A Facile Route to Flavone-3-Carboxamides and Flavone-3-Carboxylates via Palladium-Catalyzed Amino- and Aryloxy-Carbonylation Reactions


Abstract

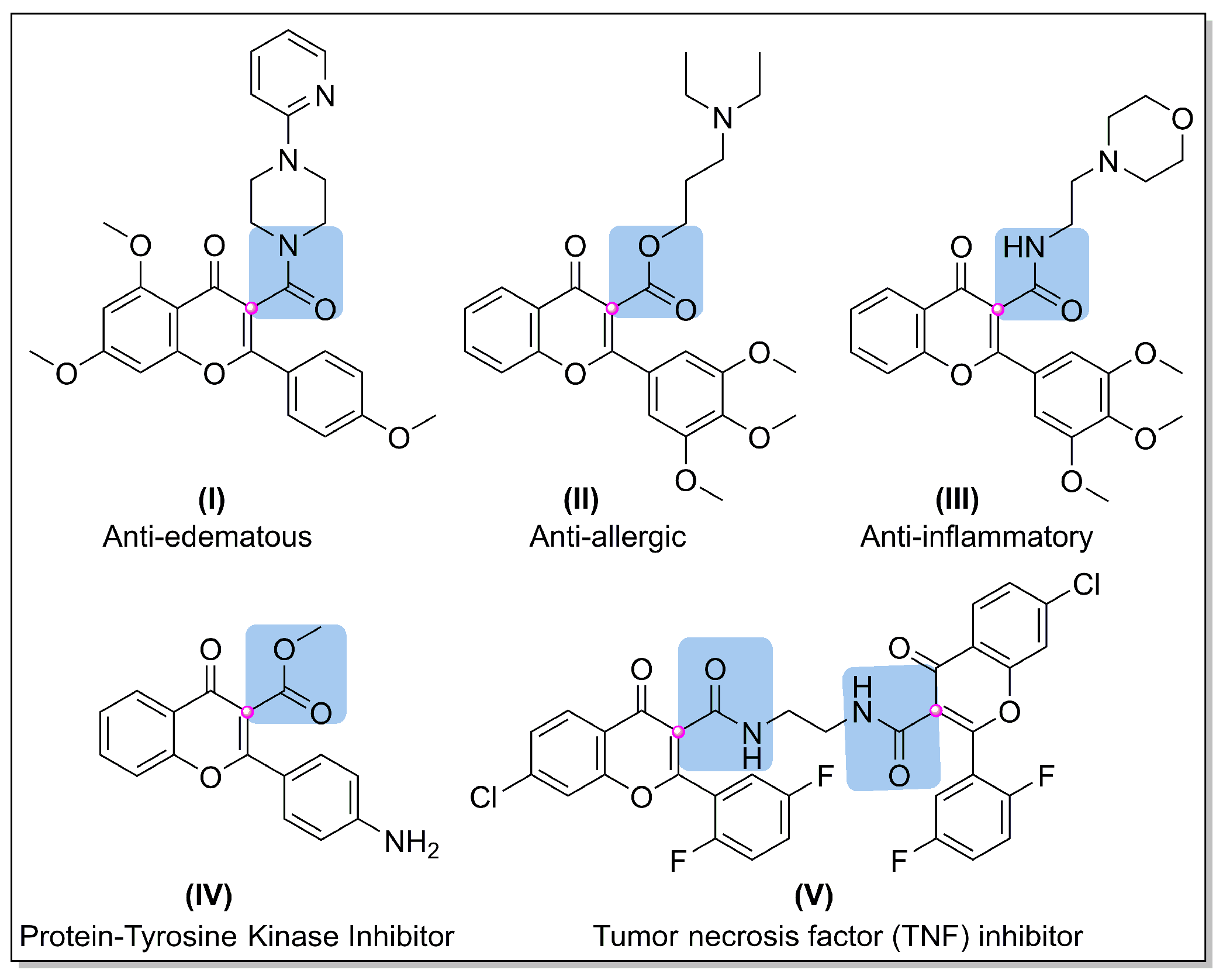
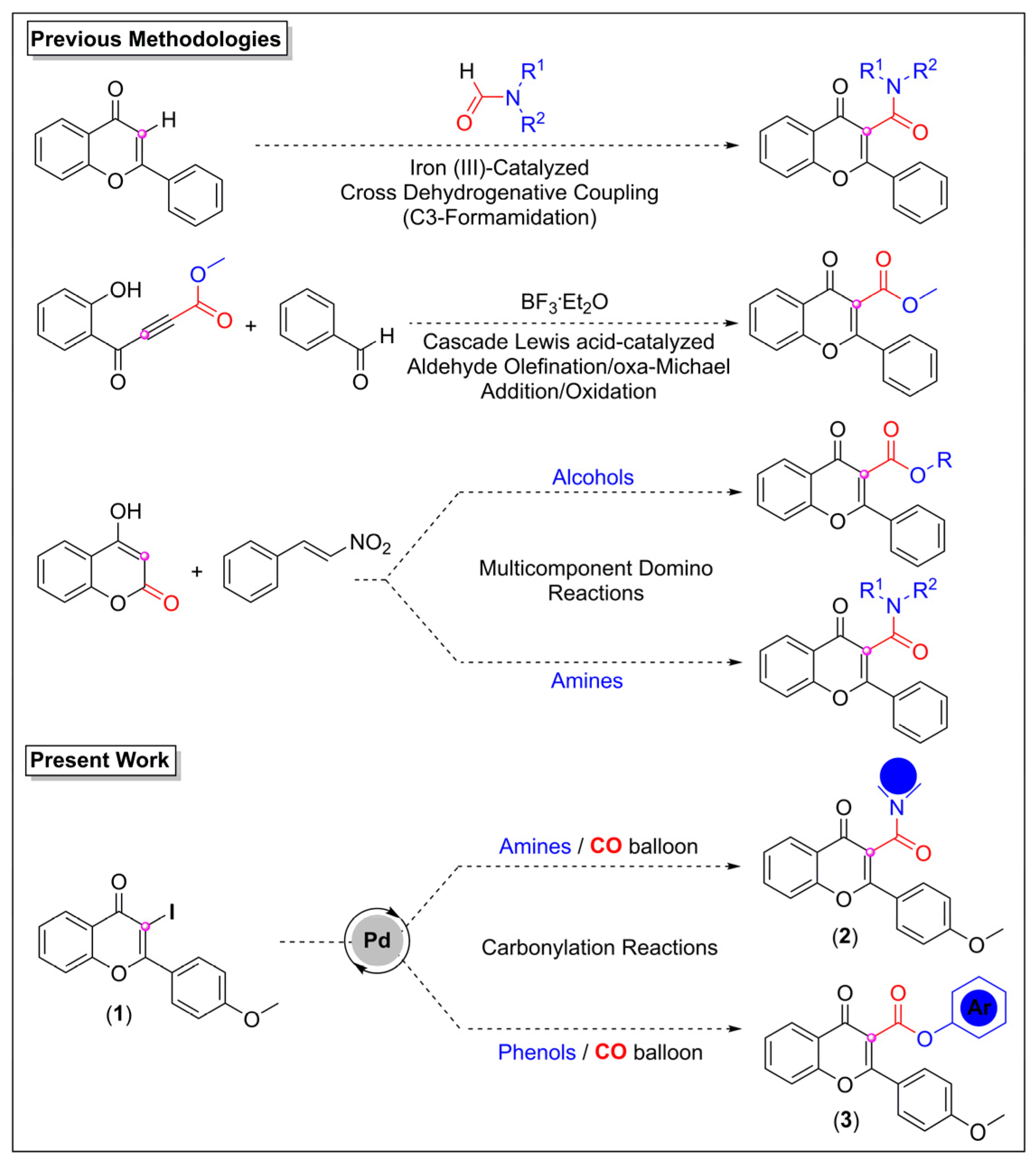
1. Introduction
2. Results and Discussions
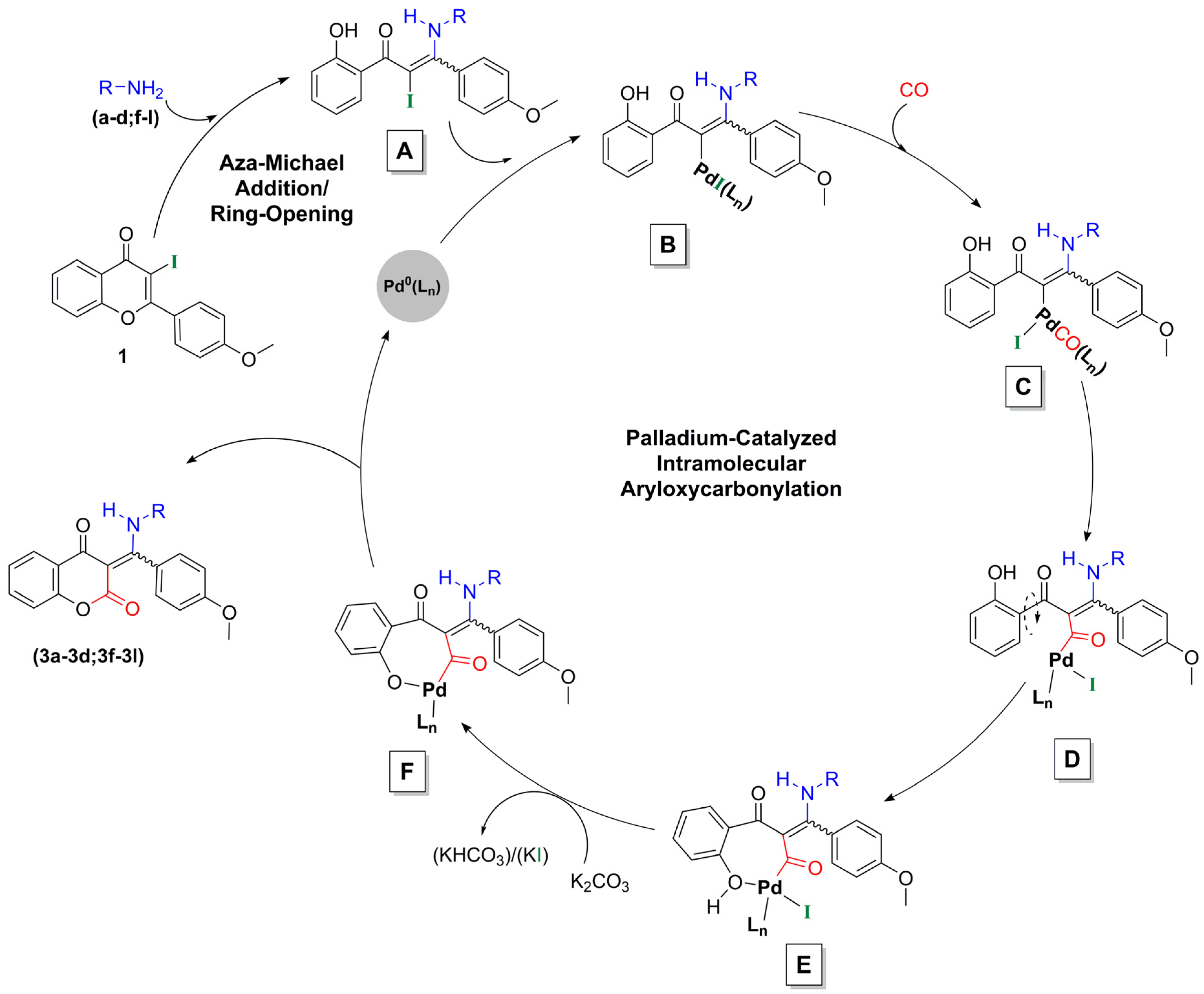
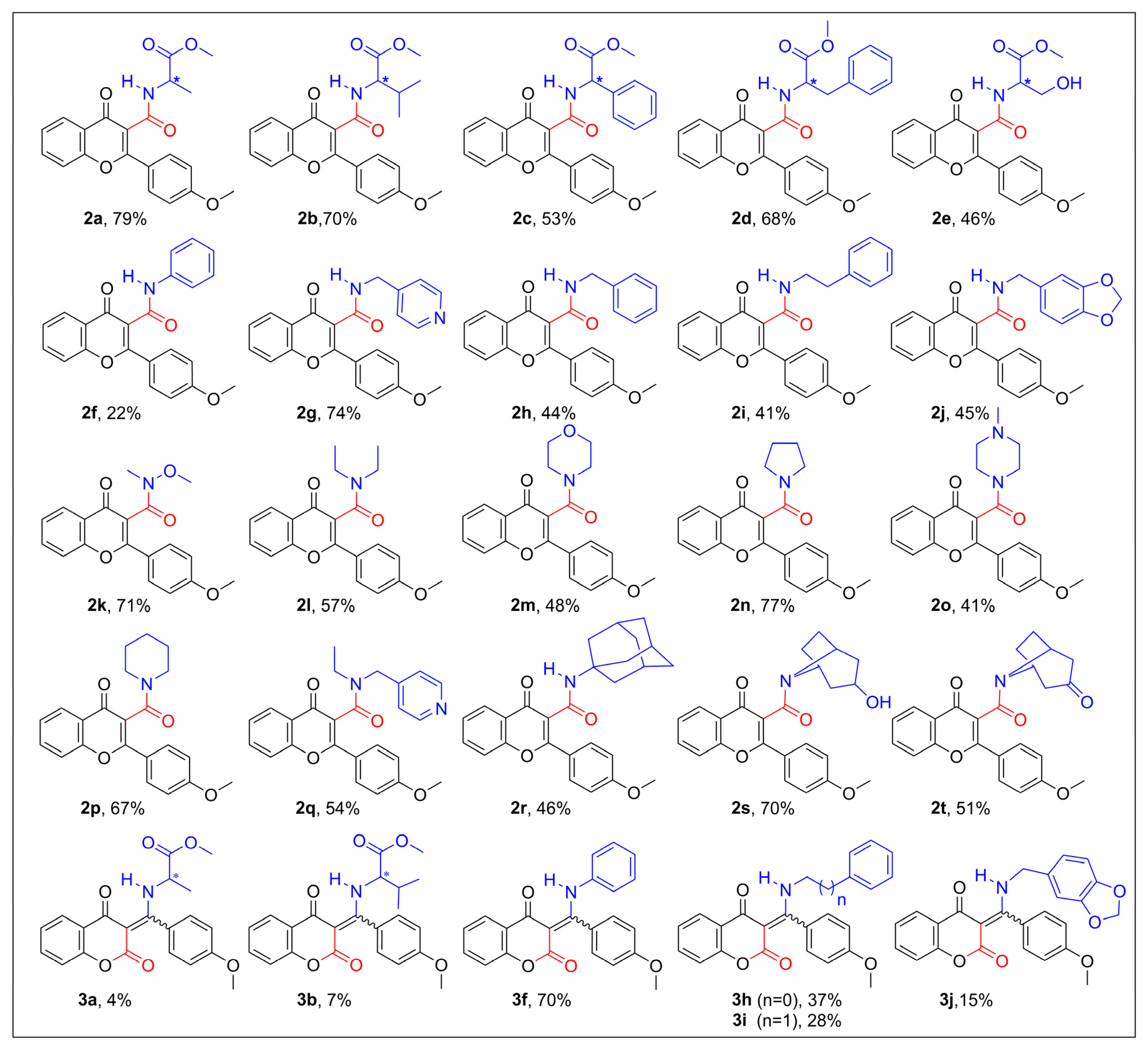
2.1. Synthesis of Flavone-3-Carboxamides via Aminocarbonylation of 3-Iodoflavone 1
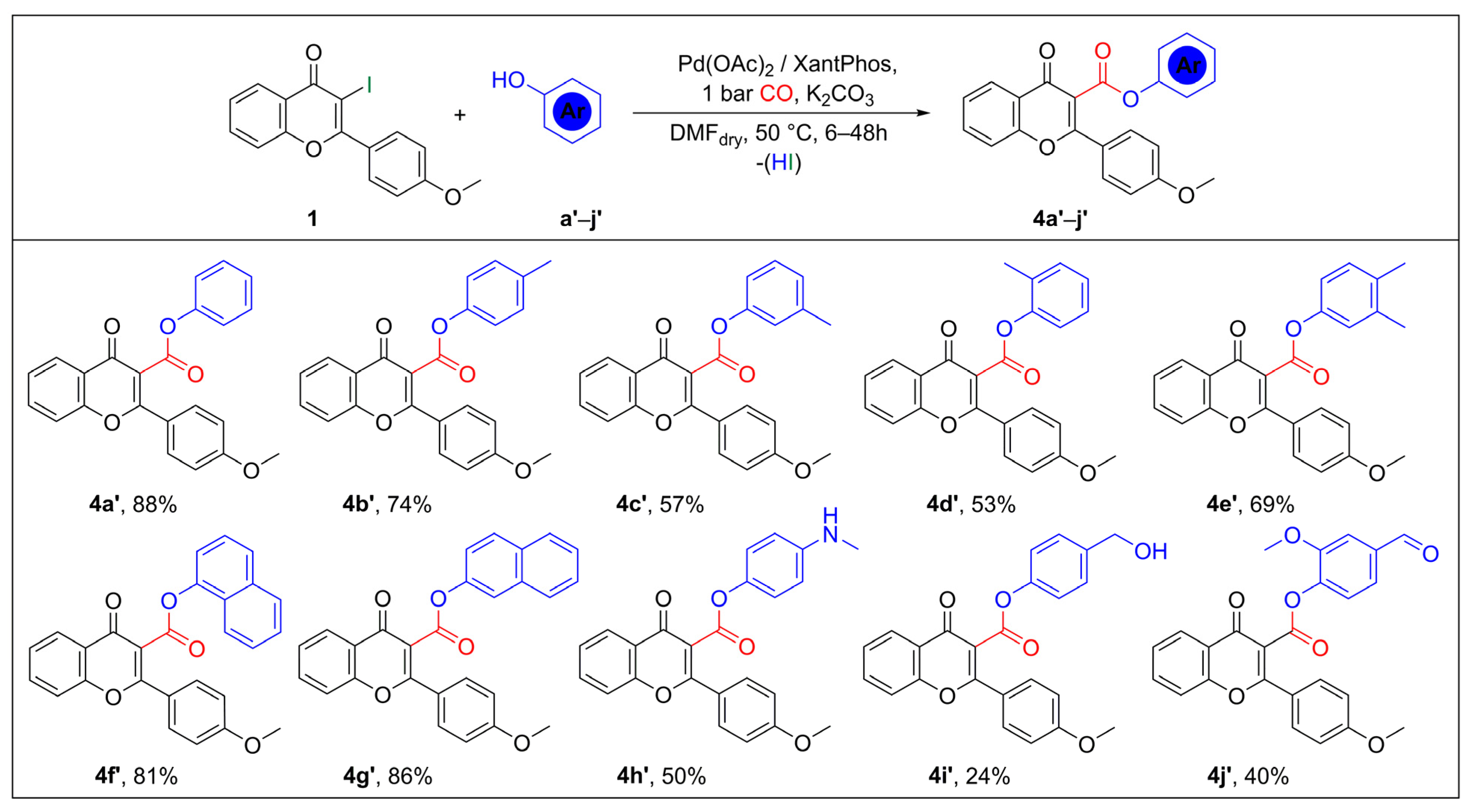
2.2. Synthesis of Flavone-3-Esters (4) via Aryloxycarbonylation of 3-Iodoflavone (1)
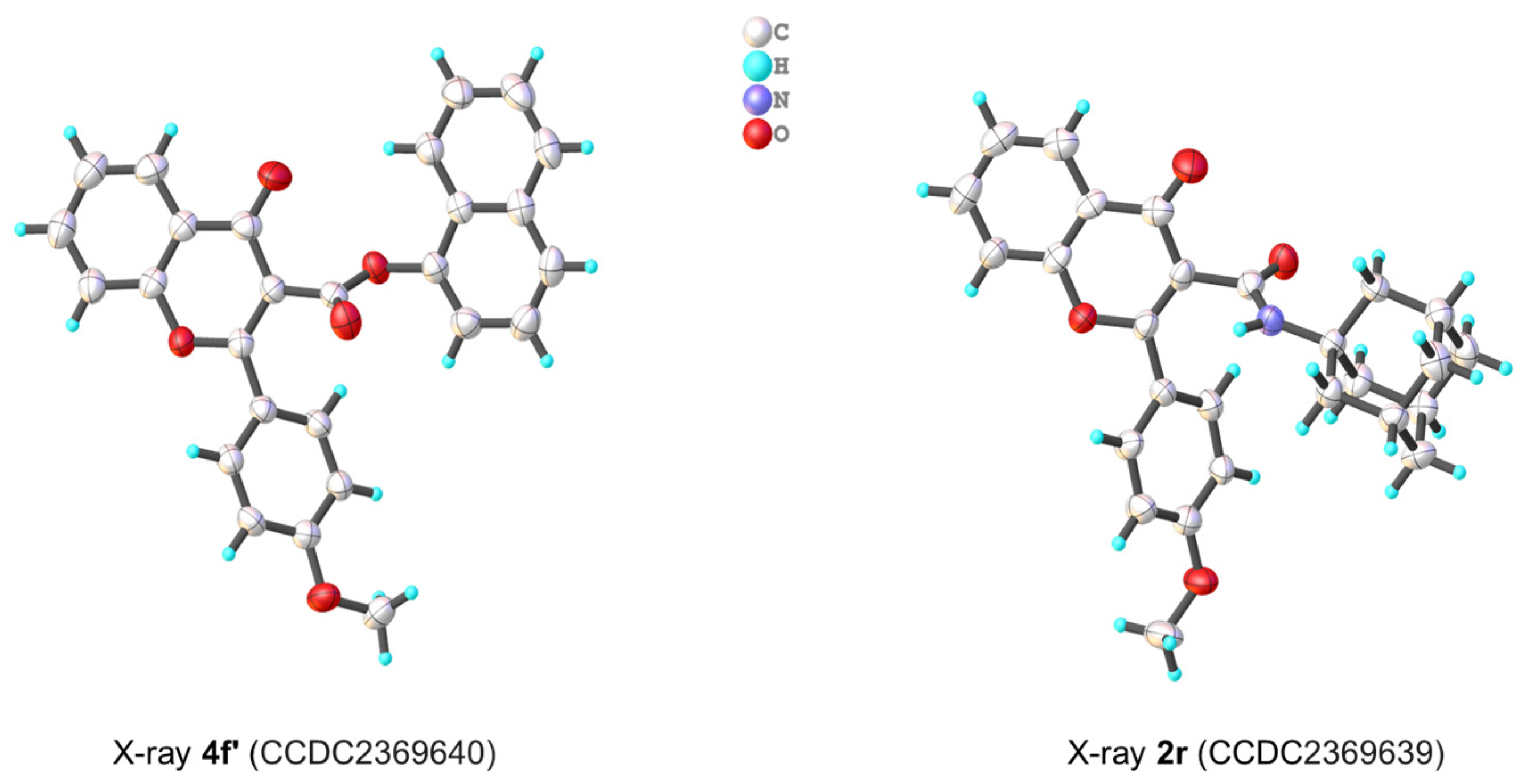
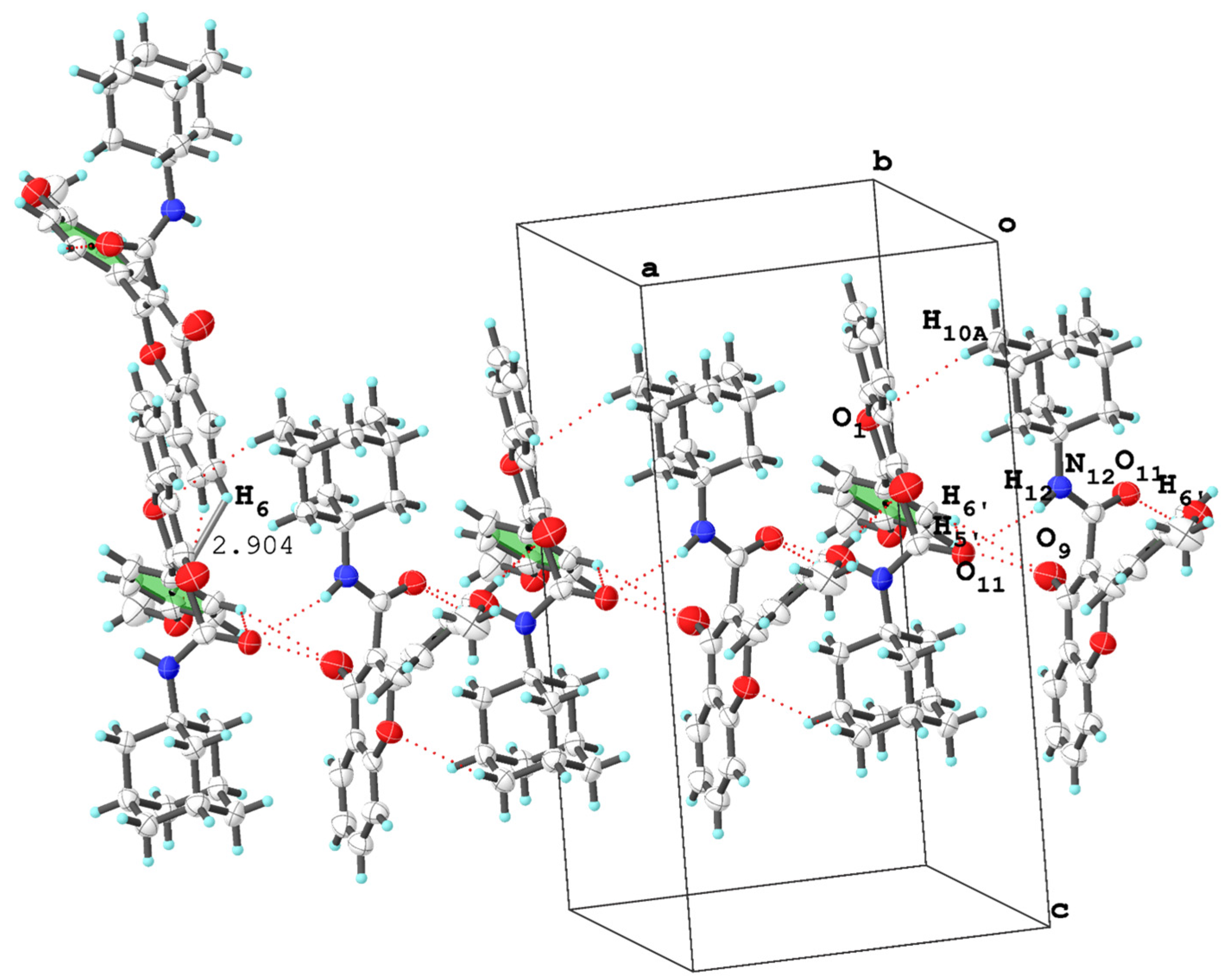
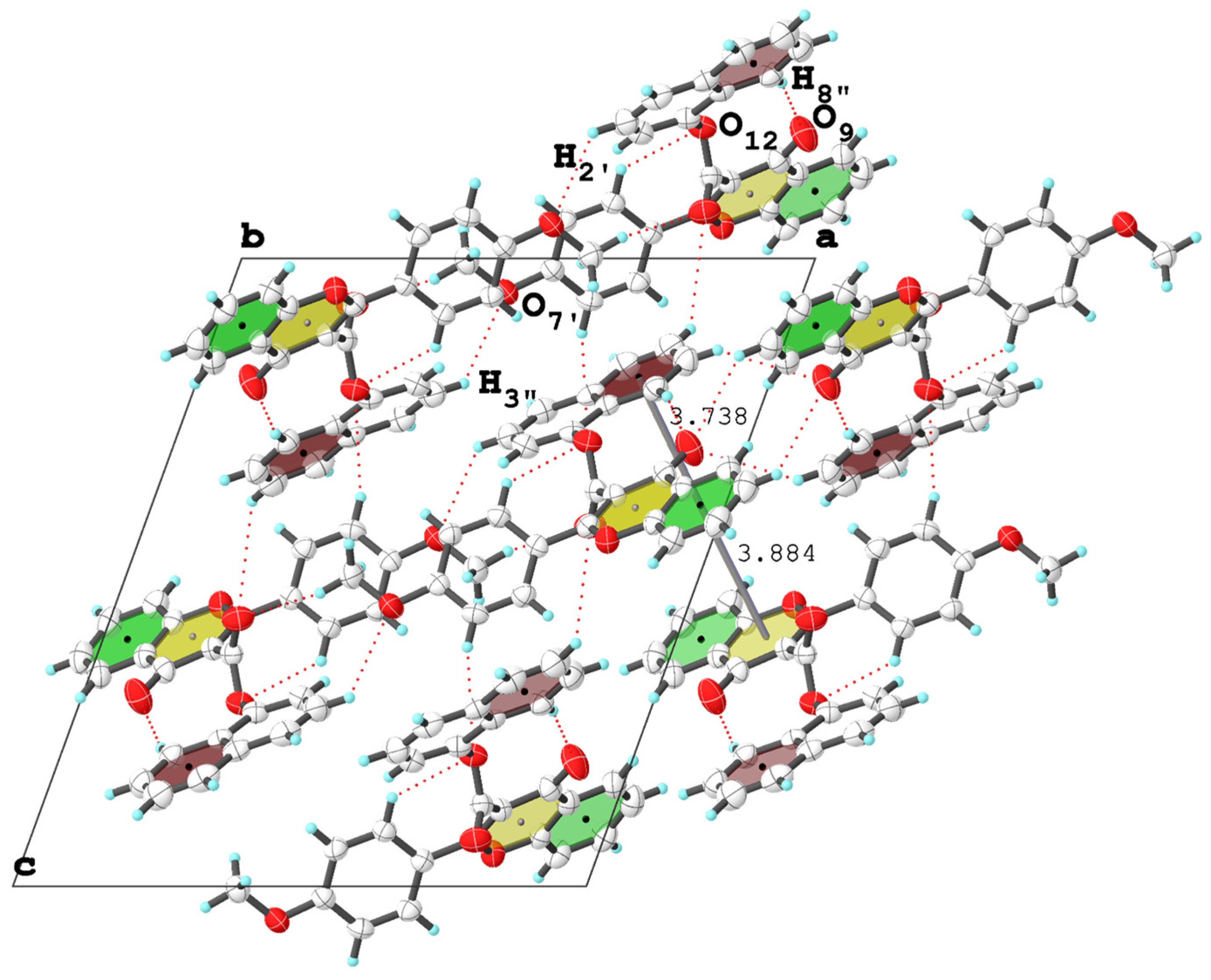
3. X-ray Crystallographic Study
3.1. Molecular Structure Analysis
3.2. Molecular Packing Analysis
4. Materials and Methods
4.1. General Procedures
4.2. Synthesis of 2-(4′-Methoxyphenyl)-3-Iodo-4H-Chromen-4-One 1
- Step 1: Claisen–Schmidt Condensation [44]
- Step 2: I2/DMSO-mediated Oxidative Cyclization [45]
- Step 3: I2/CAN-promoted Iodination [46]
4.3. General Method for the Synthesis of Flavone-3-Carboxamides 2 and Flavone-3-Esters 4 under Atmospheric Conditions
4.4. General Method of High-Pressure Aminocarbonylation Reactions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Havsteen, B. The biochemistry and medical significance of the flavonoids. Pharmacol. Ther. 2002, 96, 67–202. [Google Scholar] [CrossRef] [PubMed]
- Leonte, D.; Ungureanu, D.; Zaharia, V. Flavones and Related Compounds: Synthesis and Biological Activity. Molecules 2023, 28, 6528. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Liu, T.; Zhu, Y.; Liu, F.; Cao, X.; Wang, Q.; Liu, L.; Xue, W. Novel 1,3,4-oxadiazole sulfonate/carboxylate flavonoid derivatives: Synthesis and biological activity. Pest Manag. Sci. 2023, 79, 274–283. [Google Scholar] [CrossRef]
- Chaves, O.; Fintelman-Rodrigues, N.; Wang, X.; Sacramento, C.; Temerozo, J.; Ferreira, A.; Mattos, M.; Pereira-Dutra, F.; Bozza, P.; Castro-Faria-Neto, H.; et al. Commercially Available Flavonols Are Better SARS-CoV-2 Inhibitors than Isoflavone and Flavones. Viruses 2022, 14, 1458. [Google Scholar] [CrossRef]
