
Polymer Physics Models Reveal Structural Folding Features of Single-Molecule Gene Chromatin Conformations

 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
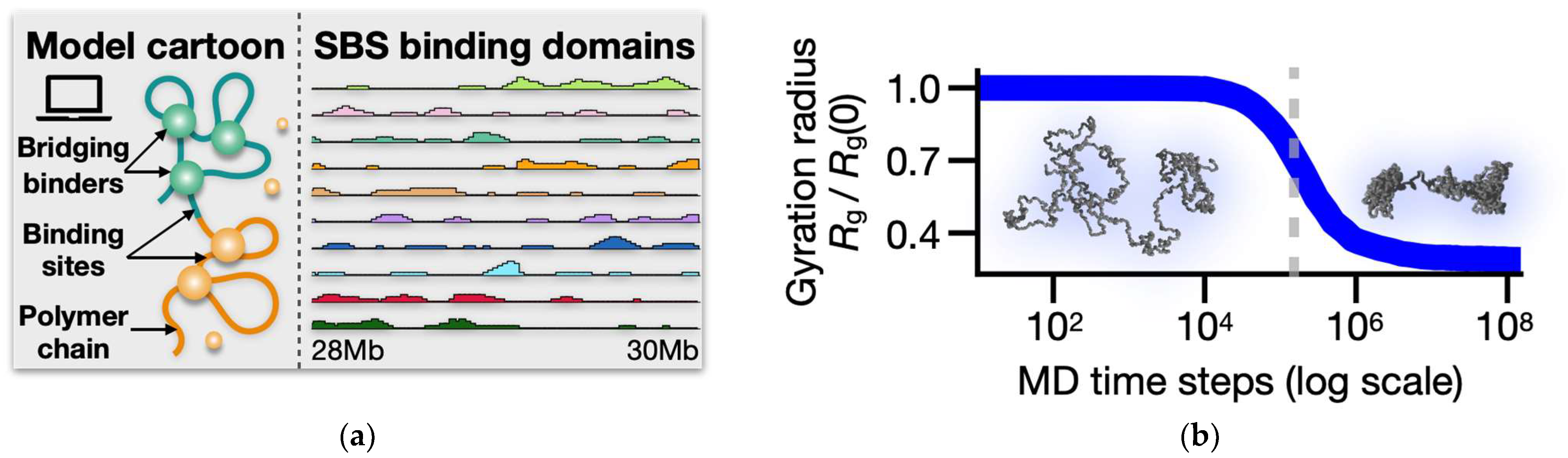
2.1. The Strings and Binders (SBS) Polymer Model
2.2. Molecular Dynamics (MD) Simulations
3. Results
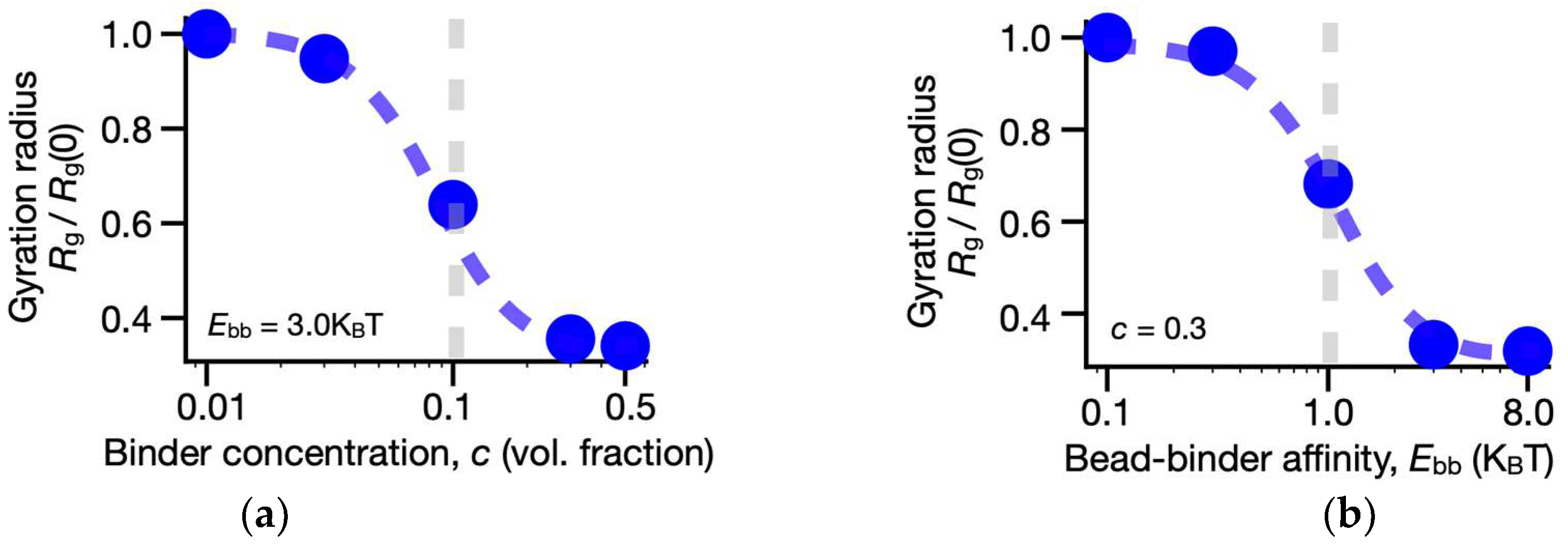
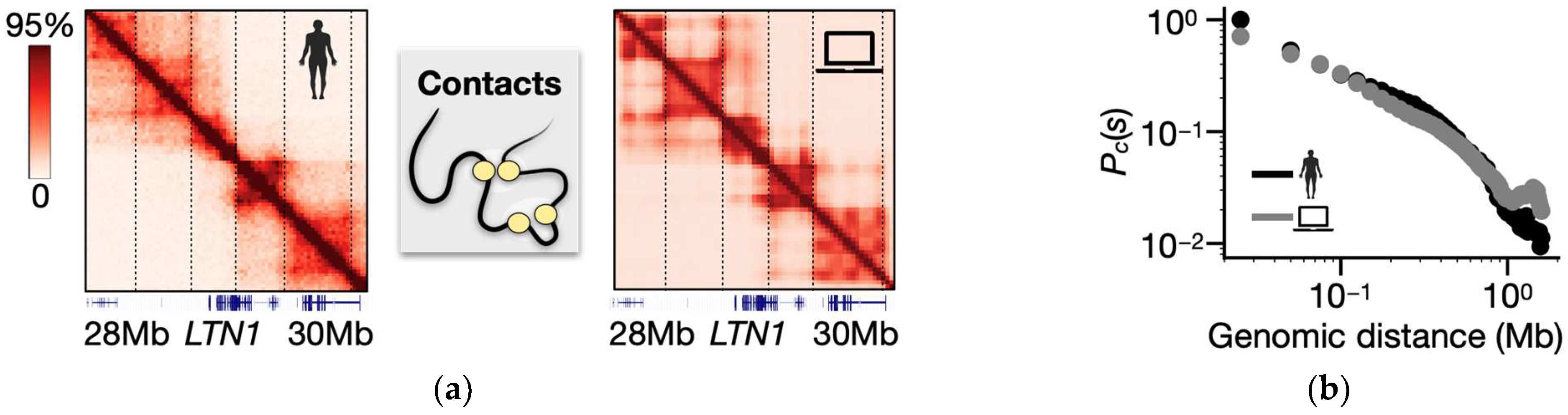
3.1. Folding of the SBS Model of the Human LTN1 Gene Locus
3.2. Structural Heterogeneity of LTN1 Single-Molecule Conformations in the Model
3.3. Shape and Size of 3D Model Single Molecules
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive Mapping of Long-Range Interactions Reveals Folding Principles of the Human Genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Belton, J.M.; McCord, R.P.; Gibcus, J.H.; Naumova, N.; Zhan, Y.; Dekker, J. Hi-C: A Comprehensive Technique to Capture the Conformation of Genomes. Methods 2012, 58, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Beagrie, R.A.; Scialdone, A.; Schueler, M.; Kraemer, D.C.A.; Chotalia, M.; Xie, S.Q.; Barbieri, M.; De Santiago, I.; Lavitas, L.M.; Branco, M.R.; et al. Complex Multi-Enhancer Contacts Captured by Genome Architecture Mapping. Nature 2017, 543, 519–524. [Google Scholar] [CrossRef]
- Winick-Ng, W.; Kukalev, A.; Harabula, I.; Zea-Redondo, L.; Szabó, D.; Meijer, M.; Serebreni, L.; Zhang, Y.; Bianco, S.; Chiariello, A.M.; et al. Cell-Type Specialization Is Encoded by Specific Chromatin Topologies. Nature 2021, 599, 684–691. [Google Scholar] [CrossRef] [PubMed]
- Quinodoz, S.A.; Ollikainen, N.; Tabak, B.; Palla, A.; Schmidt, J.M.; Detmar, E.; Lai, M.M.; Shishkin, A.A.; Bhat, P.; Takei, Y.; et al. Higher-Order Inter-Chromosomal Hubs Shape 3D Genome Organization in the Nucleus. Cell 2018, 174, 744–757.e24. [Google Scholar] [CrossRef] [PubMed]
- Quinodoz, S.A.; Bhat, P.; Chovanec, P.; Jachowicz, J.W.; Ollikainen, N.; Detmar, E.; Soehalim, E.; Guttman, M. SPRITE: A Genome-Wide Method for Mapping Higher-Order 3D Interactions in the Nucleus Using Combinatorial Split-and-Pool Barcoding. Nat. Protoc. 2022, 17, 36–75. [Google Scholar] [CrossRef] [PubMed]
- Bickmore, W.A.; van Steensel, B. Genome Architecture: Domain Organization of Interphase Chromosomes. Cell 2013, 152, 1270–1284. [Google Scholar] [CrossRef]
- Dekker, J.; Mirny, L. The 3D Genome as Moderator of Chromosomal Communication. Cell 2016, 164, 1110–1121. [Google Scholar] [CrossRef]
- Dixon, J.R.; Gorkin, D.U.; Ren, B. Chromatin Domains: The Unit of Chromosome Organization. Mol. Cell 2016, 62, 668–680. [Google Scholar] [CrossRef]
- Spielmann, M.; Lupiáñez, D.G.; Mundlos, S. Structural Variation in the 3D Genome. Nat. Rev. Genet. 2018, 19, 453–467. [Google Scholar] [CrossRef]
- Akgol Oksuz, B.; Yang, L.; Abraham, S.; Venev, S.V.; Krietenstein, N.; Parsi, K.M.; Ozadam, H.; Oomen, M.E.; Nand, A.; Mao, H.; et al. Systematic Evaluation of Chromosome Conformation Capture Assays. Nat. Methods 2021, 18, 1046–1055. [Google Scholar] [CrossRef] [PubMed]
- Bickmore, W.A. The Spatial Organization of the Human Genome. Annu. Rev. Genom. Hum. Genet. 2013, 14, 67–84. [Google Scholar] [CrossRef] [PubMed]
- Bonev, B.; Mendelson Cohen, N.; Szabo, Q.; Fritsch, L.; Papadopoulos, G.L.; Lubling, Y.; Xu, X.; Lv, X.; Hugnot, J.-P.; Tanay, A.; et al. Multiscale 3D Genome Rewiring during Mouse Neural Development. Cell 2017, 171, 557–572.e24. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.S.P.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [PubMed]
- Nora, E.P.; Lajoie, B.R.; Schulz, E.G.; Giorgetti, L.; Okamoto, I.; Servant, N.; Piolot, T.; van Berkum, N.L.; Meisig, J.; Sedat, J.; et al. Spatial Partitioning of the Regulatory Landscape of the X-Inactivation Centre. Nature 2012, 485, 381–385. [Google Scholar] [CrossRef]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological Domains in Mammalian Genomes Identified by Analysis of Chromatin Interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef]
