH and ZFPM2/FOG2 May Contribute towards the Broad Phenotype Observed in 46,XY DSD Patients with Heterozygous Variants of NR5A1. Int. J. Mol. Sci. 2020, 21, 8554. [Google Scholar] [CrossRef]
- Zidoune, H.; Ladjouze, A.; Chellat-Rezgoune, D.; Boukri, A.; Dib, S.A.; Nouri, N.; Tebibel, M.; Sifi, K.; Abadi, N.; Satta, D.; et al. Novel Genomic Variants, Atypical Phenotypes and Evidence of a Digenic/Oligogenic Contribution to Disorders/Differences of Sex Development in a Large North African Cohort. Front. Genet. 2022, 13, 900574. [Google Scholar]
- Wang, H.; Zhang, L.; Wang, N.; Zhu, H.; Han, B.; Sun, F.; Yao, H.; Zhang, Q.; Zhu, W.; Cheng, T.; et al. Next-generation sequencing reveals genetic landscape in 46, XY disorders of sexual development patients with variable phenotypes. Hum. Genet. 2018, 137, 265–277. [Google Scholar]
- Gach, A.; Pinkier, I.; Wysocka, U.; Salacinska, K.; Salachna, D.; Szarras-Czapnik, M.; Pietrzyk, A.; Sakowicz, A.; Nykel, A.; Rutkowska, L.; et al. New findings in oligogenic inheritance of congenital hypogonadotropic hypogonadism. Arch. Med. Sci. 2022, 18, 353–364. [Google Scholar]
- de Filippis, T.; Gelmini, G.; Paraboschi, E.; Vigone, M.C.; Di Frenna, M.; Marelli, F.; Bonomi, M.; Cassio, A.; Larizza, D.; Moro, M.; et al. A frequent oligogenic involvement in congenital hypothyroidism. Hum. Mol. Genet. 2017, 26, 2507–2514. [Google Scholar] [PubMed]
- Oliver-Petit, I.; Edouard, T.; Jacques, V.; Bournez, M.; Cartault, A.; Grunenwald, S.; Savagner, F. Next-Generation Sequencing Analysis Reveals Frequent Familial Origin and Oligogenism in Congenital Hypothyroidism With Dyshormonogenesis. Front. Endocrinol. 2021, 12, 657913. [Google Scholar]
- Sykiotis, G.P.; Plummer, L.; Hughes, V.A.; Au, M.; Durrani, S.; Nayak-Young, S.; Dwyer, A.A.; Quinton, R.; Hall, J.E.; Gusella, J.F.; et al. Oligogenic basis of isolated gonadotropin-releasing hormone deficiency. Proc. Natl. Acad. Sci. USA 2010, 107, 15140–15144. [Google Scholar]
- Hughes, L.A.; McKay-Bounford, K.; Webb, E.A.; Dasani, P.; Clokie, S.; Chandran, H.; McCarthy, L.; Mohamed, Z.; Kirk, J.M.W.; Krone, N.P.; et al. Next generation sequencing (NGS) to improve the diagnosis and management of patients with disorders of sex development (DSD). Endocr. Connect. 2019, 8, 100–110. [Google Scholar]
- Werner, R.; Monig, I.; Lunstedt, R.; Wunsch, L.; Thorns, C.; Reiz, B.; Krause, A.; Schwab, K.O.; Binder, G.; Holterhus, P.M.; et al. New NR5A1 mutations and phenotypic variations of gonadal dysgenesis. PLoS ONE 2017, 12, e0176720. [Google Scholar]
- Mazen, I.; Abdel-Hamid, M.; Mekkawy, M.; Bignon-Topalovic, J.; Boudjenah, R.; El Gammal, M.; Essawi, M.; Bashamboo, A.; McElreavey, K. Identification of NR5A1 Mutations and Possible Digenic Inheritance in 46,XY Gonadal Dysgenesis. Sex. Dev. 2016, 10, 147–151. [Google Scholar] [PubMed]
- de Oliveira, F.R.; Mazzola, T.N.; de Mello, M.P.; Francese-Santos, A.P.; Lemos-Marini, S.H.V.; Maciel-Guerra, A.T.; Hiort, O.; Werner, R.; Guerra-Junior, G.; Fabbri-Scallet, H. DHX37 and NR5A1 Variants Identified in Patients with 46,XY Partial Gonadal Dysgenesis. Life 2023, 13, 1093. [Google Scholar] [CrossRef] [PubMed]
- Audi, L.; Ahmed, S.F.; Krone, N.; Cools, M.; McElreavey, K.; Holterhus, P.M.; Greenfield, A.; Bashamboo, A.; Hiort, O.; Wudy, S.A.; et al. Genetics in Endocrinology: Approaches to molecular genetic diagnosis in the management of differences/disorders of sex development (DSD): Position paper of EU COST Action BM 1303 ‘DSDnet’. Eur. J. Endocrinol. 2018, 179, R197–R206. [Google Scholar] [PubMed]
