
Accumulation of Cerebrospinal Fluid, Ventricular Enlargement, and Cerebral Folate Metabolic Errors Unify a Diverse Group of Neuropsychiatric Conditions Affecting Adult Neocortical Functions



Abstract
1. Introduction
2. Results
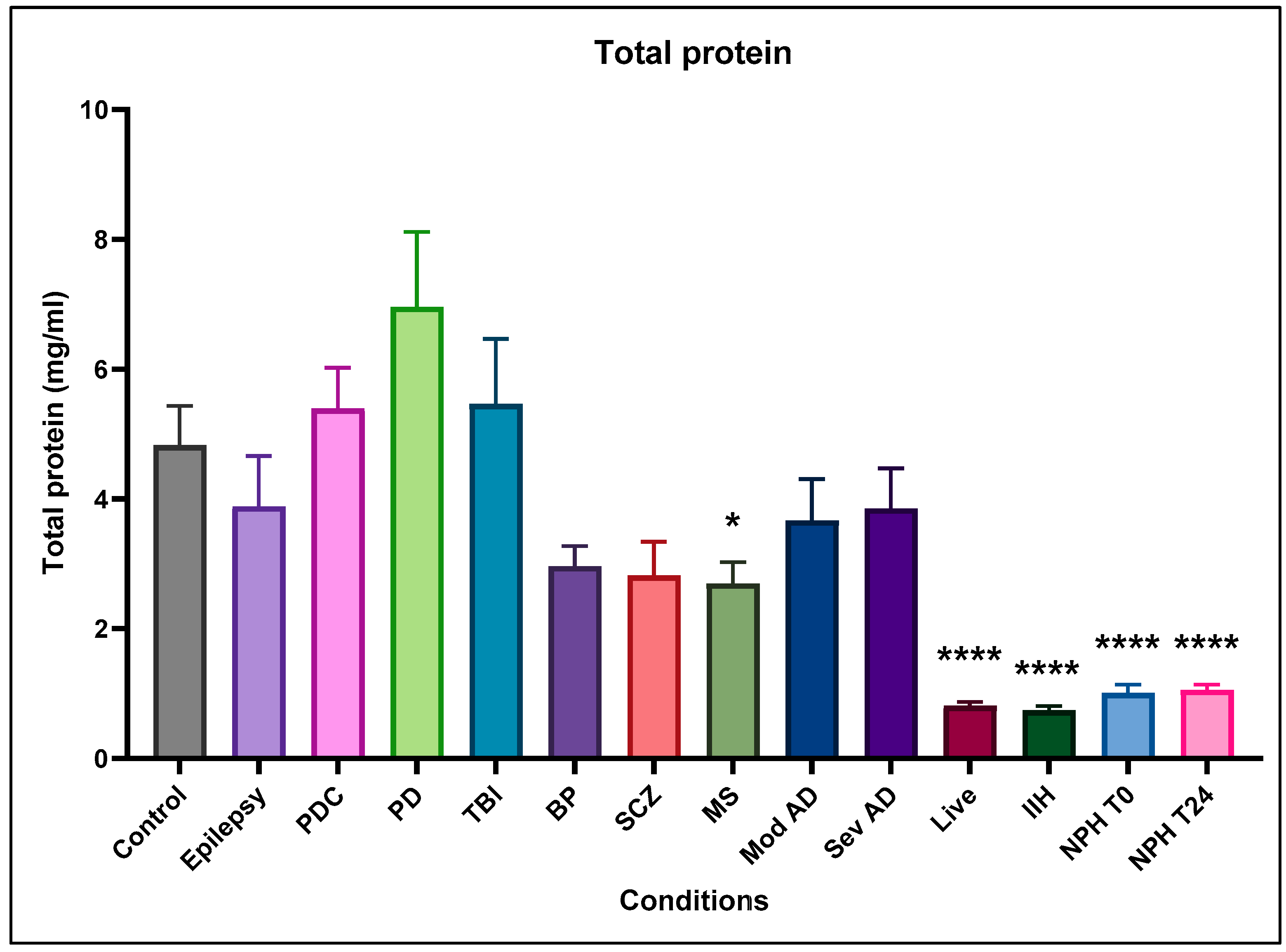
2.1. Total Protein
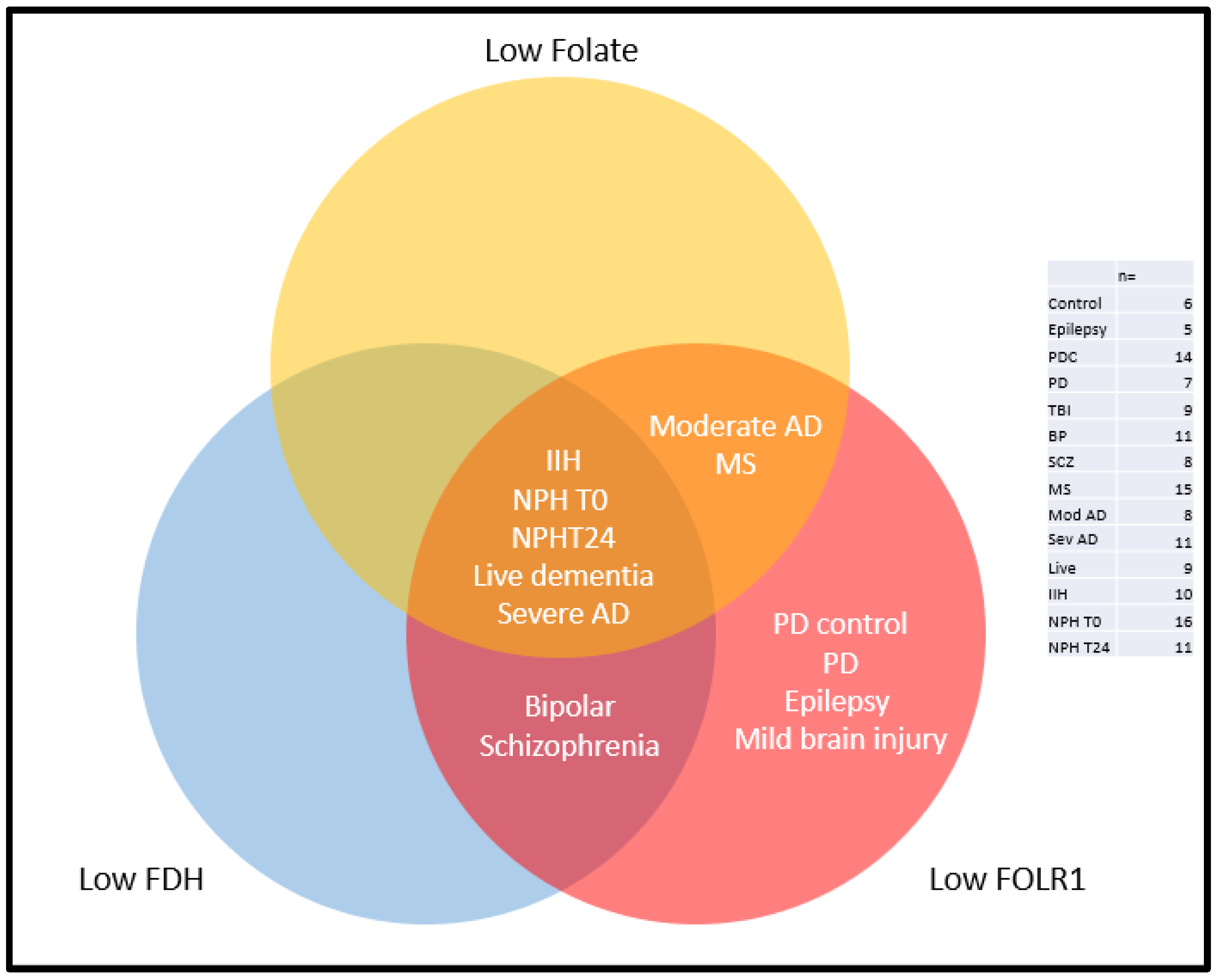
2.2. Folate
2.3. Folate Receptor Alpha (FOLR1)
2.4. 10-Formyl Tetrahydrofolate Dehydrogenase (FDH and ALDH1L1)
3. Discussion
4. Materials and Methods
4.1. Ethics
4.2. Tissue Supply
4.3. Western and Dot Blotting
4.4. Total Protein Measurement
4.5. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jones, P.B.; Harvey, I.; Lewis, S.W.; Toone, B.K.; Van Os, J.; Williams, M.; Murray, R.M. Cerebral ventricle dimensions as risk factors for schizophrenia and affective psychosis: An epidemiological approach to analysis. Psychol. Med. 1994, 24, 995–1011. [Google Scholar] [CrossRef]
- Harvey, I.; McGuffin, P.; Williams, M.; Toone, B.K. The ventricle-brain ratio (VBR) in functional psychoses: An admixture analysis. Psychiatry Res. 1990, 35, 61–69. [Google Scholar] [CrossRef]
- Saijo, T.; Abe, T.; Someya, Y.; Sassa, T.; Sudo, Y.; Suhara, T.; Shuno, T.; Asai, K.; Okubo, Y. Ten year progressive ventricular enlargement in schizophrenia: An MRI morphometrical study. Psychiatry Clin. Neurosci. 2001, 55, 41–47. [Google Scholar] [CrossRef]
- Strakowski, S.M.; DelBello, M.P.; Zimmerman, M.E.; Getz, G.E.; Mills, N.P.; Ret, J.; Shear, P.; Adler, C.M. Ventricular and periventricular structural volumes in first- versus multiple-episode bipolar disorder. Am. J. Psychiatry 2002, 159, 1841–1847. [Google Scholar] [CrossRef] [PubMed]
- Movsas, T.Z.; Pinto-Martin, J.A.; Whitaker, A.H.; Feldman, J.F.; Lorenz, J.M.; Korzeniewski, S.J.; Levy, S.E.; Paneth, N. Autism spectrum disorder is associated with ventricular enlargement in a low birth weight population. J. Pediatr. 2013, 163, 73–78. [Google Scholar] [CrossRef]
- Shen, M.D.; Nordahl, C.W.; Li, D.D.; Lee, A.; Angkustsiri, K.; Emerson, R.W.; Rogers, S.J.; Ozonoff, S.; Amaral, D.G. Extra-axial cerebrospinal fluid in high-risk and normal-risk children with autism aged 2–4 years: A case-control study. Lancet Psychiatry 2018, 5, 895–904. [Google Scholar] [CrossRef]
- Shen, M.D. Cerebrospinal fluid and the early brain development of autism. J. Neurodev. Disord. 2018, 10, 39. [Google Scholar] [CrossRef]
- Shen, M.D.; Kim, S.H.; McKinstry, R.C.; Gu, H.; Hazlett, H.C.; Nordahl, C.W.; Emerson, R.W.; Shaw, D.; Elison, J.T.; Swanson, M.R.; et al. Increased Extra-axial Cerebrospinal Fluid in High-Risk Infants Who Later Develop Autism. Biol. Psychiatry 2017, 82, 186–193. [Google Scholar] [CrossRef]
- Shen, M.D.; Nordahl, C.W.; Young, G.S.; Wootton-Gorges, S.L.; Lee, A.; Liston, S.E.; Harrington, K.R.; Ozonoff, S.; Amaral, D.G. Early brain enlargement and elevated extra-axial fluid in infants who develop autism spectrum disorder. Brain 2013, 136, 2825–2835. [Google Scholar] [CrossRef]
- Mak, E.; Su, L.; Williams, G.B.; Firbank, M.J.; Lawson, R.A.; Yarnall, A.J.; Duncan, G.W.; Mollenhauer, B.; Owen, A.M.; Khoo, T.K.; et al. Longitudinal whole-brain atrophy and ventricular enlargement in nondemented Parkinson’s disease. Neurobiol. Aging 2017, 55, 78–90. [Google Scholar] [CrossRef]
- Dalaker, T.O.; Zivadinov, R.; Ramasamy, D.P.; Beyer, M.K.; Alves, G.; Bronnick, K.S.; Tysnes, O.B.; Aarsland, D.; Larsen, J.P. Ventricular enlargement and mild cognitive impairment in early Parkinson’s disease. Mov. Disord. 2011, 26, 297–301. [Google Scholar] [CrossRef]
- Sapkota, S.; Gee, M.; Sabino, J.; Emery, D.; Camicioli, R. Association of homocysteine with ventricular dilatation and brain atrophy in Parkinson’s disease. Mov. Disord. 2014, 29, 368–374. [Google Scholar] [CrossRef]
- Tondelli, M.; Vaudano, A.E.; Sisodiya, S.M.; Meletti, S. Valproate Use Is Associated with Posterior Cortical Thinning and Ventricular Enlargement in Epilepsy Patients. Front. Neurol. 2020, 11, 622. [Google Scholar] [CrossRef]
- Mercimek-Mahmutoglu, S.; Stockler-Ipsiroglu, S. Cerebral folate deficiency and folinic acid treatment in hypomyelination with atrophy of the basal ganglia and cerebellum (H-ABC) syndrome. Tohoku J. Exp. Med. 2007, 211, 95–96, author reply 97. [Google Scholar] [CrossRef]
- Sinnecker, T.; Ruberte, E.; Schadelin, S.; Canova, V.; Amann, M.; Naegelin, Y.; Penner, I.K.; Muller, J.; Kuhle, J.; Decard, B.; et al. New and enlarging white matter lesions adjacent to the ventricle system and thalamic atrophy are independently associated with lateral ventricular enlargement in multiple sclerosis. J. Neurol. 2020, 267, 192–202. [Google Scholar] [CrossRef]
- Pontillo, G.; Cocozza, S.; Di Stasi, M.; Carotenuto, A.; Paolella, C.; Cipullo, M.B.; Perillo, T.; Vola, E.A.; Russo, C.; Masullo, M.; et al. 2D linear measures of ventricular enlargement may be relevant markers of brain atrophy and long-term disability progression in multiple sclerosis. Eur. Radiol. 2020, 30, 3813–3822. [Google Scholar] [CrossRef]