- Fraser, J.; Ferrai, C.; Chiariello, A.M.; Schueler, M.; Rito, T.; Laudanno, G.; Barbieri, M.; Moore, B.L.; Kraemer, D.C.; Aitken, S.; et al. Hierarchical Folding and Reorganization of Chromosomes Are Linked to Transcriptional Changes in Cellular Differentiation. Mol. Syst. Biol. 2015, 11, 852. [Google Scholar] [CrossRef]
- van Steensel, B.; Belmont, A.S. Lamina-Associated Domains: Links with Chromosome Architecture, Heterochromatin, and Gene Repression. Cell 2017, 169, 780–791. [Google Scholar] [CrossRef]
- Cremer, T.; Cremer, C. Chromosome Territories, Nuclear Architecture and Gene Regulation in Mammalian Cells. Nat. Rev. Genet. 2001, 2, 292–301. [Google Scholar] [CrossRef]
- Sexton, T.; Cavalli, G. The Role of Chromosome Domains in Shaping the Functional Genome. Cell 2015, 160, 1049–1059. [Google Scholar] [CrossRef]
- Schoenfelder, S.; Fraser, P. Long-Range Enhancer–Promoter Contacts in Gene Expression Control. Nat. Rev. Genet. 2019, 20, 437–455. [Google Scholar] [CrossRef] [PubMed]
- Dowen, J.M.; Fan, Z.P.; Hnisz, D.; Ren, G.; Abraham, B.J.; Zhang, L.N.; Weintraub, A.S.; Schuijers, J.; Lee, T.I.; Zhao, K.; et al. Control of Cell Identity Genes Occurs in Insulated Neighborhoods in Mammalian Chromosomes. Cell 2014, 159, 374–387. [Google Scholar] [CrossRef] [PubMed]
- Bonev, B.; Cavalli, G. Organization and Function of the 3D Genome. Nat. Rev. Genet. 2016, 17, 661–678. [Google Scholar] [CrossRef] [PubMed]
- Kubo, N.; Ishii, H.; Xiong, X.; Bianco, S.; Meitinger, F.; Hu, R.; Hocker, J.D.; Conte, M.; Gorkin, D.; Yu, M.; et al. Promoter-Proximal CTCF Binding Promotes Distal Enhancer-Dependent Gene Activation. Nat. Struct. Mol. Biol. 2021, 28, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zhu, Q.; Jussila, A.; Han, Y.; Bintu, B.; Kern, C.; Conte, M.; Zhang, Y.; Bianco, S.; Chiariello, A.M.; et al. CTCF Mediates Dosage- and Sequence-Context-Dependent Transcriptional Insulation by Forming Local Chromatin Domains. Nat. Genet. 2021, 53, 1064–1074. [Google Scholar] [CrossRef] [PubMed]
- Willemin, A.; Szabó, D.; Pombo, A. Epigenetic Regulatory Layers in the 3D Nucleus. Mol. Cell 2024, 84, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Salamon, I.; Serio, S.; Bianco, S.; Pagiatakis, C.; Crasto, S.; Chiariello, A.M.; Conte, M.; Cattaneo, P.; Fiorillo, L.; Felicetta, A.; et al. Divergent Transcription of the Nkx2-5 Locus Generates Two Enhancer RNAs with Opposing Functions. iScience 2020, 23, 101539. [Google Scholar] [CrossRef]
- Misteli, T. The Self-Organizing Genome: Principles of Genome Architecture and Function. Cell 2020, 183, 28–45. [Google Scholar] [CrossRef]
- Gorkin, D.U.; Leung, D.; Ren, B. The 3D Genome in Transcriptional Regulation and Pluripotency. Cell Stem Cell 2014, 14, 762–775. [Google Scholar] [CrossRef]
- Cavalli, G.; Misteli, T. Functional Implications of Genome Topology. Nat. Struct. Mol. Biol. 2013, 20, 290–299. [Google Scholar] [CrossRef]
- Symmons, O.; Uslu, V.V.; Tsujimura, T.; Ruf, S.; Nassari, S.; Schwarzer, W.; Ettwiller, L.; Spitz, F. Functional and Topological Characteristics of Mammalian Regulatory Domains. Genome Res. 2014, 24, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Lupiáñez, D.G.; Kraft, K.; Heinrich, V.; Krawitz, P.; Brancati, F.; Klopocki, E.; Horn, D.; Kayserili, H.; Opitz, J.M.; Laxova, R.; et al. Disruptions of Topological Chromatin Domains Cause Pathogenic Rewiring of Gene-Enhancer Interactions. Cell 2015, 161, 1012–1025. [Google Scholar] [CrossRef] [PubMed]