- Zhao, B.; Wang, J.; Wang, L.; Wang, Z.; Lu, A. Discovery of Flavone Derivatives Containing Carboxamide Fragments as Novel Antiviral Agents. Molecules 2023, 28, 2179. [Google Scholar] [CrossRef]
- Al-Rooqi, M.; Mughal, E.; Raja, Q.; Hussein, E.; Naeem, N.; Sadiq, A.; Asghar, B.; Moussa, Z.; Ahmed, S. Flavonoids and related privileged scaffolds as potential urease inhibitors: A review. RSC Adv. 2023, 13, 3210–3233. [Google Scholar] [CrossRef]
- Tian, S.; Luo, T.; Zhu, Y.; Wan, J. Recent advances in the diversification of chromones and flavones by direct C-H bond activation or functionalization. Chin. Chem. Lett. 2020, 31, 3073–3082. [Google Scholar] [CrossRef]
- Li, X.; Zhang, C.; Guo, S.; Rajaram, P.; Lee, M.; Chen, G.; Fong, R.; Gonzalez, A.; Zhang, Q.; Zheng, S.; et al. Structure-activity relationship and pharmacokinetic studies of 3-O-substitutedflavonols as anti-prostate cancer agents. Eur. J. Med. Chem. 2018, 157, 978–993. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.; Anwar, F.; Naz, F.; Mehmood, T.; Saari, N. Anti-Helicobacter pylori and Urease Inhibition Activities of Some Traditional Medicinal Plants. Molecules 2013, 18, 2135–2149. [Google Scholar] [CrossRef]
- Gomes, A.; Neuwirth, O.; Freitas, M.; Couto, D.; Ribeiro, D.; Figueiredo, A.; Silva, A.; Seixas, R.; Pinto, D.; Tomé, A.; et al. Synthesis and antioxidant properties of new chromone derivatives. Bioorg. Med. Chem. 2009, 17, 7218–7226. [Google Scholar] [CrossRef]
- Yoo, H.; Kim, S.; Lee, J.; Kim, H.; Seo, S.; Chung, B.; Jin, C.; Lee, Y. Synthesis and antioxidant activity of 3-methoxyflavones. Bull. Korean Chem. Soc. 2005, 26, 2057–2060. [Google Scholar] [CrossRef]
- Walenta, R.; Müller-Peddinghaus, R.; Ban, I.; Wurl, M.; Preuschoff, U. Flavone-3-Carboxylic-Acid Compounds, and Process, Intermediates for Their Preparation and Medicines Containing These Compounds. EP0290915B1, 14 July 1993. [Google Scholar]
- Cushman, M.; Nagarathnam, D.; Burg, D.; Geahlen, R. Synthesis and protein-tyrosine kinase inhibitory activities of flavonoid analogs. J. Med. Chem. 1991, 34, 798–806. [Google Scholar] [CrossRef] [PubMed]
- Park, K.Y. Novel TNF Activity Inhibitor Compound, and Pharmaceutically Acceptable Salt Thereof. U.S. Patent 20230271925A1, 31 August 2023. [Google Scholar]