- Dalton, C.M.; Brex, P.A.; Jenkins, R.; Fox, N.C.; Miszkiel, K.A.; Crum, W.R.; O’Riordan, J.I.; Plant, G.T.; Thompson, A.J.; Miller, D.H. Progressive ventricular enlargement in patients with clinically isolated syndromes is associated with the early development of multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2002, 73, 141–147. [Google Scholar] [CrossRef]
- Turner, B.; Ramli, N.; Blumhardt, L.D.; Jaspan, T. Ventricular enlargement in multiple sclerosis: A comparison of three-dimensional and linear MRI estimates. Neuroradiology 2001, 43, 608–614. [Google Scholar] [CrossRef]
- Chou, Y.Y.; Lepore, N.; Avedissian, C.; Madsen, S.K.; Parikshak, N.; Hua, X.; Shaw, L.M.; Trojanowski, J.Q.; Weiner, M.W.; Toga, A.W.; et al. Mapping correlations between ventricular expansion and CSF amyloid and tau biomarkers in 240 subjects with Alzheimer’s disease, mild cognitive impairment and elderly controls. Neuroimage 2009, 46, 394–410. [Google Scholar] [CrossRef]
- Madsen, S.K.; Gutman, B.A.; Joshi, S.H.; Toga, A.W.; Jack, C.R., Jr.; Weiner, M.W.; Thompson, P.M. Mapping Dynamic Changes in Ventricular Volume onto Baseline Cortical Surfaces in Normal Aging, MCI, and Alzheimer’s Disease. Multimodal Brain Image Anal. 2013, 8159, 84–94. [Google Scholar] [CrossRef]
- Palavicini, J.P.; Ding, L.; Pan, M.; Qiu, S.; Wang, H.; Shen, Q.; Dupree, J.L.; Han, X. Sulfatide Deficiency, an Early Alzheimer’s Lipidomic Signature, Causes Brain Ventricular Enlargement in the Absence of Classical Neuropathological Hallmarks. Int. J. Mol. Sci. 2022, 24, 233. [Google Scholar] [CrossRef]
- Kuroda, T.; Honma, M.; Mori, Y.; Futamura, A.; Sugimoto, A.; Yano, S.; Kinno, R.; Murakami, H.; Ono, K. Increased Presence of Cerebral Microbleeds Correlates with Ventricular Enlargement and Increased White Matter Hyperintensities in Alzheimer’s Disease. Front. Aging Neurosci. 2020, 12, 13. [Google Scholar] [CrossRef]
- Ye, B.S.; Lee, Y.; Kwak, K.; Park, Y.H.; Ham, J.H.; Lee, J.J.; Shin, N.Y.; Lee, J.M.; Sohn, Y.H.; Lee, P.H. Posterior Ventricular Enlargement to Differentiate Dementia with Lewy Bodies from Alzheimer’s Disease. J. Alzheimer’s Dis. JAD 2016, 52, 1237–1243. [Google Scholar] [CrossRef]
- Nestor, S.M.; Rupsingh, R.; Borrie, M.; Smith, M.; Accomazzi, V.; Wells, J.L.; Fogarty, J.; Bartha, R.; Alzheimer’s Disease Neuroimaging, I. Ventricular enlargement as a possible measure of Alzheimer’s disease progression validated using the Alzheimer’s disease neuroimaging initiative database. Brain 2008, 131, 2443–2454. [Google Scholar] [CrossRef]
- Koivisto, A.M.; Kurki, M.I.; Alafuzoff, I.; Sutela, A.; Rummukainen, J.; Savolainen, S.; Vanninen, R.; Jaaskelainen, J.E.; Soininen, H.; Leinonen, V. High Risk of Dementia in Ventricular Enlargement with Normal Pressure Hydrocephalus Related Symptoms1. J. Alzheimer’s Dis. JAD 2016, 52, 497–507. [Google Scholar] [CrossRef]
- Zhuang, H.; Cho, J.; Chiang, G.C.; Kovanlikaya, I.; Heier, L.A.; Dyke, J.P.; Wang, Y. Cerebral oxygen extraction fraction declines with ventricular enlargement in patients with normal pressure hydrocephalus. Clin. Imaging 2023, 97, 22–27. [Google Scholar] [CrossRef]
- Yamada, S.; Ishikawa, M.; Nozaki, K. Exploring mechanisms of ventricular enlargement in idiopathic normal pressure hydrocephalus: A role of cerebrospinal fluid dynamics and motile cilia. Fluids Barriers CNS 2021, 18, 20. [Google Scholar] [CrossRef]
- Krishnamurthy, S.; Li, J.; Schultz, L.; Jenrow, K.A. Increased CSF osmolarity reversibly induces hydrocephalus in the normal rat brain. Fluids Barriers CNS 2012, 9, 13. [Google Scholar] [CrossRef]
- Oreskovic, D.; Rados, M.; Klarica, M. New Concepts of Cerebrospinal Fluid Physiology and Development of Hydrocephalus. Pediatr. Neurosurg. 2017, 52, 417–425. [Google Scholar] [CrossRef]
- Oreskovic, D.; Rados, M.; Klarica, M. Role of choroid plexus in cerebrospinal fluid hydrodynamics. Neuroscience 2017, 354, 69–87. [Google Scholar] [CrossRef]
- Minns, R.A.; Brown, J.K.; Engleman, H.M. CSF production rate: “real time” estimation. Z. Kinderchir. 1987, 42 (Suppl. S1), 36–40. [Google Scholar]
- Naz, N.; Naqvi, S.F.; Hohn, N.; Whelan, K.; Littler, P.; Roncaroli, F.; Robinson, A.C.; Miyan, J.A. Cerebral Folate Metabolism in Post-Mortem Alzheimer’s Disease Tissues: A Small Cohort Study. Int. J. Mol. Sci. 2022, 24, 660. [Google Scholar] [CrossRef]
- Miyan, J.; Buttercase, C.; Beswick, E.; Miyan, S.; Moshkdanian, G.; Naz, N. Folate Related Pathway Gene Analysis Reveals a Novel Metabolic Variant Associated with Alzheimer’s Disease with a Change in Metabolic Profile. Metabolites 2022, 12, 475. [Google Scholar] [CrossRef]
- Requena-Jimenez, A.; Nabiuni, M.; Miyan, J.A. Profound changes in cerebrospinal fluid proteome and metabolic profile are associated with congenital hydrocephalus. J. Cereb. Blood Flow Metab. 2021, 41, 3400–3414. [Google Scholar] [CrossRef]