- Lupiáñez, D.G.; Spielmann, M.; Mundlos, S. Breaking TADs: How Alterations of Chromatin Domains Result in Disease. Trends Genet. 2016, 32, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Weischenfeldt, J.; Dubash, T.; Drainas, A.P.; Mardin, B.R.; Chen, Y.; Stütz, A.M.; Waszak, S.M.; Bosco, G.; Halvorsen, A.R.; Raeder, B.; et al. Pan-Cancer Analysis of Somatic Copy-Number Alterations Implicates IRS4 and IGF2 in Enhancer Hijacking. Nat. Genet. 2017, 49, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Kraft, K.; Magg, A.; Heinrich, V.; Riemenschneider, C.; Schöpflin, R.; Markowski, J.; Ibrahim, D.M.; Acuna-Hidalgo, R.; Despang, A.; Andrey, G.; et al. Serial Genomic Inversions Induce Tissue-Specific Architectural Stripes, Gene Misexpression and Congenital Malformations. Nat. Cell Biol. 2019, 21, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Hnisz, D.; Weintraub, A.S.; Day, D.S.; Valton, A.-L.; Bak, R.O.; Li, C.H.; Goldmann, J.; Lajoie, B.R.; Fan, Z.P.; Sigova, A.A.; et al. Activation of Proto-Oncogenes by Disruption of Chromosome Neighborhoods. Science 2016, 351, 1454–1458. [Google Scholar] [CrossRef]
- Franke, M.; Ibrahim, D.M.; Andrey, G.; Schwarzer, W.; Heinrich, V.; Schöpflin, R.; Kraft, K.; Kempfer, R.; Jerković, I.; Chan, W.-L.; et al. Formation of New Chromatin Domains Determines Pathogenicity of Genomic Duplications. Nature 2016, 538, 265–269. [Google Scholar] [CrossRef]
- Sati, S.; Bonev, B.; Szabo, Q.; Jost, D.; Bensadoun, P.; Serra, F.; Loubiere, V.; Papadopoulos, G.L.; Rivera-Mulia, J.-C.; Fritsch, L.; et al. 4D Genome Rewiring during Oncogene-Induced and Replicative Senescence. Mol. Cell 2020, 78, 522–538.e9. [Google Scholar] [CrossRef]
- Anania, C.; Lupiáñez, D.G. Order and Disorder: Abnormal 3D Chromatin Organization in Human Disease. Brief. Funct. Genom. 2020, 19, 128–138. [Google Scholar] [CrossRef]
- Norton, H.K.; Phillips-Cremins, J.E. Crossed Wires: 3D Genome Misfolding in Human Disease. J. Cell Biol. 2017, 216, 3441–3452. [Google Scholar] [CrossRef]
- Ibn-Salem, J.; Köhler, S.; Love, M.I.; Chung, H.-R.; Huang, N.; Hurles, M.E.; Haendel, M.; Washington, N.L.; Smedley, D.; Mungall, C.J.; et al. Deletions of Chromosomal Regulatory Boundaries Are Associated with Congenital Disease. Genome Biol. 2014, 15, 423. [Google Scholar] [CrossRef] [PubMed]
- Flavahan, W.A.; Drier, Y.; Liau, B.B.; Gillespie, S.M.; Venteicher, A.S.; Stemmer-Rachamimov, A.O.; Suvà, M.L.; Bernstein, B.E. Insulator Dysfunction and Oncogene Activation in IDH Mutant Gliomas. Nature 2016, 529, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Lee, C.; Zichner, T.; Stütz, A.M.; Erkek, S.; Kawauchi, D.; Shih, D.J.H.; Hovestadt, V.; Zapatka, M.; Sturm, D.; et al. Enhancer Hijacking Activates GFI1 Family Oncogenes in Medulloblastoma. Nature 2014, 511, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Finn, E.H.; Misteli, T. Molecular Basis and Biological Function of Variability in Spatial Genome Organization. Science 2019, 365, eaaw9498. [Google Scholar] [CrossRef]
- Bintu, B.; Mateo, L.J.; Su, J.-H.; Sinnott-Armstrong, N.A.; Parker, M.; Kinrot, S.; Yamaya, K.; Boettiger, A.N.; Zhuang, X. Super-Resolution Chromatin Tracing Reveals Domains and Cooperative Interactions in Single Cells. Science 2018, 362, eaau1783. [Google Scholar] [CrossRef]
- Su, J.-H.; Zheng, P.; Kinrot, S.S.; Bintu, B.; Zhuang, X. Genome-Scale Imaging of the 3D Organization and Transcriptional Activity of Chromatin. Cell 2020, 182, 1641–1659.e26. [Google Scholar] [CrossRef]
- Mateo, L.J.; Murphy, S.E.; Hafner, A.; Cinquini, I.S.; Walker, C.A.; Boettiger, A.N. Visualizing DNA Folding and RNA in Embryos at Single-Cell Resolution. Nature 2019, 568, 49–54. [Google Scholar] [CrossRef]
- Wang, S.; Su, J.H.; Beliveau, B.J.; Bintu, B.; Moffitt, J.R.; Wu, C.T.; Zhuang, X. Spatial Organization of Chromatin Domains and Compartments in Single Chromosomes. Science 2016, 353. [Google Scholar] [CrossRef]