- Nagaraj, A.; Rao, G. Synthesis and biological evaluation of novel tetrazole analogues of flavones as antimicrobial agents. Int. J. Pharm. Sci. Res. 2023, 14, 3444–3451. [Google Scholar] [CrossRef]
- Mir, B.; Banerjee, A.; Santra, S.; Rajamanickam, S.; Patel, B. Iron(III)-Catalyzed Peroxide-Mediated C-3 Functionalization of Flavones. Adv. Synth. Catal. 2016, 358, 3471–3476. [Google Scholar] [CrossRef]
- Wang, N.; Cai, S.; Zhou, C.; Lu, P.; Wang, Y. One-pot synthesis of 2-aryl-3-alkoxycarbonyl chromones through a cascade Lewis acid-catalyzed aldehyde olefination/oxa-Michael addition/oxidation. Tetrahedron 2013, 69, 647–652. [Google Scholar] [CrossRef]
- Yoshida, M.; Saito, K.; Fujino, Y.; Doi, T. A concise total synthesis of biologically active frutinones via tributylphosphine-catalyzed tandem acyl transfer-cyclization. Tetrahedron 2014, 70, 3452–3458. [Google Scholar] [CrossRef]
- Dong, Q.; Shen, H.; Jiang, M. Efficient Efficient synthesis of functionalized chromones via a two-base mediated formal [3 + 3] cycloaddition. Tetrahedron Lett. 2016, 57, 2116–2120. [Google Scholar] [CrossRef]
- Zanwar, M.; Raihan, M.; Gawande, S.; Kayala, V.; Janreddy, D.; Kuo, C.; Ambre, R.; Yao, C. Alcohol Mediated Synthesis of 4-Oxo-2-aryl-4H-chromene-3-carboxylate Derivatives from 4-Hydroxycoumarins. J. Org. Chem. 2012, 77, 6495–6504. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Khan, A. Synthesis of 3-substituted carboxylate/carboxamide flavone derivatives from 4-hydroxycoumarin, β-nitrostyrene and alcohol/amine using multicomponent reaction. Tetrahedron Lett. 2016, 57, 1831–1834. [Google Scholar] [CrossRef]
- Bartolo, G. Carbon Monoxide in Organic Synthesis:Carbonylation Chemistry; Wiley-VCH GmbH: Weinheim, Germany, 2021. [Google Scholar]
- Mucsi, Z.; Chass, G.; Viskolcz, B.; Csizmadia, I. Quantitative scale for the extent of conjugation of carbonyl groups: “Carbonylicity” percentage as a chemical driving force. J. Phys. Chem. A 2008, 112, 9153–9165. [Google Scholar] [CrossRef]
- Tong, Q.; Xiu, R.; Chen, J.; Zhang, Y.; Cui, B.; Wan, N.; Chen, Y.; Han, W. Toward the Generation of 2-Amino-3-Formyl Difunctionalized Chromones via Pd-Enabled Rearrangement Strategy. ACS Catal. 2023, 13, 12692–12699. [Google Scholar] [CrossRef]