- Naz, N.; Jimenez, A.R.; Sanjuan-Vilaplana, A.; Gurney, M.; Miyan, J. Neonatal hydrocephalus is a result of a block in folate handling and metabolism involving 10-formyltetrahydrofolate dehydrogenase. J. Neurochem. 2016, 138, 610–623. [Google Scholar] [CrossRef]
- Cains, S.; Shepherd, A.; Nabiuni, M.; Owen-Lynch, P.J.; Miyan, J. Addressing a folate imbalance in fetal cerebrospinal fluid can decrease the incidence of congenital hydrocephalus. J. Neuropathol. Exp. Neurol. 2009, 68, 404–416. [Google Scholar] [CrossRef]
- Mafi, S.; Laroche-Raynaud, C.; Chazelas, P.; Lia, A.S.; Derouault, P.; Sturtz, F.; Baaj, Y.; Froget, R.; Rio, M.; Benoist, J.F.; et al. Pharmacoresistant Epilepsy in Childhood: Think of the Cerebral Folate Deficiency, a Treatable Disease. Brain Sci. 2020, 10, 762. [Google Scholar] [CrossRef]
- Duarte, S.; Cruz Martins, R.; Rodrigues, M.; Lourenco, E.; Moreira, I.; Alonso, I.; Magalhaes, M. Association of cerebral folate deficiency and hereditary spastic paraplegia. Neurologia 2020, 36, 550–552. [Google Scholar] [CrossRef]
- Masingue, M.; Benoist, J.F.; Roze, E.; Moussa, F.; Sedel, F.; Lubetzki, C.; Nadjar, Y. Cerebral folate deficiency in adults: A heterogeneous potentially treatable condition. J. Neurol. Sci. 2019, 396, 112–118. [Google Scholar] [CrossRef]
- Krsicka, D.; Geryk, J.; Vlckova, M.; Havlovicova, M.; Macek, M., Jr.; Pourova, R. Identification of likely associations between cerebral folate deficiency and complex genetic- and metabolic pathogenesis of autism spectrum disorders by utilization of a pilot interaction modeling approach. Autism Res. 2017, 10, 1424–1435. [Google Scholar] [CrossRef]
- Thome, U.; Klima, P.; Moosa, A.N.; Gupta, A.; Parikh, S.; Pestana Knight, E.M. Electrographic status epilepticus in sleep in an adult with cerebral folate deficiency. Neurol. Clin. Pract. 2016, 6, e4–e7. [Google Scholar] [CrossRef]
- Nicolai, J.; van Kempen, M.J.; Postma, A.A. Teaching NeuroImages: White matter hypomyelination and progressive calcifications in cerebral folate deficiency. Neurology 2016, 87, e4–e5. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, P.; Luco, S.M.; Sawyer, S.L.; Davila, J.; Boycott, K.M.; Dyment, D.A. Late diagnosis of cerebral folate deficiency: Fewer seizures with folinic acid in adult siblings. Neurol. Genet. 2016, 2, e38. [Google Scholar] [CrossRef]
- Sadighi, Z.; Butler, I.J.; Koenig, M.K. Adult-onset cerebral folate deficiency. Arch. Neurol. 2012, 69, 778–779. [Google Scholar] [CrossRef]
- Leuzzi, V.; Mastrangelo, M.; Celato, A.; Carducci, C.; Carducci, C. A new form of cerebral folate deficiency with severe self-injurious behaviour. Acta Paediatr. 2012, 101, e482–e483. [Google Scholar] [CrossRef] [PubMed]
- Wijesekara, N. Dihydrofolate reductase mutations-associated megaloblastic anemia and cerebral folate deficiency. Clin. Genet. 2011, 79, 507–508. [Google Scholar] [CrossRef]
- Ho, A.; Michelson, D.; Aaen, G.; Ashwal, S. Cerebral folate deficiency presenting as adolescent catatonic schizophrenia: A case report. J. Child. Neurol. 2010, 25, 898–900. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Cazorla, A.; Quadros, E.V.; Nascimento, A.; Garcia-Silva, M.T.; Briones, P.; Montoya, J.; Ormazabal, A.; Artuch, R.; Sequeira, J.M.; Blau, N.; et al. Mitochondrial diseases associated with cerebral folate deficiency. Neurology 2008, 70, 1360–1362. [Google Scholar] [CrossRef]
- Moretti, P.; Sahoo, T.; Hyland, K.; Bottiglieri, T.; Peters, S.; del Gaudio, D.; Roa, B.; Curry, S.; Zhu, H.; Finnell, R.H.; et al. Cerebral folate deficiency with developmental delay, autism, and response to folinic acid. Neurology 2005, 64, 1088–1090. [Google Scholar] [CrossRef]
- Fame, R.M.; Cortes-Campos, C.; Sive, H.L. Brain Ventricular System and Cerebrospinal Fluid Development and Function: Light at the End of the Tube: A Primer with Latest Insights. Bioessays 2020, 42, e1900186. [Google Scholar] [CrossRef]
- Miyan, J.; Cains, S.; Larcombe, S.; Naz, N.; Jimenez, A.R.; Bueno, D.; Gato, A. Subarachnoid cerebrospinal fluid is essential for normal development of the cerebral cortex. Semin. Cell Dev. Biol. 2020, 102, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Gato, A.; Alonso, M.I.; Lamus, F.; Miyan, J. Neurogenesis: A process ontogenically linked to brain cavities and their content, CSF. Semin. Cell Dev. Biol. 2020, 102, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Alonso, M.I.; Santiago, C.; Moro, J.A.; De la Mano, A.; Carretero, R.; Gato, A. Early embryonic brain development in rats requires the trophic influence of cerebrospinal fluid. Int. J. Dev. Neurosci. 2009, 27, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Parada, C.; Martin, C.; Alonso, M.I.; Moro, J.A.; Bueno, D.; Gato, A. Embryonic cerebrospinal fluid collaborates with the isthmic organizer to regulate mesencephalic gene expression. J. Neurosci. Res. 2005, 82, 333–345. [Google Scholar] [CrossRef]