- Nguyen, H.Q.; Chattoraj, S.; Castillo, D.; Nguyen, S.C.; Nir, G.; Lioutas, A.; Hershberg, E.A.; Martins, N.M.C.; Reginato, P.L.; Hannan, M.; et al. 3D Mapping and Accelerated Super-Resolution Imaging of the Human Genome Using in Situ Sequencing. Nat. Methods 2020, 17, 822–832. [Google Scholar] [CrossRef]
- Takei, Y.; Yun, J.; Zheng, S.; Ollikainen, N.; Pierson, N.; White, J.; Shah, S.; Thomassie, J.; Suo, S.; Eng, C.-H.L.; et al. Integrated Spatial Genomics Reveals Global Architecture of Single Nuclei. Nature 2021, 590, 344–350. [Google Scholar] [CrossRef]
- Takei, Y.; Zheng, S.; Yun, J.; Shah, S.; Pierson, N.; White, J.; Schindler, S.; Tischbirek, C.H.; Yuan, G.-C.; Cai, L. Single-Cell Nuclear Architecture across Cell Types in the Mouse Brain. Science 2021, 374, 586–594. [Google Scholar] [CrossRef] [PubMed]
- Szabo, Q.; Donjon, A.; Jerković, I.; Papadopoulos, G.L.; Cheutin, T.; Bonev, B.; Nora, E.P.; Bruneau, B.G.; Bantignies, F.; Cavalli, G. Regulation of Single-Cell Genome Organization into TADs and Chromatin Nanodomains. Nat. Genet. 2020, 52, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Finn, E.H.; Pegoraro, G.; Brandão, H.B.; Valton, A.-L.; Oomen, M.E.; Dekker, J.; Mirny, L.; Misteli, T. Extensive Heterogeneity and Intrinsic Variation in Spatial Genome Organization. Cell 2019, 176, 1502–1515.e10. [Google Scholar] [CrossRef] [PubMed]
- Boettiger, A.N.; Bintu, B.; Moffitt, J.R.; Wang, S.; Beliveau, B.J.; Fudenberg, G.; Imakaev, M.; Mirny, L.A.; Wu, C.; Zhuang, X. Super-Resolution Imaging Reveals Distinct Chromatin Folding for Different Epigenetic States. Nature 2016, 529, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Götz, M.; Messina, O.; Espinola, S.; Fiche, J.-B.; Nollmann, M. Multiple Parameters Shape the 3D Chromatin Structure of Single Nuclei at the Doc Locus in Drosophila. Nat. Commun. 2022, 13, 5375. [Google Scholar] [CrossRef] [PubMed]
- Cardozo Gizzi, A.M.; Cattoni, D.I.; Fiche, J.-B.; Espinola, S.M.; Gurgo, J.; Messina, O.; Houbron, C.; Ogiyama, Y.; Papadopoulos, G.L.; Cavalli, G.; et al. Microscopy-Based Chromosome Conformation Capture Enables Simultaneous Visualization of Genome Organization and Transcription in Intact Organisms. Mol. Cell 2019, 74, 212–222.e5. [Google Scholar] [CrossRef]
- Nir, G.; Farabella, I.; Pérez Estrada, C.; Ebeling, C.G.; Beliveau, B.J.; Sasaki, H.M.; Lee, S.H.; Nguyen, S.C.; McCole, R.B.; Chattoraj, S.; et al. Walking along Chromosomes with Super-Resolution Imaging, Contact Maps, and Integrative Modeling. PLoS Genet. 2018, 14, e1007872. [Google Scholar] [CrossRef]
- Jerkovic’, I.; Cavalli, G. Understanding 3D Genome Organization by Multidisciplinary Methods. Nat. Rev. Mol. Cell Biol. 2021, 22, 511–528. [Google Scholar] [CrossRef]
- Li, Q.; Tjong, H.; Li, X.; Gong, K.; Zhou, X.J.; Chiolo, I.; Alber, F. The Three-Dimensional Genome Organization of Drosophila Melanogaster through Data Integration. Genome Biol. 2017, 18, 145. [Google Scholar] [CrossRef]
- Tjong, H.; Li, W.; Kalhor, R.; Dai, C.; Hao, S.; Gong, K.; Zhou, Y.; Li, H.; Zhou, X.J.; Le Gros, M.A.; et al. Population-Based 3D Genome Structure Analysis Reveals Driving Forces in Spatial Genome Organization. Proc. Natl. Acad. Sci. USA 2016, 113, E1663–E1672. [Google Scholar] [CrossRef]
- Chiariello, A.M.; Annunziatella, C.; Bianco, S.; Esposito, A.; Nicodemi, M. Polymer Physics of Chromosome Large-Scale 3D Organisation. Sci. Rep. 2016, 6, 29775. [Google Scholar] [CrossRef] [PubMed]
- Chiariello, A.M.; Bianco, S.; Esposito, A.; Fiorillo, L.; Conte, M.; Irani, E.; Musella, F.; Abraham, A.; Prisco, A.; Nicodemi, M. Physical Mechanisms of Chromatin Spatial Organization. FEBS J. 2022, 289, 1180–1190. [Google Scholar] [CrossRef] [PubMed]
- Brackley, C.A.; Taylor, S.; Papantonis, A.; Cook, P.R.; Marenduzzo, D. Nonspecific Bridging-Induced Attraction Drives Clustering of DNA-Binding Proteins and Genome Organization. Proc. Natl. Acad. Sci. USA 2013, 110, E3605–E3611. [Google Scholar] [CrossRef] [PubMed]