- Alhomaidah, D.; McGowan, R.; Ahmed, S.F. The current state of diagnostic genetics for conditions affecting sex development. Clin. Genet. 2017, 91, 157–162. [Google Scholar]
- Delot, E.C.; Vilain, E. Towards improved genetic diagnosis of human differences of sex development. Nat. Rev. Genet. 2021, 22, 588–602. [Google Scholar]
- Papadimitriou, S.; Gravel, B.; Nachtegael, C.; De Baere, E.; Loeys, B.; Vikkula, M.; Smits, G.; Lenaerts, T. Toward reporting standards for the pathogenicity of variant combinations involved in multilocus/oligogenic diseases. HGG Adv. 2023, 4, 100165. [Google Scholar]
- Camats, N.; Pandey, A.V.; Fernandez-Cancio, M.; Andaluz, P.; Janner, M.; Toran, N.; Moreno, F.; Bereket, A.; Akcay, T.; Garcia-Garcia, E.; et al. Ten novel mutations in the NR5A1 gene cause disordered sex development in 46,XY and ovarian insufficiency in 46,XX individuals. J. Clin. Endocrinol. Metab. 2012, 97, E1294–E1306. [Google Scholar]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [PubMed]
- Lin, L.; Achermann, J.C. Steroidogenic factor-1 (SF-1, Ad4BP, NR5A1) and disorders of testis development. Sex. Dev. 2008, 2, 200–209. [Google Scholar]
- Bashamboo, A.; McElreavey, K. Human sex-determination and disorders of sex-development (DSD). Semin. Cell Dev. Biol. 2015, 45, 77–83. [Google Scholar]
- Budefeld, T.; Tobet, S.A.; Majdic, G. Altered position of cell bodies and fibers in the ventromedial region in SF-1 knockout mice. Exp. Neurol. 2011, 232, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Dahlman-Wright, K. Liver X receptor in cholesterol metabolism. J. Endocrinol. 2010, 204, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K.M.; Schuster, G.U.; Steffensen, K.R.; Hovatta, O.; Meaney, S.; Hultenby, K.; Johansson, L.C.; Svechnikov, K.; Soder, O.; Gustafsson, J.A. The liver X receptor-beta is essential for maintaining cholesterol homeostasis in the testis. Endocrinology 2005, 146, 2519–2530. [Google Scholar] [CrossRef]
- Nilsson, M.; Stulnig, T.M.; Lin, C.Y.; Yeo, A.L.; Nowotny, P.; Liu, E.T.; Steffensen, K.R. Liver X receptors regulate adrenal steroidogenesis and hypothalamic-pituitary-adrenal feedback. Mol. Endocrinol. 2007, 21, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Rondanino, C.; Ouchchane, L.; Chauffour, C.; Marceau, G.; Dechelotte, P.; Sion, B.; Pons-Rejraji, H.; Janny, L.; Volle, D.H.; Lobaccaro, J.M.; et al. Levels of liver X receptors in testicular biopsies of patients with azoospermia. Fertil. Steril. 2014, 102, 361–371. [Google Scholar] [CrossRef]
- Jarvis, S.; Williamson, C.; Bevan, C.L. Liver X Receptors and Male (In)fertility. Int. J. Mol. Sci. 2019, 20, 5379. [Google Scholar] [CrossRef]
- Barakat, B.; Itman, C.; Mendis, S.H.; Loveland, K.L. Activins and inhibins in mammalian testis development: New models, new insights. Mol. Cell Endocrinol. 2012, 359, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Arslan Ates, E.; Eltan, M.; Sahin, B.; Gurpinar Tosun, B.; Seven Menevse, T.; Geckinli, B.B.; Greenfield, A.; Turan, S.; Bereket, A.; Guran, T. Homozygosity for a novel INHA mutation in two male siblings with hypospadias, primary hypogonadism, and high-normal testicular volume. Eur. J. Endocrinol. 2022, 186, K25–K31. [Google Scholar] [PubMed]