- Gato, A.; Moro, J.A.; Alonso, M.I.; Bueno, D.; De La Mano, A.; Martin, C. Embryonic cerebrospinal fluid regulates neuroepithelial survival, proliferation, and neurogenesis in chick embryos. Anat. Rec. A Discov. Mol. Cell Evol. Biol. 2005, 284, 475–484. [Google Scholar] [CrossRef]
- Miyan, J.A.; Nabiyouni, M.; Zendah, M. Development of the brain: A vital role for cerebrospinal fluid. Can. J. Physiol. Pharmacol. 2003, 81, 317–328. [Google Scholar] [CrossRef]
- Miyan, J.A.; Sobkowiak, C.; Draper, C.E. Humanity Lost: The cost of cortical maldevelopment. Is there light ahead? Eur. J. Pediatr. Surg. 2001, 11, S4–S7. [Google Scholar] [CrossRef]
- Albargothy, N.J.; Johnston, D.A.; MacGregor-Sharp, M.; Weller, R.O.; Verma, A.; Hawkes, C.A.; Carare, R.O. Convective influx/glymphatic system: Tracers injected into the CSF enter and leave the brain along separate periarterial basement membrane pathways. Acta Neuropathol. 2018, 136, 139–152. [Google Scholar] [CrossRef]
- Iliff, J.; Simon, M. CrossTalk proposal: The glymphatic system supports convective exchange of cerebrospinal fluid and brain interstitial fluid that is mediated by perivascular aquaporin-4. J. Physiol. 2019, 597, 4417–4419. [Google Scholar] [CrossRef]
- Piantino, J.; Lim, M.M.; Newgard, C.D.; Iliff, J. Linking Traumatic Brain Injury, Sleep Disruption and Post-Traumatic Headache: A Potential Role for Glymphatic Pathway Dysfunction. Curr. Pain Headache Rep. 2019, 23, 62. [Google Scholar] [CrossRef]
- Braun, M.; Iliff, J.J. The impact of neurovascular, blood-brain barrier, and glymphatic dysfunction in neurodegenerative and metabolic diseases. Int. Rev. Neurobiol. 2020, 154, 413–436. [Google Scholar] [CrossRef] [PubMed]
- Harrison, I.F.; Ismail, O.; Machhada, A.; Colgan, N.; Ohene, Y.; Nahavandi, P.; Ahmed, Z.; Fisher, A.; Meftah, S.; Murray, T.K.; et al. Impaired glymphatic function and clearance of tau in an Alzheimer’s disease model. Brain 2020, 143, 2576–2593. [Google Scholar] [CrossRef] [PubMed]
- Bohr, T.; Hjorth, P.G.; Holst, S.C.; Hrabetova, S.; Kiviniemi, V.; Lilius, T.; Lundgaard, I.; Mardal, K.A.; Martens, E.A.; Mori, Y.; et al. The glymphatic system: Current understanding and modeling. iScience 2022, 25, 104987. [Google Scholar] [CrossRef] [PubMed]
- Ke, Z.; Mo, Y.; Li, J.; Yang, D.; Huang, L.; Yang, Z.; Qin, R.; Mao, C.; Lv, W.; Huang, Y.; et al. Glymphatic Dysfunction Mediates the Influence of White Matter Hyperintensities on Episodic Memory in Cerebral Small Vessel Disease. Brain Sci. 2022, 12, 1611. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ruan, C.; Wu, Y.; Musa, M.; Zibrila, A.I.; Zhang, Z.; Salimeen, M. Variances of quantifying of Virchow-Robin spaces detecting the different functional status of glymphatic system in simple febrile seizures affected by seizures duration. Medicine 2022, 101, e32606. [Google Scholar] [CrossRef]
- Li, X.; Ruan, C.; Zibrila, A.I.; Musa, M.; Wu, Y.; Zhang, Z.; Liu, H.; Salimeen, M. Children with autism spectrum disorder present glymphatic system dysfunction evidenced by diffusion tensor imaging along the perivascular space. Medicine 2022, 101, e32061. [Google Scholar] [CrossRef]
- Piantino, J.A.; Iliff, J.J.; Lim, M.M. The Bidirectional Link Between Sleep Disturbances and Traumatic Brain Injury Symptoms: A Role for Glymphatic Dysfunction? Biol. Psychiatry 2022, 91, 478–487. [Google Scholar] [CrossRef]
- Shen, T.; Yue, Y.; Ba, F.; He, T.; Tang, X.; Hu, X.; Pu, J.; Huang, C.; Lv, W.; Zhang, B.; et al. Diffusion along perivascular spaces as marker for impairment of glymphatic system in Parkinson’s disease. NPJ Parkinsons Dis. 2022, 8, 174. [Google Scholar] [CrossRef]
- Simon, M.; Wang, M.X.; Ismail, O.; Braun, M.; Schindler, A.G.; Reemmer, J.; Wang, Z.; Haveliwala, M.A.; O’Boyle, R.P.; Han, W.Y.; et al. Loss of perivascular aquaporin-4 localization impairs glymphatic exchange and promotes amyloid beta plaque formation in mice. Alzheimers Res. Ther. 2022, 14, 59. [Google Scholar] [CrossRef]
- Verghese, J.P.; Terry, A.; de Natale, E.R.; Politis, M. Research Evidence of the Role of the Glymphatic System and Its Potential Pharmacological Modulation in Neurodegenerative Diseases. J. Clin. Med. 2022, 11, 6964. [Google Scholar] [CrossRef]
- Garic, D.; McKinstry, R.C.; Rutsohn, J.; Slomowitz, R.; Wolff, J.; MacIntyre, L.C.; Weisenfeld, L.A.H.; Kim, S.H.; Pandey, J.; St John, T.; et al. Enlarged Perivascular Spaces in Infancy and Autism Diagnosis, Cerebrospinal Fluid Volume, and Later Sleep Problems. JAMA Netw. Open 2023, 6, e2348341. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Dai, S.; Guo, T.; Si, X.; Lv, D.; Wang, Z.; Lu, J.; Fang, Y.; Guan, X.; Zhou, C.; et al. Noninvasive neuroimaging provides evidence for deterioration of the glymphatic system in Parkinson’s disease relative to essential tremor. Park. Relat. Disord. 2023, 107, 105254. [Google Scholar] [CrossRef] [PubMed]