- Brackley, C.A.; Liebchen, B.; Michieletto, D.; Mouvet, F.; Cook, P.R.; Marenduzzo, D. Ephemeral Protein Binding to DNA Shapes Stable Nuclear Bodies and Chromatin Domains. Biophys. J. 2017, 112, 1085–1093. [Google Scholar] [CrossRef] [PubMed]
- Di Pierro, M.; Zhang, B.; Aiden, E.L.; Wolynes, P.G.; Onuchic, J.N. Transferable Model for Chromosome Architecture. Proc. Natl. Acad. Sci. USA 2016, 113, 12168–12173. [Google Scholar] [CrossRef]
- Rosa, A.; Everaers, R. Structure and Dynamics of Interphase Chromosomes. PLoS Comput. Biol. 2008, 4, e1000153. [Google Scholar] [CrossRef]
- Esposito, A.; Bianco, S.; Fiorillo, L.; Conte, M.; Abraham, A.; Musella, F.; Nicodemi, M.; Prisco, A.; Chiariello, A.M. Polymer Models Are a Versatile Tool to Study Chromatin 3D Organization. Biochem. Soc. Trans. 2021, 49, 1675–1684. [Google Scholar] [CrossRef]
- Bianco, S.; Chiariello, A.M.; Conte, M.; Esposito, A.; Fiorillo, L.; Musella, F.; Nicodemi, M. Computational Approaches from Polymer Physics to Investigate Chromatin Folding. Curr. Opin. Cell Biol. 2020, 64, 10–17. [Google Scholar] [CrossRef]
- Neguembor, M.V.; Arcon, J.P.; Buitrago, D.; Lema, R.; Walther, J.; Garate, X.; Martin, L.; Romero, P.; AlHaj Abed, J.; Gut, M.; et al. MiOS, an Integrated Imaging and Computational Strategy to Model Gene Folding with Nucleosome Resolution. Nat. Struct. Mol. Biol. 2022, 29, 1011–1023. [Google Scholar] [CrossRef]
- Racko, D.; Benedetti, F.; Dorier, J.; Stasiak, A. Transcription-Induced Supercoiling as the Driving Force of Chromatin Loop Extrusion during Formation of TADs in Interphase Chromosomes. Nucleic Acids Res. 2018, 46, 1648–1660. [Google Scholar] [CrossRef]
- Serra, F.; Baù, D.; Goodstadt, M.; Castillo, D.; Filion, G.J.; Marti-Renom, M.A. Automatic Analysis and 3D-Modelling of Hi-C Data Using TADbit Reveals Structural Features of the Fly Chromatin Colors. PLoS Comput. Biol. 2017, 13, e1005665. [Google Scholar] [CrossRef] [PubMed]
- Sanborn, A.L.; Rao, S.S.P.; Huang, S.C.; Durand, N.C.; Huntley, M.H.; Jewett, A.I.; Bochkov, I.D.; Chinnappan, D.; Cutkosky, A.; Li, J.; et al. Chromatin Extrusion Explains Key Features of Loop and Domain Formation in Wild-Type and Engineered Genomes. Proc. Natl. Acad. Sci. USA 2015, 112, E6456–E6465. [Google Scholar] [CrossRef] [PubMed]
- Fudenberg, G.; Imakaev, M.; Lu, C.; Goloborodko, A.; Abdennur, N.; Mirny, L.A. Formation of Chromosomal Domains by Loop Extrusion. Cell Rep. 2016, 15, 2038–2049. [Google Scholar] [CrossRef] [PubMed]
- Yildirim, A.; Boninsegna, L.; Zhan, Y.; Alber, F. Uncovering the Principles of Genome Folding by 3D Chromatin Modeling. Cold Spring Harb. Perspect. Biol. 2022, 14, a039693. [Google Scholar] [CrossRef] [PubMed]
- Boninsegna, L.; Yildirim, A.; Polles, G.; Zhan, Y.; Quinodoz, S.A.; Finn, E.H.; Guttman, M.; Zhou, X.J.; Alber, F. Integrative Genome Modeling Platform Reveals Essentiality of Rare Contact Events in 3D Genome Organizations. Nat. Methods 2022, 19, 938–949. [Google Scholar] [CrossRef]
- Esposito, A.; Abraham, A.; Conte, M.; Vercellone, F.; Prisco, A.; Bianco, S.; Chiariello, A.M. The Physics of DNA Folding: Polymer Models and Phase-Separation. Polymers 2022, 14, 1918. [Google Scholar] [CrossRef]
- Conte, M.; Irani, E.; Chiariello, A.M.; Abraham, A.; Bianco, S.; Esposito, A.; Nicodemi, M. Loop-Extrusion and Polymer Phase-Separation Can Co-Exist at the Single-Molecule Level to Shape Chromatin Folding. Nat. Commun. 2022, 13, 4070. [Google Scholar] [CrossRef]
- Fiorillo, L.; Bianco, S.; Esposito, A.; Conte, M.; Sciarretta, R.; Musella, F.; Chiariello, A.M. A Modern Challenge of Polymer Physics: Novel Ways to Study, Interpret, and Reconstruct Chromatin Structure. WIREs Comput. Mol. Sci. 2020, 10, e1454. [Google Scholar] [CrossRef]