- Sundblad, V.; Chiauzzi, V.A.; Andreone, L.; Campo, S.; Charreau, E.H.; Dain, L. Controversial role of inhibin alpha-subunit gene in the aetiology of premature ovarian failure. Hum. Reprod. 2006, 21, 1154–1160. [Google Scholar] [CrossRef]
- Huhtaniemi, I. The first report on homozygous INHA inactivation in humans. Eur. J. Endocrinol. 2022, 187, C1–C2. [Google Scholar] [CrossRef]
- Matzuk, M.M.; Finegold, M.J.; Mather, J.P.; Krummen, L.; Lu, H.; Bradley, A. Development of cancer cachexia-like syndrome and adrenal tumors in inhibin-deficient mice. Proc. Natl. Acad. Sci. USA 1994, 91, 8817–8821. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Park, Y.; Weck, J.; Mayo, K.E.; Jameson, J.L. Synergistic activation of the inhibin alpha-promoter by steroidogenic factor-1 and cyclic adenosine 3′,5′-monophosphate. Mol. Endocrinol. 2000, 14, 66–81. [Google Scholar] [PubMed]
- Korinek, V.; Barker, N.; Moerer, P.; van Donselaar, E.; Huls, G.; Peters, P.J.; Clevers, H. Depletion of epithelial stem-cell compartments in the small intestine of mice lacking Tcf-4. Nat. Genet. 1998, 19, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Nateri, A.S.; Spencer-Dene, B.; Behrens, A. Interaction of phosphorylated c-Jun with TCF4 regulates intestinal cancer development. Nature 2005, 437, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Lili, L.N.; Farkas, A.E.; Gerner-Smidt, C.; Overgaard, C.E.; Moreno, C.S.; Parkos, C.A.; Capaldo, C.T.; Nusrat, A. Claudin-based barrier differentiation in the colonic epithelial crypt niche involves Hopx/Klf4 and Tcf7l2/Hnf4-alpha cascades. Tissue Barriers 2016, 4, e1214038. [Google Scholar] [CrossRef]
- Angus-Hill, M.L.; Elbert, K.M.; Hidalgo, J.; Capecchi, M.R. T-cell factor 4 functions as a tumor suppressor whose disruption modulates colon cell proliferation and tumorigenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 4914–4919. [Google Scholar] [CrossRef]
- Boj, S.F.; van Es, J.H.; Huch, M.; Li, V.S.; Jose, A.; Hatzis, P.; Mokry, M.; Haegebarth, A.; van den Born, M.; Chambon, P.; et al. Diabetes risk gene and Wnt effector Tcf7l2/TCF4 controls hepatic response to perinatal and adult metabolic demand. Cell 2012, 151, 1595–1607. [Google Scholar] [CrossRef]
- Helgason, A.; Palsson, S.; Thorleifsson, G.; Grant, S.F.; Emilsson, V.; Gunnarsdottir, S.; Adeyemo, A.; Chen, Y.; Chen, G.; Reynisdottir, I.; et al. Refining the impact of TCF7L2 gene variants on type 2 diabetes and adaptive evolution. Nat. Genet. 2007, 39, 218–225. [Google Scholar] [CrossRef]
- Freathy, R.M.; Weedon, M.N.; Bennett, A.; Hypponen, E.; Relton, C.L.; Knight, B.; Shields, B.; Parnell, K.S.; Groves, C.J.; Ring, S.M.; et al. Type 2 diabetes TCF7L2 risk genotypes alter birth weight: A study of 24,053 individuals. Am. J. Hum. Genet. 2007, 80, 1150–1161. [Google Scholar] [CrossRef]
- Gummow, B.M.; Winnay, J.N.; Hammer, G.D. Convergence of Wnt signaling and steroidogenic factor-1 (SF-1) on transcription of the rat inhibin alpha gene. J. Biol. Chem. 2003, 278, 26572–26579. [Google Scholar] [CrossRef]
- Diana, P.; Carvalheira, G.M.G. NIBAN1, Exploring its Roles in Cell Survival Under Stress Context. Front. Cell Dev. Biol. 2022, 10, 867003. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.D.; Kobayashi, T.; Abe, M.; Tada, N.; Adachi, H.; Shiota, A.; Totsuka, Y.; Hino, O. The endoplasmic reticulum stress-inducible protein Niban regulates eIF2alpha and S6K1/4E-BP1 phosphorylation. Biochem. Biophys. Res. Commun. 2007, 360, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Esmaeili, R.; Mohammadi, S.; Jafarbeik-Iravani, N.; Yadegari, F.; Olfatbakhsh, A.; Mazaheri, M.; Kaviani, A.; Rezaee, M.; Majidzadeh, A.K. Expression of SCUBE2 and BCL2 Predicts Favorable Response in ERalpha Positive Breast Cancer. Arch. Iran. Med. 2021, 24, 209–217. [Google Scholar] [CrossRef]