- Yang, Q.; Wang, R.; Han, J.; Li, C.; Wang, T.; Liang, Y.; Zhang, Z. Photo-induced tandem cyclization of 3-iodoflavones with electron rich five-membered heteroarenes. RSC Adv. 2017, 7, 43206–43211. [Google Scholar] [CrossRef]
- Yang, J.; Yoshikai, N. Cobalt-Catalyzed Annulation of Salicylaldehydes and Alkynes to form Chromones and 4-Chromanones. Angew. Chem. Int. Ed. 2016, 55, 2870–2874. [Google Scholar] [CrossRef] [PubMed]
- Tomé, S.; Silva, A.; Santos, C. Synthesis and Transformation of Halochromones. Curr. Org. Synth. 2014, 11, 317–341. [Google Scholar] [CrossRef]
- Chniti, S.; Pongrácz, P.; Kollár, L.; Bényei, A.; Dörnyei, A.; Takács, A. Synthesis of Chroman-2,4-diones via Ring-Opening/Ring-Closing Reaction Involving Palladium-Catalyzed Intramolecular Aryloxycarbonylation. J. Org. Chem. 2024, 89, 1175–1183. [Google Scholar] [CrossRef]
- Chniti, S.; Kollár, L.; Bényei, A.; Dörnyei, Á.; Takács, A. Highly Chemoselective One-Step Synthesis of Novel N-Substituted-Pyrrolo[3,4-b]quinoline-1,3-diones via Palladium-Catalyzed Aminocarbonylation/Carbonylative Cyclisation Sequence. Eur. J. Org. Chem. 2023, 26, e202201374. [Google Scholar] [CrossRef]
- Chniti, S.; Kollár, L.; Bényei, A.; Takács, A. Highly Selective Synthesis of 6-Glyoxylamidoquinoline Derivatives via Palladium-Catalyzed Aminocarbonylation. Molecules 2022, 27, 4. [Google Scholar] [CrossRef]
- Uzunlu, N.; Pongrácz, P.; Kollár, L.; Takács, A. Alkyl Levulinates and 2-Methyltetrahydrofuran: Possible Biomass-Based Solvents in Palladium-Catalyzed Aminocarbonylation. Molecules 2023, 28, 442. [Google Scholar] [CrossRef]
- Kollár, L.; Takács, A.; Molnár, C.; Kovács, A.; Mika, L.; Pongrácz, P. Palladium-Catalyzed Selective Amino- and Alkoxycarbonylation of Iodoarenes with Aliphatic Aminoalcohols as Heterobifunctional O,N-Nucleophiles. J. Org. Chem. 2023, 88, 5172–5179. [Google Scholar] [CrossRef]
- Kégl, T.; Mika, L.; Kégl, T. 27 Years of Catalytic Carbonylative Coupling Reactions in Hungary (1994–2021). Molecules 2022, 27, 460. [Google Scholar] [CrossRef]