- Esposito, A.; Chiariello, A.M.; Conte, M.; Fiorillo, L.; Musella, F.; Sciarretta, R.; Bianco, S. Higher-Order Chromosome Structures Investigated by Polymer Physics in Cellular Morphogenesis and Differentiation. J. Mol. Biol. 2020, 432, 701–711. [Google Scholar] [CrossRef]
- Zhang, B.; Wolynes, P.G. Topology, Structures, and Energy Landscapes of Human Chromosomes. Proc. Natl. Acad. Sci. USA 2015, 112, 6062–6067. [Google Scholar] [CrossRef]
- Shi, G.; Thirumalai, D. From Hi-C Contact Map to Three-Dimensional Organization of Interphase Human Chromosomes. Phys. Rev. X 2021, 11, 011051. [Google Scholar] [CrossRef]
- Lin, D.; Bonora, G.; Yardimci, G.G.; Noble, W.S. Computational Methods for Analyzing and Modeling Genome Structure and Organization. Wiley Interdiscip. Rev. Syst. Biol. Med. 2018, 11, e1435. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.; Liu, L.; Hyeon, C.; Thirumalai, D. Interphase Human Chromosome Exhibits out of Equilibrium Glassy Dynamics. Nat. Commun. 2018, 9, 3161. [Google Scholar] [CrossRef] [PubMed]
- Plewczynski, D.; Kadlof, M. Computational Modelling of Three-Dimensional Genome Structure. Methods 2020, 181–182, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Banigan, E.J.; Mirny, L.A. Loop Extrusion: Theory Meets Single-Molecule Experiments. Curr. Opin. Cell Biol. 2020, 64, 124–138. [Google Scholar] [CrossRef]
- Nicodemi, M.; Prisco, A. Thermodynamic Pathways to Genome Spatial Organization in the Cell Nucleus. Biophys. J. 2009, 96, 2168–2177. [Google Scholar] [CrossRef]
- Barbieri, M.; Chotalia, M.; Fraser, J.; Lavitas, L.-M.; Dostie, J.; Pombo, A.; Nicodemi, M. Complexity of Chromatin Folding Is Captured by the Strings and Binders Switch Model. Proc. Natl. Acad. Sci. USA 2012, 109, 16173–16178. [Google Scholar] [CrossRef]
- Crippa, M.; Zhan, Y.; Tiana, G. Effective Model of Loop Extrusion Predicts Chromosomal Domains. Phys. Rev. E 2020, 102, 032414. [Google Scholar] [CrossRef]
- Conte, M.; Esposito, A.; Vercellone, F.; Abraham, A.; Bianco, S. Unveiling the Machinery behind Chromosome Folding by Polymer Physics Modeling. Int. J. Mol. Sci. 2023, 24, 3660. [Google Scholar] [CrossRef]
- Buckle, A.; Brackley, C.A.; Boyle, S.; Marenduzzo, D.; Gilbert, N. Polymer Simulations of Heteromorphic Chromatin Predict the 3D Folding of Complex Genomic Loci. Mol. Cell 2018, 72, 786–797.e11. [Google Scholar] [CrossRef]
- Brackley, C.A.; Johnson, J.; Michieletto, D.; Morozov, A.N.; Nicodemi, M.; Cook, P.R.; Marenduzzo, D. Extrusion without a Motor: A New Take on the Loop Extrusion Model of Genome Organization. Nucleus 2018, 9, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Conte, M.; Esposito, A.; Fiorillo, L.; Annunziatella, C.; Corrado, A.; Musella, F.; Sciarretta, R.; Chiariello, A.M.; Bianco, S. Hybrid Machine Learning and Polymer Physics Approach to Investigate 3D Chromatin Structure. In Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics, Proceedings of theEuro-Par 2019: Parallel Processing Workshops: Euro-Par 2019 International Workshops, Göttingen, Germany, 26–30 August 2019; Lecture Notes in Computer Science; Springer: Berlin/Heidelberg, Germany, 2020; Volume 11997. [Google Scholar]
- Di Stefano, M.; Paulsen, J.; Lien, T.G.; Hovig, E.; Micheletti, C. Hi-C-Constrained Physical Models of Human Chromosomes Recover Functionally-Related Properties of Genome Organization. Sci. Rep. 2016, 6, 35985. [Google Scholar] [CrossRef] [PubMed]
- Erdel, F.; Rippe, K. Formation of Chromatin Subcompartments by Phase Separation. Biophys. J. 2018, 114, 2262–2270. [Google Scholar] [CrossRef] [PubMed]