- Lin, Y.C.; Chen, C.C.; Cheng, C.J.; Yang, R.B. Domain and functional analysis of a novel breast tumor suppressor protein, SCUBE2. J. Biol. Chem. 2011, 286, 27039–27047. [Google Scholar] [CrossRef]
- Lin, Y.C.; Roffler, S.R.; Yan, Y.T.; Yang, R.B. Disruption of Scube2 Impairs Endochondral Bone Formation. J. Bone Miner. Res. 2015, 30, 1255–1267. [Google Scholar] [CrossRef]
- Krycer, J.R.; Brown, A.J. Cross-talk between the androgen receptor and the liver X receptor: Implications for cholesterol homeostasis. J. Biol. Chem. 2011, 286, 20637–20647. [Google Scholar] [CrossRef]
- Jorgensen, J.S.; Nilson, J.H. AR suppresses transcription of the LHbeta subunit by interacting with steroidogenic factor-1. Mol. Endocrinol. 2001, 15, 1505–1516. [Google Scholar] [PubMed]
- Mazen, I.; Mekkawy, M.; Kamel, A.; Essawi, M.; Hassan, H.; Abdel-Hamid, M.; Amr, K.; Soliman, H.; El-Ruby, M.; Torky, A.; et al. Advances in genomic diagnosis of a large cohort of Egyptian patients with disorders of sex development. Am. J. Med. Genet. A 2021, 185, 1666–1677. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, A.E.; De Kretser, D.M. Inhibins in normal male physiology. Semin. Reprod. Med. 2004, 22, 177–185. [Google Scholar] [CrossRef]
- Majdic, G.; McNeilly, A.S.; Sharpe, R.M.; Evans, L.R.; Groome, N.P.; Saunders, P.T. Testicular expression of inhibin and activin subunits and follistatin in the rat and human fetus and neonate and during postnatal development in the rat. Endocrinology 1997, 138, 2136–2147. [Google Scholar] [CrossRef]
- Matzuk, M.M.; Finegold, M.J.; Su, J.G.; Hsueh, A.J.; Bradley, A. Alpha-inhibin is a tumour-suppressor gene with gonadal specificity in mice. Nature 1992, 360, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Walton, K.L.; Makanji, Y.; Robertson, D.M.; Harrison, C.A. The synthesis and secretion of inhibins. Vitam. Horm. 2011, 85, 149–184. [Google Scholar] [PubMed]
- Walton, K.L.; Kelly, E.K.; Johnson, K.E.; Robertson, D.M.; Stanton, P.G.; Harrison, C.A. A Novel, More Efficient Approach to Generate Bioactive Inhibins. Endocrinology 2016, 157, 2799–2809. [Google Scholar] [CrossRef]
- Anawalt, B.D.; Bebb, R.A.; Matsumoto, A.M.; Groome, N.P.; Illingworth, P.J.; McNeilly, A.S.; Bremner, W.J. Serum inhibin B levels reflect Sertoli cell function in normal men and men with testicular dysfunction. J. Clin. Endocrinol. Metab. 1996, 81, 3341–3345. [Google Scholar] [PubMed]
- Duval, D.L.; Nelson, S.E.; Clay, C.M. A binding site for steroidogenic factor-1 is part of a complex enhancer that mediates expression of the murine gonadotropin-releasing hormone receptor gene. Biol. Reprod. 1997, 56, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Ngan, E.S.; Cheng, P.K.; Leung, P.C.; Chow, B.K. Steroidogenic factor-1 interacts with a gonadotrope-specific element within the first exon of the human gonadotropin-releasing hormone receptor gene to mediate gonadotrope-specific expression. Endocrinology 1999, 140, 2452–2462. [Google Scholar] [CrossRef]