- Gumeler, E.; Aygun, E.; Tezer, F.I.; Saritas, E.U.; Oguz, K.K. Assessment of glymphatic function in narcolepsy using DTI-ALPS index. Sleep. Med. 2023, 101, 522–527. [Google Scholar] [CrossRef]
- Hagiwara, A.; Tomizawa, Y.; Hoshino, Y.; Yokoyama, K.; Kamagata, K.; Sekine, T.; Takabayashi, K.; Nakaya, M.; Maekawa, T.; Akashi, T.; et al. Glymphatic System Dysfunction in Myelin Oligodendrocyte Glycoprotein Immunoglobulin G Antibody-Associated Disorders: Association with Clinical Disability. AJNR Am. J. Neuroradiol. 2023, 45, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Lee, J.; Chen, X.; Ziontz, J.; Ward, T.; Landau, S.M.; Baker, S.L.; Harrison, T.M.; Jagust, W.J. Global brain activity and its coupling with cerebrospinal fluid flow is related to tau pathology. bioRxiv 2023. [Google Scholar] [CrossRef]
- Osuna-Ramos, J.F.; Camberos-Barraza, J.; Torres-Mondragon, L.E.; Rabago-Monzon, A.R.; Camacho-Zamora, A.; Valdez-Flores, M.A.; Angulo-Rojo, C.E.; Guadron-Llanos, A.M.; Picos-Cardenas, V.J.; Calderon-Zamora, L.; et al. Interplay between the Glymphatic System and the Endocannabinoid System: Implications for Brain Health and Disease. Int. J. Mol. Sci. 2023, 24, 17458. [Google Scholar] [CrossRef] [PubMed]
- Pu, W.; Wei, S.; Qiu, M.; Chen, X.; Zou, W.; Ge, Y.; Qiu, W. Dysfunction of the glymphatic system in childhood absence epilepsy. Front. Neurosci. 2023, 17, 1312676. [Google Scholar] [CrossRef]
- Ye, F.; Keep, R.F.; Hua, Y.; Garton, H.J.L.; Xi, G. Glymphatic System and Post-hemorrhagic Hydrocephalus. Brain Hemorrhages 2023, 4, 44–46. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, S.; Wu, Y.; Tang, Z.; Wu, Y.; Qi, Y.; Dong, F.; Wang, Y. Enlarged Perivascular Space and Index for Diffusivity Along the Perivascular Space as Emerging Neuroimaging Biomarkers of Neurological Diseases. Cell. Mol. Neurobiol. 2023, 44, 14. [Google Scholar] [CrossRef]
- Hsu, S.L.; Liao, Y.C.; Wu, C.H.; Chang, F.C.; Chen, Y.L.; Lai, K.L.; Chung, C.P.; Chen, S.P.; Lee, Y.C. Impaired cerebral interstitial fluid dynamics in cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy. Brain Commun. 2024, 6, fcad349. [Google Scholar] [CrossRef]
- Huang, S.Y.; Zhang, Y.R.; Guo, Y.; Du, J.; Ren, P.; Wu, B.S.; Feng, J.F.; Alzheimer’s Disease Neuroimaging, I.; Cheng, W.; Yu, J.T. Glymphatic system dysfunction predicts amyloid deposition, neurodegeneration, and clinical progression in Alzheimer’s disease. Alzheimers Dement. 2024, 20, 3251–3269. [Google Scholar] [CrossRef] [PubMed]
- Ishida, K.; Yamada, K. Detection of Glymphatic Outflow of Tau from Brain to Cerebrospinal Fluid in Mice. Methods Mol. Biol. 2024, 2754, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Joseph, C.R.; Lim, J.K.; Grohol, B.N.; Zivcevska, M.; Lencke, J.; Rich, E.D.; Arrasmith, C.J.; Dorman, I.S.; Clark, B.W.; Love, K.; et al. Identifying delay in glymphatic clearance of labeled protons post-acute head trauma utilizing 3D ASL MRI (arterial spin labeling): A pilot study. Sci. Rep. 2024, 14, 6188. [Google Scholar] [CrossRef]
- Lopes, D.M.; Wells, J.A.; Ma, D.; Wallis, L.; Park, D.; Llewellyn, S.K.; Ahmed, Z.; Lythgoe, M.F.; Harrison, I.F. Glymphatic inhibition exacerbates tau propagation in an Alzheimer’s disease model. Alzheimers Res. Ther. 2024, 16, 71. [Google Scholar] [CrossRef]
- Margoni, M.; Pagani, E.; Meani, A.; Preziosa, P.; Mistri, D.; Gueye, M.; Moiola, L.; Filippi, M.; Rocca, M.A. Cognitive Impairment Is Related to Glymphatic System Dysfunction in Pediatric Multiple Sclerosis. Ann. Neurol. 2024, 95, 1080–1092. [Google Scholar] [CrossRef] [PubMed]
- Moum, S.J.; Gadde, J.A. Beyond the AJR: Updates on the Glymphatic System, Perivascular Spaces, and Autism During Infancy. AJR Am. J. Roentgenol. 2024. [Google Scholar] [CrossRef]
- Tu, Y.; Fang, Y.; Li, G.; Xiong, F.; Gao, F. Glymphatic System Dysfunction Underlying Schizophrenia Is Associated with Cognitive Impairment. Schizophr. Bull. 2024, 50, 1223–1231. [Google Scholar] [CrossRef]
- Xie, Y.; Zhu, H.; Yao, Y.; Liu, C.; Wu, S.; Zhang, Y.; Zhu, W. Enlarged choroid plexus in relapsing-remitting multiple sclerosis may lead to brain structural changes through the glymphatic impairment. Mult. Scler. Relat. Disord. 2024, 85, 105550. [Google Scholar] [CrossRef]
- Yamamoto, E.A.; Koike, S.; Luther, M.; Dennis, L.; Lim, M.M.; Raskind, M.; Pagulayan, K.; Iliff, J.; Peskind, E.; Piantino, J.A. Perivascular Space Burden and Cerebrospinal Fluid Biomarkers in US Veterans with Blast-Related Mild Traumatic Brain Injury. J. Neurotrauma 2024, 41, 13–14. [Google Scholar] [CrossRef]
- Yue, Y.; Zhang, X.; Lv, W.; Lai, H.Y.; Shen, T. Interplay between the glymphatic system and neurotoxic proteins in Parkinson’s disease and related disorders: Current knowledge and future directions. Neural Regen. Res. 2024, 19, 1973–1980. [Google Scholar] [CrossRef]