- Csákai, Z.; Skoda-Földes, R.; Kollár, L. NMR investigation of Pd(II)–Pd(0) reduction in the presence of mono- and ditertiary phosphines. Inorg. Chim. Acta 1999, 286, 93–97. [Google Scholar] [CrossRef]
- Amatore, C.; Carre, E.; Jutand, A.; Mbarki, M.; Meyer, G. Evidence for the Ligation of Palladium(0) Complexes by Acetate Ions: Consequences on the Mechanism of Their Oxidative Addition with Phenyl Iodide and PhPd(OAc)(PPh3)2 as Intermediate in the Heck Reaction. Organometallics 1995, 14, 5605–5614. [Google Scholar] [CrossRef]
- Amatore, C.; Jutand, A.; Mbarki, M. Evidence of the formation of zerovalentpalladium from Pd(OAc)2 and triphenylphosphine. Organometallics 1992, 11, 3009–3013. [Google Scholar] [CrossRef]
- Parveen, I.; Ahmed, N. A Route to Highly Functionalized Stereospecific trans-Aminated Aurones from 3-Bromoflavones with Aniline and N-Phenylurea via a Domino Aza-Michael Ring Opening and Cyclization Reactions. Synthesis 2019, 51, 960–970. [Google Scholar] [CrossRef]
- van Leeuwen, P.W.N.M.; Kamer, P. Featuring Xantphos. Catal. Sci. Technol. 2018, 8, 26–113. [Google Scholar] [CrossRef]
- Martinelli, J.; Watson, D.; Freckmann, D.; Barder, T.; Buchwald, S. Palladium-catalyzed carbonylation reactions of aryl bromides at atmospheric pressure: A general system based on xantphos. J. Org. Chem. 2008, 73, 7102–7107. [Google Scholar] [CrossRef]
- Parveen, I.; Khan, D.; Ahmed, N. Regioselective Hydrodehalogenation of Aromatic α- and β-Halo carbonyl Compounds by CuI in Isopropanol. Eur. J. Org. Chem. 2019, 2019, 759–764. [Google Scholar] [CrossRef]
- Stimac, A.; Sekutor, M.; Mlinaric-Majerski, K.; Frkanec, L.; Frkanec, R. Adamantane in Drug Delivery Systems and Surface Recognition. Molecules 2017, 22, 297. [Google Scholar] [CrossRef]
- Wanka, L.; Iqbal, K.; Schreiner, P. The Lipophilic Bullet Hits the Targets: Medicinal Chemistry of Adamantane Derivatives. Chem. Rev. 2013, 113, 3516–3604. [Google Scholar] [CrossRef]
- Hsieh, W.; Manjappa, K.; Yang, D. Structural tuning enables piezochromic and photochemical properties in N-aryl-β-enaminones. RSC Adv. 2019, 9, 34088–34094. [Google Scholar] [CrossRef]
- Syam, S.; Abdelwahab, S.; Al-Mamary, M.; Mohan, S. Synthesis of Chalcones with Anticancer Activities. Molecules 2012, 17, 6179–6195. [Google Scholar] [CrossRef] [PubMed]