- Brackley, C.A.; Johnson, J.; Kelly, S.; Cook, P.R.; Marenduzzo, D. Simulated Binding of Transcription Factors to Active and Inactive Regions Folds Human Chromosomes into Loops, Rosettes and Topological Domains. Nucleic Acids Res. 2016, 44, 3503–3512. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, M.; Rosa, A.; Belcastro, V.; di Bernardo, D.; Micheletti, C. Colocalization of Coregulated Genes: A Steered Molecular Dynamics Study of Human Chromosome 19. PLoS Comput. Biol. 2013, 9, e1003019. [Google Scholar] [CrossRef]
- Di Stefano, M.; Stadhouders, R.; Farabella, I.; Castillo, D.; Serra, F.; Graf, T.; Marti-Renom, M.A. Transcriptional Activation during Cell Reprogramming Correlates with the Formation of 3D Open Chromatin Hubs. Nat. Commun. 2020, 11, 2564. [Google Scholar] [CrossRef]
- Lesne, A.; Riposo, J.; Roger, P.; Cournac, A.; Mozziconacci, J. 3D Genome Reconstruction from Chromosomal Contacts. Nat. Methods 2014, 11, 1141–1143. [Google Scholar] [CrossRef]
- Brackey, C.A.; Marenduzzo, D.; Gilbert, N. Mechanistic Modeling of Chromatin Folding to Understand Function. Nat. Methods 2020, 17, 767–775. [Google Scholar] [CrossRef]
- Zhang, S.; Chasman, D.; Knaack, S.; Roy, S. In Silico Prediction of High-Resolution Hi-C Interaction Matrices. Nat. Commun. 2019, 10, 5449. [Google Scholar] [CrossRef]
- Ghosh, S.K.; Jost, D. How Epigenome Drives Chromatin Folding and Dynamics, Insights from Efficient Coarse-Grained Models of Chromosomes. PLoS Comput. Biol. 2018, 14, e1006159. [Google Scholar] [CrossRef]
- Brackley, C.A.; Brown, J.M.; Waithe, D.; Babbs, C.; Davies, J.; Hughes, J.R.; Buckle, V.J.; Marenduzzo, D. Predicting the Three-Dimensional Folding of Cis-Regulatory Regions in Mammalian Genomes Using Bioinformatic Data and Polymer Models. Genome Biol. 2016, 17, 59. [Google Scholar] [CrossRef] [PubMed]
- Di Pierro, M.; Cheng, R.R.; Lieberman Aiden, E.; Wolynes, P.G.; Onuchic, J.N. De Novo Prediction of Human Chromosome Structures: Epigenetic Marking Patterns Encode Genome Architecture. Proc. Natl. Acad. Sci. USA 2017, 114, 12126–12131. [Google Scholar] [CrossRef] [PubMed]
- Belokopytova, P.; Viesná, E.; Chiliński, M.; Qi, Y.; Salari, H.; Di Stefano, M.; Esposito, A.; Conte, M.; Chiariello, A.M.; Teif, V.B.; et al. 3DGenBench: A Web-Server to Benchmark Computational Models for 3D Genomics. Nucleic Acids Res. 2022, 50, W4–W12. [Google Scholar] [CrossRef]
- Chiang, M.; Michieletto, D.; Brackley, C.A.; Rattanavirotkul, N.; Mohammed, H.; Marenduzzo, D.; Chandra, T. Polymer Modeling Predicts Chromosome Reorganization in Senescence. Cell Rep. 2019, 28, 3212–3223.e6. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Qi, Y.; Latham, A.P.; Zhang, B. Multiscale Modeling of Genome Organization with Maximum Entropy Optimization. J. Chem. Phys. 2021, 155, 010901. [Google Scholar] [CrossRef]
- Michieletto, D.; Orlandini, E.; Marenduzzo, D. Polymer Model with Epigenetic Recoloring Reveals a Pathway for the de Novo Establishment and 3D Organization of Chromatin Domains. Phys. Rev. X 2016, 6, 041047. [Google Scholar] [CrossRef]
- Laghmach, R.; Di Pierro, M.; Potoyan, D.A. Mesoscale Liquid Model of Chromatin Recapitulates Nuclear Order of Eukaryotes. Biophys. J. 2020, 118, 2130–2140. [Google Scholar] [CrossRef]
- Salari, H.; Di Stefano, M.; Jost, D. Spatial Organization of Chromosomes Leads to Heterogeneous Chromatin Motion and Drives the Liquid- or Gel-like Dynamical Behavior of Chromatin. Genome Res. 2022, 32, 28–43. [Google Scholar] [CrossRef]
- Jost, D.; Carrivain, P.; Cavalli, G.; Vaillant, C. Modeling Epigenome Folding: Formation and Dynamics of Topologically Associated Chromatin Domains. Nucleic Acids Res. 2014, 42, 9553–9561. [Google Scholar] [CrossRef]
- Tortora, M.M.; Salari, H.; Jost, D. Chromosome Dynamics during Interphase: A Biophysical Perspective. Curr. Opin. Genet. Dev. 2020, 61, 37–43. [Google Scholar] [CrossRef]
- Bohn, M.; Heermann, D.W. Diffusion-Driven Looping Provides a Consistent Framework for Chromatin Organization. PLoS ONE 2010, 5, e12218. [Google Scholar] [CrossRef] [PubMed]