- Fernandez-Vazquez, G.; Kaiser, U.B.; Albarracin, C.T.; Chin, W.W. Transcriptional activation of the gonadotropin-releasing hormone receptor gene by activin A. Mol. Endocrinol. 1996, 10, 356–366. [Google Scholar]
- Bilotta, M.T.; Petillo, S.; Santoni, A.; Cippitelli, M. Liver X Receptors: Regulators of Cholesterol Metabolism, Inflammation, Autoimmunity, and Cancer. Front. Immunol. 2020, 11, 584303. [Google Scholar] [CrossRef]
- Annicotte, J.S.; Schoonjans, K.; Auwerx, J. Expression of the liver X receptor alpha and beta in embryonic and adult mice. Anat. Rec. A Discov. Mol. Cell Evol. Biol. 2004, 277, 312–316. [Google Scholar] [CrossRef]
- Volle, D.H.; Mouzat, K.; Duggavathi, R.; Siddeek, B.; Dechelotte, P.; Sion, B.; Veyssiere, G.; Benahmed, M.; Lobaccaro, J.M. Multiple roles of the nuclear receptors for oxysterols liver X receptor to maintain male fertility. Mol. Endocrinol. 2007, 21, 1014–1027. [Google Scholar] [CrossRef]
- Lee, J.H.; Gong, H.; Khadem, S.; Lu, Y.; Gao, X.; Li, S.; Zhang, J.; Xie, W. Androgen deprivation by activating the liver X receptor. Endocrinology 2008, 149, 3778–3788. [Google Scholar] [PubMed]
- Naamneh Elzenaty, R.; du Toit, T.; Fluck, C.E. Basics of androgen synthesis and action. Best. Pract. Res. Clin. Endocrinol. Metab. 2022, 36, 101665. [Google Scholar] [CrossRef] [PubMed]
- Hornig, N.C.; Holterhus, P.M. Molecular basis of androgen insensitivity syndromes. Mol. Cell Endocrinol. 2021, 523, 111146. [Google Scholar] [CrossRef] [PubMed]
- Knerr, J.; Werner, R.; Schwan, C.; Wang, H.; Gebhardt, P.; Grotsch, H.; Caliebe, A.; Spielmann, M.; Holterhus, P.M.; Grosse, R.; et al. Formin-mediated nuclear actin at androgen receptors promotes transcription. Nature 2023, 617, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Hornig, N.C.; Rodens, P.; Dorr, H.; Hubner, N.C.; Kulle, A.E.; Schweikert, H.U.; Welzel, M.; Bens, S.; Hiort, O.; Werner, R.; et al. Epigenetic Repression of Androgen Receptor Transcription in Mutation-Negative Androgen Insensitivity Syndrome (AIS Type II). J. Clin. Endocrinol. Metab. 2018, 103, 4617–4627. [Google Scholar] [CrossRef]
- Fabbri-Scallet, H.; Werner, R.; Guaragna, M.S.; de Andrade, J.G.R.; Maciel-Guerra, A.T.; Hornig, N.C.; Hiort, O.; Guerra-Junior, G.; de Mello, M.P. Can Non-Coding NR5A1 Gene Variants Explain Phenotypes of Disorders of Sex Development? Sex. Dev. 2022, 16, 252–260. [Google Scholar] [CrossRef]
- Gonen, N.; Eozenou, C.; Mitter, R.; Elzaiat, M.; Stevant, I.; Aviram, R.; Bernardo, A.S.; Chervova, A.; Wankanit, S.; Frachon, E.; et al. In vitro cellular reprogramming to model gonad development and its disorders. Sci. Adv. 2023, 9, eabn9793. [Google Scholar] [CrossRef]
- Rodriguez Gutierrez, D.; Eid, W.; Biason-Lauber, A. A Human Gonadal Cell Model From Induced Pluripotent Stem Cells. Front. Genet. 2018, 9, 498. [Google Scholar] [CrossRef]
- Renaux, A.; Papadimitriou, S.; Versbraegen, N.; Nachtegael, C.; Boutry, S.; Nowe, A.; Smits, G.; Lenaerts, T. ORVAL: A novel platform for the prediction and exploration of disease-causing oligogenic variant combinations. Nucleic Acids Res. 2019, 47, W93–W98. [Google Scholar] [CrossRef]
- Udhane, S.S.; Pandey, A.V.; Hofer, G.; Mullis, P.E.; Fluck, C.E. Retinoic acid receptor beta and angiopoietin-like protein 1 are involved in the regulation of human androgen biosynthesis. Sci. Rep. 2015, 5, 10132. [Google Scholar] [CrossRef]