- Patonay, T.; Cavaleiro, J.; Levai, A.; Silva, A. Dehydrogenation by iodine-dimethylsulfoxide system: A general route to substituted chromones and thiochromones. Heterocycl. Commun. 1997, 3, 223–229. [Google Scholar] [CrossRef]
- Zhang, F.; Li, Y. Synthesis of 3-Iodo Derivatives of Flavones, Thioflavones, and Thiochromones. Synthesis 1993, 565–567. [Google Scholar] [CrossRef]
- Manjappa, K.; Yang, Y.; Mysore, S.; Yang, D. Nitroalkane-Mediated Multicomponent Synthesis of β-Enaminones. ChemistrySelect 2018, 3, 10701–10705. [Google Scholar] [CrossRef]
- Saha, A.; Payra, S.; Akhtar, S.; Banerjee, S. Fabrication of Nano-CuO@ZnO for the Synthesis of Functionalized β-Enaminone Derivatives from β-Nitrostyrenes, Aliphatic/Aromatic Amines and 1,3-Dicarbonyl/4-Hydroxy Coumarin. ChemistrySelect 2017, 2, 7319–7324. [Google Scholar] [CrossRef]
- Sheldrick, G. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Westrip, S. publCIF: Software for editing, validating and formatting crystallographic information files. J. Appl. Crystallogr. 2010, 43, 920–925. [Google Scholar] [CrossRef]
- Dolomanov, O.; Bourhis, L.; Gildea, R.; Howard, J.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Spek, A. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
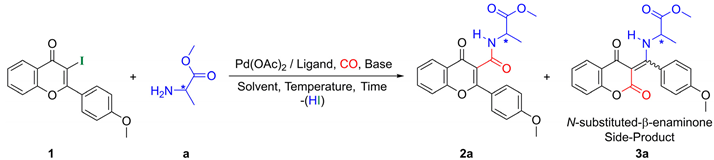 | |||||||||
| Entry | Base | Ligand | Solvent | Temp. [°C] | P(CO) [bar] | Time [h] | Conv. | Ratio (b) (2a/3a) | Yield (c) (2a/3a) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Et3N | PPh3 | DMF | 50 | 1 | 72 | 50 | 100/0 | 15/0 |
| 2 | Et3N | PPh3 | DMF | 80 | 1 | 72 | 100 | 81/19 | 42/10 |
| 3 | Et3N | PPh3 | DMF | 50 | 40 | 72 | 20 | 100/0 | 5/0 |
| 4 | Et3N | PPh3 | DMF | 80 | 40 | 72 | 85 | 100/0 | 25/0 |
| 5 | Et3N | dppf | DMF | 80 | 1 | 24 | 100 | 100/0 | 49/0 |
| 6 | Et3N | dppp | DMF | 80 | 1 | 72 | 80 | 100/0 | 30/0 |
| 7 | Et3N | XantPhos | DMF | 80 | 1 | 12 | 100 | 78/22 | 56/16 |
| 8 | Et3N | XantPhos | Toluene | 80 | 1 | 72 | 100 | 100/0 | 40/0 |
| 9 | Et3N | XantPhos | ACN | 80 | 1 | 48 | 100 | 75/25 | 55/18 |
| 10 | Et3N | XantPhos | Dioxane | 80 | 1 | 72 | 80 | 100/0 | 32/0 |
| 11 | K2CO3 | XantPhos | DMF | 80 | 1 | 12 | 100 | 96/4 | 72/3 |