- Zhi, Z.; Liang, X.; Huang, M.; Wu, L.; Zhou, F. The association between glymphatic system dysfunction and alterations in cerebral function and structure in patients with white matter hyperintensities. Neuroreport 2024, 35, 476–485. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Mo, J.; Liu, K.; Chen, Q.; Li, Z.; He, Y.; Chang, Y.; Lin, C.; Yu, M.; Xu, Y.; et al. Glymphatic System Impairment Contributes to the Formation of Brain Edema After Ischemic Stroke. Stroke 2024, 55, 5. [Google Scholar] [CrossRef]
- Zou, K.; Deng, Q.; Zhang, H.; Huang, C. Glymphatic system: A gateway for neuroinflammation. Neural Regen. Res. 2024, 19, 2661–2672. [Google Scholar] [CrossRef] [PubMed]
- Shah, T.; Leurgans, S.E.; Mehta, R.I.; Yang, J.; Galloway, C.A.; de Mesy Bentley, K.L.; Schneider, J.A.; Mehta, R.I. Arachnoid granulations are lymphatic conduits that communicate with bone marrow and dura-arachnoid stroma. J. Exp. Med. 2023, 220, e20220618. [Google Scholar] [CrossRef]
- Sokolowski, W.; Barszcz, K.; Kupczynska, M.; Czubaj, N.; Skibniewski, M.; Purzyc, H. Lymphatic drainage of cerebrospinal fluid in mammals—are arachnoid granulations the main route of cerebrospinal fluid outflow? Biologia 2018, 73, 563–568. [Google Scholar] [CrossRef]
- Rekate, H.L.; Nadkarni, T.D.; Wallace, D. The importance of the cortical subarachnoid space in understanding hydrocephalus. J. Neurosurg. Pediatr. 2008, 2, 1–11. [Google Scholar] [CrossRef]
- Mehta, B.C.; Holman, D.W.; Grzybowski, D.M.; Chalmers, J.J. Characterization of arachnoidal cells cultured on three-dimensional nonwoven PET matrix. Tissue Eng. 2007, 13, 1269–1279. [Google Scholar] [CrossRef]
- Grzybowski, D.M.; Herderick, E.E.; Kapoor, K.G.; Holman, D.W.; Katz, S.E. Human arachnoid granulations Part I: A technique for quantifying area and distribution on the superior surface of the cerebral cortex. Cerebrospinal Fluid. Res. 2007, 4, 6. [Google Scholar] [CrossRef]
- Koh, L.; Zakharov, A.; Nagra, G.; Armstrong, D.; Friendship, R.; Johnston, M. Development of cerebrospinal fluid absorption sites in the pig and rat: Connections between the subarachnoid space and lymphatic vessels in the olfactory turbinates. Anat. Embryol. 2006, 211, 335–344. [Google Scholar] [CrossRef]
- Grzybowski, D.M.; Holman, D.W.; Katz, S.E.; Lubow, M. In vitro model of cerebrospinal fluid outflow through human arachnoid granulations. Investig. Ophthalmol. Vis. Sci. 2006, 47, 3664–3672. [Google Scholar] [CrossRef]
- Sandberg, D.I.; McComb, J.G.; Krieger, M.D. Chemical analysis of fluid obtained from intracranial arachnoid cysts in pediatric patients. J. Neurosurg. 2005, 103, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Zakharov, A.; Papaiconomou, C.; Djenic, J.; Midha, R.; Johnston, M. Lymphatic cerebrospinal fluid absorption pathways in neonatal sheep revealed by subarachnoid injection of Microfil. Neuropathol. Appl. Neurobiol. 2003, 29, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Boulton, M.; Flessner, M.; Armstrong, D.; Mohamed, R.; Hay, J.; Johnston, M. Contribution of extracranial lymphatics and arachnoid villi to the clearance of a CSF tracer in the rat. Am. J. Physiol. 1999, 276, R818–R823. [Google Scholar] [CrossRef] [PubMed]
- Boulton, M.; Armstrong, D.; Flessner, M.; Hay, J.; Szalai, J.P.; Johnston, M. Raised intracranial pressure increases CSF drainage through arachnoid villi and extracranial lymphatics. Am. J. Physiol. 1998, 275, R889–R896. [Google Scholar] [CrossRef]
- Frye, R.E.; Melnyk, S.; Fuchs, G.; Reid, T.; Jernigan, S.; Pavliv, O.; Hubanks, A.; Gaylor, D.W.; Walters, L.; James, S.J. Effectiveness of methylcobalamin and folinic Acid treatment on adaptive behavior in children with autistic disorder is related to glutathione redox status. Autism Res. Treat. 2013, 2013, 609705. [Google Scholar] [CrossRef]
- Turgut, M.; Erdogan, S.; Ergin, K.; Serter, M. Melatonin ameliorates blood-brain barrier permeability, glutathione, and nitric oxide levels in the choroid plexus of the infantile rats with kaolin-induced hydrocephalus. Brain Res. 2007, 1175, 117–125. [Google Scholar] [CrossRef]
- Chanson, A.; Rock, E.; Martin, J.F.; Liotard, A.; Brachet, P. Preferential response of glutathione-related enzymes to folate-dependent changes in the redox state of rat liver. Eur. J. Nutr. 2007, 46, 204–212. [Google Scholar] [CrossRef]
- Child, D.F.; Hudson, P.R.; Jones, H.; Davies, G.K.; De, P.; Mukherjee, S.; Brain, A.M.; Williams, C.P.; Harvey, J.N. The effect of oral folic acid on glutathione, glycaemia and lipids in Type 2 diabetes. Diabetes Nutr. Metab. 2004, 17, 95–102. [Google Scholar]
- Naz, N.; Moshkdanian, G.; Miyan, S.; Eljabri, S.; James, C.; Miyan, J. A Paternal Methylation Error in the Congenital Hydrocephalic Texas (H-Tx) Rat Is Partially Rescued with Natural Folate Supplements. Int. J. Mol. Sci. 2023, 24, 1638. [Google Scholar] [CrossRef]