- Eggers, S.; Sadedin, S.; van den Bergen, J.A.; Robevska, G.; Ohnesorg, T.; Hewitt, J.; Lambeth, L.; Bouty, A.; Knarston, I.M.; Tan, T.Y.; et al. Disorders of sex development: Insights from targeted gene sequencing of a large international patient cohort. Genome Biol. 2016, 17, 243. [Google Scholar] [CrossRef] [PubMed]
- Allali, S.; Muller, J.-B.; Brauner, R.; Lourenço, D.; Boudjenah, R.; Karageorgou, V.; Trivin, C.; Lottmann, H.; Lortat-Jacob, S.; Nihoul-Fékété, C.; et al. Mutation Analysis of NR5A1 Encoding Steroidogenic Factor 1 in 77 Patients with 46, XY Disorders of Sex Development (DSD) Including Hypospadias. PLoS ONE 2011, 6, e24117. [Google Scholar] [CrossRef] [PubMed]
- Robevska, G.; van den Bergen, J.A.; Ohnesorg, T.; Eggers, S.; Hanna, C.; Hersmus, R.; Thompson, E.M.; Baxendale, A.; Verge, C.F.; Lafferty, A.R.; et al. Functional characterization of novel NR5A1 variants reveals multiple complex roles in disorders of sex development. Hum. Mutat. 2018, 39, 124–139. [Google Scholar] [CrossRef]
- Köhler, B.; Lin, L.; Ferraz-de-Souza, B.; Wieacker, P.; Heidemann, P.; Schröder, V.; Biebermann, H.; Schnabel, D.; Grüters, A.; Achermann, J.C. Five novel mutations in steroidogenic factor 1 (SF1, NR5A1) in 46,XY patients with severe underandrogenization but without adrenal insufficiency. Hum. Mutat. 2008, 29, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Eggers, S.; Smith, K.R.; Bahlo, M.; Looijenga, L.H.J.; Drop, S.L.S.; Juniarto, Z.A.; Harley, V.R.; Koopman, P.; Faradz, S.M.H.; Sinclair, A.H. Whole exome sequencing combined with linkage analysis identifies a novel 3 bp deletion in NR5A1. Eur. J. Hum. Genet. 2015, 23, 486–493. [Google Scholar] [CrossRef]
- Bashamboo, A.; Brauner, R.; Bignon-Topalovic, J.; Lortat-Jacob, S.; Karageorgou, V.; Lourenco, D.; Guffanti, A.; McElreavey, K. Mutations in the FOG2/ZFPM2 gene are associated with anomalies of human testis determination. Hum. Mol. Genet. 2014, 23, 3657–3665. [Google Scholar] [CrossRef]
- Sreenivasan, R.; Bell, K.; van den Bergen, J.; Robevska, G.; Belluoccio, D.; Dahiya, R.; Leong, G.M.; Dulon, J.; Touraine, P.; Tucker, E.J.; et al. Whole exome sequencing reveals copy number variants in individuals with disorders of sex development. Mol. Cell Endocrinol. 2022, 546, 111570. [Google Scholar] [CrossRef]
- Cheng, Y.; Chen, J.; Zhou, X.; Yang, J.; Ji, Y.; Xu, C. Characteristics and possible mechanisms of 46, XY differences in sex development caused by novel compound variants in NR5A1 and MAP3K1. Orphanet. J. Rare Dis. 2021, 16, 268. [Google Scholar] [CrossRef]
- Laan, M.; Kasak, L.; Timinskas, K.; Grigorova, M.; Venclovas, C.; Renaux, A.; Lenaerts, T.; Punab, M. NR5A1 c.991-1G > C splice-site variant causes familial 46,XY partial gonadal dysgenesis with incomplete penetrance. Clin. Endocrinol. (Oxf.) 2021, 94, 656–666. [Google Scholar] [CrossRef]
- Cannarella, R.; Condorelli, R.A.; Paolacci, S.; Barbagallo, F.; Guerri, G.; Bertelli, M.; La Vignera, S.; Calogero, A.E. Next-generation sequencing: Toward an increase in the diagnostic yield in patients with apparently idiopathic spermatogenic failure. Asian J. Androl. 2021, 23, 24–29. [Google Scholar] [CrossRef]
- Giannakopoulos, A.; Sertedaki, A.; Chrysis, D. A human paradigm of LHX4 and NR5A1 developmental gene interaction in the pituitary gland and ovary? Eur. J. Hum. Genet. 2022, 30, 1191–1194. [Google Scholar] [CrossRef] [PubMed]