| 12 | K2CO3 | XantPhos | DMF | 50 | 1 | 24 | 100 | 95/5 | 79/4 |
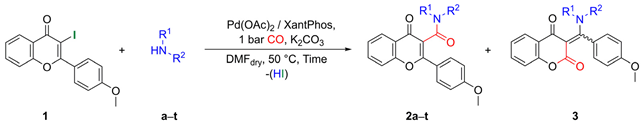 | ||||
| Entry | Amines | Time [h] | Flavone-3-carboxamides (2) N-Substituted β-Enaminones (3) | |
|---|---|---|---|---|
| Ratio (b) (2/3) | Yield (c) (2/3) | |||
| 1 | L-Alanine methyl ester (a) | 24 | 95/5 | 79/4 |
| 2 | L-Valine methyl ester (b) | 24 | 90/10 | 70/7 |
| 3 | (S)-2-Phenylglycine methyl ester (c) | 24 | 92/8 | 53/5 (d) |
| 4 | L-Phenylalanine methyl ester (d) | 24 | 96/4 | 68/3 (d) |
| 5 | L-Serine methyl ester (e) | 24 | 100/0 | 46/0 |
| 6 | Aniline (f) | 24 | 24/76 | 22/70 |
| 7 | 4-Picolylamine (g) | 48 | 86/14 | 36/6 (d) |
| 8 | Benzylamine (h) | 48 | 55/45 | 44/37 |
| 9 | Phenethylamine (i) | 48 | 60/40 | 41/28 |
| 10 | Piperonylamine (j) | 48 | 75/25 | 45/15 |
| 11 | N,O-dimethylhydroxylamine (k) | 48 | 95/5 | 71/4 (d) |
| 12 | Diethylamine (l) | 24 | 95/5 | 57/3 (d) |
| 13 | Morpholine (m) | 24 | 100/0 | 48/0 |
| 14 | Pyrrolidine (n) | 24 | 100/0 | 77/0 |
| 15 | N-Methylpiperazine (o) | 24 | 100/0 | 41/0 |
| 16 | Piperidine (p) | 48 | 100/0 | 67/0 |
| 17 | N-Ethyl-4-picolylamine (q) | 24 | 100/0 | 54/0 |
| 18 | 1-Adamantylamine (r) | 72 | 100/0 | 46/0 |
| 19 | Nortropine (s) | 48 | 100/0 | 70/0 |
| 20 | Nortropinone (t) | 24 | 100/0 | 51/0 |
| Compounds | 2r | 4f′ |
|---|---|---|
| Chemical formula | C27H27NO4 | C27H18NO5 |
| Mr | 429.49 | 422.41 |
| Crystal system | Orthorhombic | Monoclinic |
| Space group | P212121 | P21/c |
| Temperature (K) | 293 | 293 |
| a, b, c (Å) | 9.898 (5), 11.606 (7), 18.521 (11) | 15.086 (17), 8.042 (8), 17.586 (16) |
| α, β, γ (°) | 90, 90, 90 | 90, 110.03 (3), 90 |
| V (Å3) | 2128 (2) | 2005 (4) |
| Z | 4 | 4 |
| D (g cm−3) | 1.341 | 1.482 |
| Radiation type | Mo Kα | |
| μ (mm−1) | 0.09 | 0.10 |
| Crystal size (mm) | 0.70 × 0.09 × 0.07 | 0.24 × 0.17 × 0.10 |
| Data collection | ||
| Diffractometer | Bruker D8 VENTURE | |
| Absorption correction | Multi-scan SADABS2016/2—Bruker AXS area detector scaling and absorption correction | |
| Tmin, Tmax | 0.82, 0.99 | 0.83, 0.99 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 10,530, 4171, 1812 | 24,369, 3653, 2407 |
| Rint | 0.179 | 0.095 |