- Lucock, M. Folic acid: Nutritional biochemistry, molecular biology, and role in disease processes. Mol. Genet. Metab. 2000, 71, 121–138. [Google Scholar] [CrossRef]
- Lucock, M. Is folic acid the ultimate functional food component for disease prevention? BMJ 2004, 328, 211–214. [Google Scholar] [CrossRef] [PubMed]
- Bueno, D.; Garcia-Fernandez, J. Evolutionary development of embryonic cerebrospinal fluid composition and regulation: An open research field with implications for brain development and function. Fluids Barriers CNS 2016, 13, 5. [Google Scholar] [CrossRef] [PubMed]
- Sangha, V.; Hoque, M.T.; Henderson, J.T.; Bendayan, R. Novel localization of folate transport systems in the murine central nervous system. Fluids Barriers CNS 2022, 19, 92. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.; Sequeira, J.M.; Quadros, E.V. The metabolic basis for developmental disorders due to defective folate transport. Biochimie 2016, 126, 31–42. [Google Scholar] [CrossRef]
- Delmelle, F.; Thony, B.; Clapuyt, P.; Blau, N.; Nassogne, M.C. Neurological improvement following intravenous high-dose folinic acid for cerebral folate transporter deficiency caused by FOLR-1 mutation. Eur. J. Paediatr. Neurol. 2016, 20, 709–713. [Google Scholar] [CrossRef]
- Grapp, M.; Just, I.A.; Linnankivi, T.; Wolf, P.; Lucke, T.; Hausler, M.; Gartner, J.; Steinfeld, R. Molecular characterization of folate receptor 1 mutations delineates cerebral folate transport deficiency. Brain 2012, 135, 2022–2031. [Google Scholar] [CrossRef]
- Steinfeld, R.; Grapp, M.; Kraetzner, R.; Dreha-Kulaczewski, S.; Helms, G.; Dechent, P.; Wevers, R.; Grosso, S.; Gartner, J. Folate receptor alpha defect causes cerebral folate transport deficiency: A treatable neurodegenerative disorder associated with disturbed myelin metabolism. Am. J. Hum. Genet. 2009, 85, 354–363. [Google Scholar] [CrossRef]
- Spector, R.; Lorenzo, A.V. Folate transport in the central nervous system. Am. J. Physiol. 1975, 229, 777–782. [Google Scholar] [CrossRef]
- Cousins, O.; Hodges, A.; Schubert, J.; Veronese, M.; Turkheimer, F.; Miyan, J.; Engelhardt, B.; Roncaroli, F. The blood-CSF-brain route of neurological disease: The indirect pathway into the brain. Neuropathol. Appl. Neurobiol. 2022, 48, e12789. [Google Scholar] [CrossRef]
- Alam, C.; Aufreiter, S.; Georgiou, C.J.; Hoque, M.T.; Finnell, R.H.; O’Connor, D.L.; Goldman, I.D.; Bendayan, R. Upregulation of reduced folate carrier by vitamin D enhances brain folate uptake in mice lacking folate receptor alpha. Proc. Natl. Acad. Sci. USA 2019, 116, 17531–17540. [Google Scholar] [CrossRef]
- Jimenez, A.R.; Naz, N.; Miyan, J.A. Altered folate binding protein expression and folate delivery are associated with congenital hydrocephalus in the hydrocephalic Texas rat. J. Cereb. Blood Flow. Metab. 2019, 39, 2061–2073. [Google Scholar] [CrossRef] [PubMed]
- Ramaekers, V.T.; Segers, K.; Sequeira, J.M.; Koenig, M.; Van Maldergem, L.; Bours, V.; Kornak, U.; Quadros, E.V. Genetic assessment and folate receptor autoantibodies in infantile-onset cerebral folate deficiency (CFD) syndrome. Mol. Genet. Metab. 2018, 124, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Perez-Duenas, B.; Ormazabal, A.; Toma, C.; Torrico, B.; Cormand, B.; Serrano, M.; Sierra, C.; De Grandis, E.; Marfa, M.P.; Garcia-Cazorla, A.; et al. Cerebral folate deficiency syndromes in childhood: Clinical, analytical, and etiologic aspects. Arch. Neurol. 2011, 68, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Ramaekers, V.T.; Sequeira, J.M.; Blau, N.; Quadros, E.V. A milk-free diet downregulates folate receptor autoimmunity in cerebral folate deficiency syndrome. Dev. Med. Child. Neurol. 2008, 50, 346–352. [Google Scholar] [CrossRef]
- Ramaekers, V.T.; Rothenberg, S.P.; Sequeira, J.M.; Opladen, T.; Blau, N.; Quadros, E.V.; Selhub, J. Autoantibodies to folate receptors in the cerebral folate deficiency syndrome. N. Engl. J. Med. 2005, 352, 1985–1991. [Google Scholar] [CrossRef]
- Karin, I.; Borggraefe, I.; Catarino, C.B.; Kuhm, C.; Hoertnagel, K.; Biskup, S.; Opladen, T.; Blau, N.; Heinen, F.; Klopstock, T. Folinic acid therapy in cerebral folate deficiency: Marked improvement in an adult patient. J. Neurol. 2017, 264, 578–582. [Google Scholar] [CrossRef]
- Mangold, S.; Blau, N.; Opladen, T.; Steinfeld, R.; Wessling, B.; Zerres, K.; Hausler, M. Cerebral folate deficiency: A neurometabolic syndrome? Mol. Genet. Metab. 2011, 104, 369–372. [Google Scholar] [CrossRef]
- Cao, X.; Wolf, A.; Kim, S.E.; Cabrera, R.M.; Wlodarczyk, B.J.; Zhu, H.; Parker, M.; Lin, Y.; Steele, J.W.; Han, X.; et al. CIC de novo loss of function variants contribute to cerebral folate deficiency by downregulating FOLR1 expression. J. Med. Genet. 2020, 58, 484–494. [Google Scholar] [CrossRef]