- Gomes, N.L.; Batista, R.L.; Nishi, M.Y.; Lerario, A.M.; Silva, T.E.; de Moraes Narcizo, A.; Benedetti, A.F.F.; de Assis Funari, M.F.; Faria Junior, J.A.; Moraes, D.R.; et al. Contribution of Clinical and Genetic Approaches for Diagnosing 209 Index Cases With 46,XY Differences of Sex Development. J. Clin. Endocrinol. Metab. 2022, 107, e1797–e1806. [Google Scholar] [CrossRef] [PubMed]
- Oral, E.; Toksoy, G.; Sofiyeva, N.; Celik, H.G.; Karaman, B.; Basaran, S.; Azami, A.; Uyguner, Z.O. Clinical and Genetic Investigation of Premature Ovarian Insufficiency Cases from Turkey. J. Gynecol. Obstet. Hum. Reprod. 2019, 48, 817–823. [Google Scholar] [CrossRef]
- Wang, N.; Zhu, W.; Han, B.; Wang, H.; Zhu, H.; Chen, Y.; Chen, Y.; Liu, J.; Liu, Y.; Zhao, S.; et al. Inherited Missense Mutation Occurring in Arginine76 of the SRY Gene Does Not Account for Familial 46, XY Sex Reversal. J. Clin. Endocrinol. Metab. 2020, 105, 1355–1365. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Hormones/Markers | Biochemical Value | Range | Units |
|---|---|---|---|
| Sex hormones | |||
| FSH | 85.1 | 0.95–11.95 | mU/mL |
| LH | 20.3 | 0.57–12.07 | mU/mL |
| Prolactin | 29.1 | 3.46–19.4 | ng/mL |
| Testosterone | 4.15 | 1–12 | ng/mL |
| AMH | 5.18 | 27–1141 | pM |
| Adrenal function | |||
| ACTH | 53.7 | 9.0–40.0 | pg/mL |
| Cortisol | 179 | 30–210 | ng/mL |
| DHEA-S | 2243 | 166–2427 | ng/mL |
| Gene Name | Gene Transcript | Variant | Chromosome Position | Type/ Consequence | ACMG Classification (Criteria) | SIFT | Polyphen | Mutation Taster | Panther | SNPs and Go | M-CAP | Mutation Assessor | REVEL | Provean | CADD Score | ORVAL—VarCoPP Score |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NR5A1 | ENST00000373588.9 | c.58G>C; p.(Val20Leu) | 9:124503338 | SNV/missense | P | Unc | B | Unc | Prdam | Dis | P | Unc | P | Unc | 23.3 | ND |
| NR1H2 | ENST00000253727.10 | c.515_516insCAA; p.(Arg171_Lys172insAsn) | 19:50378563 | Ins/In-frame insertion | VUS | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| INHA | ENST00000243786.3 | c.675T>G; p.(Ser225Arg) | 2:219575100 | SNV/missense | B | B | B | B | ND | Dis | Unc | Unc | B | B | 15.4 | 0.5825 |
| TCF7L2 | ENST00000355995.9 | c.1535C>G; p.(Pro512Arg) | 10:113165647 | SNV/missense | LB | B | B | Unc | Prben | Neu | Unc | B | B | B | 23.5 | 0.9825 |
| NIBAN1 | ENST00000367511.4 | c.929G>A; p.(Arg310His) | 1:184823223 | SNV/missense | VUS | Unc | Prdam | Unc | Prdam | Neu | B | Unc | B | Unc | 25.2 | 0.8500 |
| SCUBE2 | ENST00000649792.2 | c.692C>T; p.(Thr231Ile) | 11:9066765 | SNV/missense | VUS | Unc | Prdam | Unc | Prdam | Neu | B | Unc | Unc | P | 24.6 | 0.8450 |
| Gene/Protein | Biological Function | Phenotype Associated with This Gene in Humans | The Phenotype Associated with This Gene in Mice Models | In Vitro Studies (NR5A1-Related) | A Possible Contribution of This Gene to the DSD Phenotype of the Patient? |
|---|---|---|---|---|---|
| NR5A1/SF-1 |
| NR5A1 homozygous and heterozygous variants are associated with disorders of sex development including adrenal insufficiency and 46,XY gonadal dysgenesis, ambiguous genitalia, hypospadias, micropenis, spermatogenic failure with normal genitalia, and primary ovarian insufficiency [29,30]. | The majority of heterozygous NR5A1/SF-1 variants located in the DNA-binding domain present with impaired functional activity on different human steroidogenic enzyme promoters, while variants located elsewhere in the SF-1 protein present with variable activity. Mostly, no genotype-phenotype correlation was found [6]. | Yes | |