| (sin θ/λ)max (Å−1) | 0.620 | 0.603 |
| Refinement | ||
| R [F2 > 2s(F2)], wR(F2), S | 0.078, 0.175, 0.99 | 0.051, 0.142, 1.09 |
| No. of reflections | 4171 | 3653 |
| No. of parameters | 294 | 291 |
| H-atom treatment | H atoms are treated by a mixture of independent and constrained refinement | |
| Δ > max, Δ > min (e Å−3) | 0.22, −0.21 | 0.26, −0.25 |
| Crystals | Inter/Intramolecular Hydrogen Bonds and | Short Interactions (Å) | |||||
|---|---|---|---|---|---|---|---|
| A···H—D | D—H | H···A | D···A | ∠D—H···A | π···π | CH···π | |
| (2r) | N12—H12···O11 i | 0.850 | 2.421 | 3.241 | 161.03 | - | 2.907 |
| C6′—H6′···O11 ii | 0.930 | 2.713 | 3.417 | 133.13 | |||
| C5′—H5′ i ···O9 ii | 0.930 | 2.898 | 3.599 | 133.21 | |||
| (4f′) | C2′—H2′···O12 i | 0.930 | 2.867 | 3.556 | 132.93 | 3.74–3.89 | - |
| C8”—H8”···O9 ii | 0.930 | 2.650 | 3.544 | 161.68 | |||
| C3”—H3” ii···O7′ i | 0.930 | 2.766 | 3.544 | 121.21 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chniti, S.; Kollár, L.; Bényei, A.; Dörnyei, Á.; Takács, A. A Facile Route to Flavone-3-Carboxamides and Flavone-3-Carboxylates via Palladium-Catalyzed Amino- and Aryloxy-Carbonylation Reactions. Int. J. Mol. Sci. 2024, 25, 10128. https://doi.org/10.3390/ijms251810128
Chniti S, Kollár L, Bényei A, Dörnyei Á, Takács A. A Facile Route to Flavone-3-Carboxamides and Flavone-3-Carboxylates via Palladium-Catalyzed Amino- and Aryloxy-Carbonylation Reactions. International Journal of Molecular Sciences. 2024; 25(18):10128. https://doi.org/10.3390/ijms251810128
Chicago/Turabian StyleChniti, Sami, László Kollár, Attila Bényei, Ágnes Dörnyei, and Attila Takács. 2024. "A Facile Route to Flavone-3-Carboxamides and Flavone-3-Carboxylates via Palladium-Catalyzed Amino- and Aryloxy-Carbonylation Reactions" International Journal of Molecular Sciences 25, no. 18: 10128. https://doi.org/10.3390/ijms251810128
APA StyleChniti, S., Kollár, L., Bényei, A., Dörnyei, Á., & Takács, A. (2024). A Facile Route to Flavone-3-Carboxamides and Flavone-3-Carboxylates via Palladium-Catalyzed Amino- and Aryloxy-Carbonylation Reactions. International Journal of Molecular Sciences, 25(18), 10128. https://doi.org/10.3390/ijms251810128