- Grapp, M.; Wrede, A.; Schweizer, M.; Huwel, S.; Galla, H.J.; Snaidero, N.; Simons, M.; Buckers, J.; Low, P.S.; Urlaub, H.; et al. Choroid plexus transcytosis and exosome shuttling deliver folate into brain parenchyma. Nat. Commun. 2013, 4, 2123. [Google Scholar] [CrossRef]
- Ramaekers, V.T.; Quadros, E.V.; Sequeira, J.M. Role of folate receptor autoantibodies in infantile autism. Mol. Psychiatry 2013, 18, 270–271. [Google Scholar] [CrossRef]
- Ramaekers, V.; Sequeira, J.M.; Quadros, E.V. Clinical recognition and aspects of the cerebral folate deficiency syndromes. Clin. Chem. Lab. Med. 2013, 51, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Krupenko, S.A.; Krupenko, N.I. Loss of ALDH1L1 folate enzyme confers a selective metabolic advantage for tumor progression. Chem. Biol. Interact. 2019, 302, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Krupenko, S.A.; Krupenko, N.I. ALDH1L1 and ALDH1L2 Folate Regulatory Enzymes in Cancer. Adv. Exp. Med. Biol. 2018, 1032, 127–143. [Google Scholar] [CrossRef] [PubMed]
- Owen-Lynch, P.J.; Draper, C.E.; Mashayekhi, F.; Bannister, C.M.; Miyan, J.A. Defective cell cycle control underlies abnormal cortical development in the hydrocephalic Texas rat. Brain 2003, 126, 623–631. [Google Scholar] [CrossRef]
- Winchenbach, J.; Duking, T.; Berghoff, S.A.; Stumpf, S.K.; Hulsmann, S.; Nave, K.A.; Saher, G. Inducible targeting of CNS astrocytes in Aldh1l1-CreERT2 BAC transgenic mice. F1000Research 2016, 5, 2934. [Google Scholar] [CrossRef]
- Kwon, J.W.; Im, J.H.; Lee, K.Y.; Yoo, B.C.; Lee, J.H.; Kim, K.H.; Kim, J.H.; Shin, S.H.; Yoo, H.; Gwak, H.S. Different Metabolomic and Proteomic Profiles of Cerebrospinal Fluid in Ventricular and Lumbar Compartments in Relation to Leptomeningeal Metastases. Metabolites 2022, 12, 80. [Google Scholar] [CrossRef]
- Combrinck, J.; Tshavhungwe, P.; Rohlwink, U.; Enslin, N.; Thango, N.; Lazarus, J.; Kriegler, K.; Castel, S.; Abdelgawad, N.; McIlleron, H.; et al. Rifampicin and protein concentrations in paired spinal versus ventricular cerebrospinal fluid samples of children with tuberculous meningitis. J. Antimicrob. Chemother. 2024, 79, 280–286. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition | (n) | Source |
|---|---|---|
| Control | 7 | Netherlands Brain Bank |
| Epilepsy | 6 | |
| Schizophrenia (SCZ) | 12 | |
| Bipolar disorder (BP) | 13 | |
| Non-Parkinson’s (PDC) | 14 | Imperial College Brain Bank |
| Parkinson’s disease (PD) | 9 | |
| Multiple sclerosis (MS) | 15 | |
| Mild traumatic brain injury (TBI) | 9 | Manchester Brain Bank—brains for dementia research |
| Moderate Alzheimer’s (AD) | 10 | |
| Severe Alzheimer’s (AD) | 12 | |
| Live dementia | 9 | Liverpool Walton Centre Biobank |
| Intracranial hypertension (IIH) | 10 | |
| Normal-pressure hydrocephalus T0 | 14 | Preston Royal Hospital |
| and T24 | 13 |
| Primary Antibody | Cat. Number | Source | Host Species | Working Concentration |
|---|---|---|---|---|
| Polyclonal anti-ALDH1L1 | 17390-1-AP | Proteintech | Rabbit | 1:2000 |
| Polyclonal anti-FOLR1 | 29472-1-AP | Proteintech | Rabbit | 1:2000 |
| Monoclonal anti- 5MTH folic acid | M5028 | Sigma-Aldrich | Mouse | 1:2000 |
| Secondary Antibody | ||||
| StarBright Blue 700 Goat anti-Rabbit IgG | 12004162 | BIORAD | Goat | 1:4000 |
| StarBright Blue 700 Goat anti-Mouse IgG | 12004158 | BIORAD | Goat | 1:4000 |
| Protein | Source | Cat. Number | Info |
|---|---|---|---|
| FOLR1 | Proteintech | Ag19959 | GST fusion protein—partial length |
| ALDH1L1 | Abnova | H00010840-q01 | 60Kd partial protein |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ikeda, L.; Capel, A.V.; Doddaballapur, D.; Miyan, J. Accumulation of Cerebrospinal Fluid, Ventricular Enlargement, and Cerebral Folate Metabolic Errors Unify a Diverse Group of Neuropsychiatric Conditions Affecting Adult Neocortical Functions. Int. J. Mol. Sci. 2024, 25, 10205. https://doi.org/10.3390/ijms251810205
Ikeda L, Capel AV, Doddaballapur D, Miyan J. Accumulation of Cerebrospinal Fluid, Ventricular Enlargement, and Cerebral Folate Metabolic Errors Unify a Diverse Group of Neuropsychiatric Conditions Affecting Adult Neocortical Functions. International Journal of Molecular Sciences. 2024; 25(18):10205. https://doi.org/10.3390/ijms251810205
Chicago/Turabian StyleIkeda, Lena, Adrià Vilaseca Capel, Dhruti Doddaballapur, and Jaleel Miyan. 2024. "Accumulation of Cerebrospinal Fluid, Ventricular Enlargement, and Cerebral Folate Metabolic Errors Unify a Diverse Group of Neuropsychiatric Conditions Affecting Adult Neocortical Functions" International Journal of Molecular Sciences 25, no. 18: 10205. https://doi.org/10.3390/ijms251810205
APA StyleIkeda, L., Capel, A. V., Doddaballapur, D., & Miyan, J. (2024). Accumulation of Cerebrospinal Fluid, Ventricular Enlargement, and Cerebral Folate Metabolic Errors Unify a Diverse Group of Neuropsychiatric Conditions Affecting Adult Neocortical Functions. International Journal of Molecular Sciences, 25(18), 10205. https://doi.org/10.3390/ijms251810205