| NR1H2/LXRβ | Plays an important role as a modulator of lipid homeostasis and inflammation throughout the human body [32]. | Diseases associated with NR1H2 include type 2 diabetes and male infertility (azoospermia) [32,33,34,35,36]. |
| LXRβ is involved in the basal expression levels of CYP11A1, StAR, and NR5A1 in NCI-H295R adrenal cells [34]. | Yes |
| INHA/Inhibin α | Antagonizes activin signaling in the reproductive hypothalamic-pituitary gonadal axis [37,38]. | Homozygous INHA variants are associated with decreased prenatal and postnatal testosterone production and infertility in males, and primary ovarian failure in women [38,39,40]. | INHA knockout mice develop mixed or incompletely differentiated gonadal stromal tumors and die from cachexia syndrome [38,41]. | Rat inhibin alpha gene expression is regulated by the synergistic activity of Nr5a1 and cAMP [42]. | Yes |
| TCF7L2/TCF-4 | TCF7L2 variants are associated with an increased risk of type 2 diabetes [47,48,49]. | Tcf7l2 knockout causes neonatal death in mice [43]. Conditional inactivation of Tcf7l2 in the adult intestinal epithelium in mice causes impaired cell proliferation in the small intestines and colon [46]. | Tcf-4 is involved in rat inhibin alpha gene expression: Tcf-4 disrupts β-catenin’s ability to synergize with Sf-1 on the inhibin alpha promoter in a dose-dependent manner [50]. | Unlikely | |
| NIBAN1/FAM129A | Plays an important role in apoptosis, preventing cell death and tumor progression under stress conditions [51,52]. | NIBAN1 expression has been described in several tumor subtypes, including microcarcinomas, papillary and follicular carcinoma, and prostate cancer, as well as in Hashimoto’s Thyroiditis [51]. | Niban1−/− mice are viable and show no obvious phenotype or any phenotypic abnormalities [52]. | Not found | Unlikely |
| SCUBE2/SCUB2 | Plays an important role as a tumor suppressor in different types of cancer [53,54]. | SCUBE2 expression is reduced in endometrial, breast, and colorectal cancers [53]. | Scube2(−/−) mice have a defective endochondral bone formation and impaired Indian hedgehog-dependent chondrocyte-mediated chondrocyte differentiation and proliferation [55]. | Not found | Unlikely |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naamneh Elzenaty, R.; Kouri, C.; Martinez de Lapiscina, I.; Sauter, K.-S.; Moreno, F.; Camats-Tarruella, N.; Flück, C.E. NR5A1/SF-1 Collaborates with Inhibin α and the Androgen Receptor. Int. J. Mol. Sci. 2024, 25, 10109. https://doi.org/10.3390/ijms251810109
Naamneh Elzenaty R, Kouri C, Martinez de Lapiscina I, Sauter K-S, Moreno F, Camats-Tarruella N, Flück CE. NR5A1/SF-1 Collaborates with Inhibin α and the Androgen Receptor. International Journal of Molecular Sciences. 2024; 25(18):10109. https://doi.org/10.3390/ijms251810109
Chicago/Turabian StyleNaamneh Elzenaty, Rawda, Chrysanthi Kouri, Idoia Martinez de Lapiscina, Kay-Sara Sauter, Francisca Moreno, Núria Camats-Tarruella, and Christa E. Flück. 2024. "NR5A1/SF-1 Collaborates with Inhibin α and the Androgen Receptor" International Journal of Molecular Sciences 25, no. 18: 10109. https://doi.org/10.3390/ijms251810109
APA StyleNaamneh Elzenaty, R., Kouri, C., Martinez de Lapiscina, I., Sauter, K.-S., Moreno, F., Camats-Tarruella, N., & Flück, C. E. (2024). NR5A1/SF-1 Collaborates with Inhibin α and the Androgen Receptor. International Journal of Molecular Sciences, 25(18), 10109. https://doi.org/10.3390/ijms251810109