- Conte, M.; Fiorillo, L.; Bianco, S.; Chiariello, A.M.; Esposito, A.; Musella, F.; Flora, F.; Abraham, A.; Nicodemi, M. A Polymer Physics Model to Dissect Genome Organization in Healthy and Pathological Phenotypes. In Methods in Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2022; Volume 2301, pp. 307–316. [Google Scholar]
- Fudenberg, G.; Kelley, D.R.; Pollard, K.S. Predicting 3D Genome Folding from DNA Sequence with Akita. Nat. Methods 2020, 17, 1111–1117. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J. Sequence-Based Modeling of Three-Dimensional Genome Architecture from Kilobase to Chromosome Scale. Nat. Genet. 2022, 54, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Yang, Y.; Póczos, B.; Ma, J. Predicting Enhancer-promoter Interaction from Genomic Sequence with Deep Neural Networks. Quant. Biol. 2019, 7, 122–137. [Google Scholar] [CrossRef]
- Bianco, S.; Lupiáñez, D.G.; Chiariello, A.M.; Annunziatella, C.; Kraft, K.; Schöpflin, R.; Wittler, L.; Andrey, G.; Vingron, M.; Pombo, A.; et al. Polymer Physics Predicts the Effects of Structural Variants on Chromatin Architecture. Nat. Genet. 2018, 50, 662–667. [Google Scholar] [CrossRef]
- Chiariello, A.M.; Abraham, A.; Bianco, S.; Esposito, A.; Fontana, A.; Vercellone, F.; Conte, M.; Nicodemi, M. Multiscale Modelling of Chromatin 4D Organization in SARS-CoV-2 Infected Cells. Nat. Commun. 2024, 15, 4014. [Google Scholar] [CrossRef]
- Schwessinger, R.; Gosden, M.; Downes, D.; Brown, R.C.; Oudelaar, A.M.; Telenius, J.; Teh, Y.W.; Lunter, G.; Hughes, J.R. DeepC: Predicting 3D Genome Folding Using Megabase-Scale Transfer Learning. Nat. Methods 2020, 17, 1118–1124. [Google Scholar] [CrossRef]
- Li, W.; Wong, W.H.; Jiang, R. DeepTACT: Predicting 3D Chromatin Contacts via Bootstrapping Deep Learning. Nucleic Acids Res. 2019, 47, e60. [Google Scholar] [CrossRef]
- Belokopytova, P.S.; Nuriddinov, M.A.; Mozheiko, E.A.; Fishman, D.; Fishman, V. Quantitative Prediction of Enhancer–Promoter Interactions. Genome Res. 2020, 30, 72–84. [Google Scholar] [CrossRef]
- Zhang, R.; Wang, Y.; Yang, Y.; Zhang, Y.; Ma, J. Predicting CTCF-Mediated Chromatin Loops Using CTCF-MP. Bioinformatics 2018, 34, i133–i141. [Google Scholar] [CrossRef]
- Tan, J.; Shenker-Tauris, N.; Rodriguez-Hernaez, J.; Wang, E.; Sakellaropoulos, T.; Boccalatte, F.; Thandapani, P.; Skok, J.; Aifantis, I.; Fenyö, D.; et al. Cell-Type-Specific Prediction of 3D Chromatin Organization Enables High-Throughput in Silico Genetic Screening. Nat. Biotechnol. 2023, 41, 1140–1150. [Google Scholar] [CrossRef] [PubMed]
- Cao, F.; Zhang, Y.; Cai, Y.; Animesh, S.; Zhang, Y.; Akincilar, S.C.; Loh, Y.P.; Li, X.; Chng, W.J.; Tergaonkar, V.; et al. Chromatin Interaction Neural Network (ChINN): A Machine Learning-Based Method for Predicting Chromatin Interactions from DNA Sequences. Genome Biol. 2021, 22, 226. [Google Scholar] [CrossRef] [PubMed]
- Fiorillo, L.; Musella, F.; Conte, M.; Kempfer, R.; Chiariello, A.M.; Bianco, S.; Kukalev, A.; Irastorza-Azcarate, I.; Esposito, A.; Abraham, A.; et al. Comparison of the Hi-C, GAM and SPRITE Methods Using Polymer Models of Chromatin. Nat. Methods 2021, 18, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Fiorillo, L.; Bianco, S.; Chiariello, A.M.; Barbieri, M.; Esposito, A.; Annunziatella, C.; Conte, M.; Corrado, A.; Prisco, A.; Pombo, A.; et al. Inference of Chromosome 3D Structures from GAM Data by a Physics Computational Approach. Methods 2020, 181–182, 70–79. [Google Scholar] [CrossRef]
- Conte, M.; Fiorillo, L.; Bianco, S.; Chiariello, A.M.; Esposito, A.; Nicodemi, M. Polymer Physics Indicates Chromatin Folding Variability across Single-Cells Results from State Degeneracy in Phase Separation. Nat. Commun. 2020, 11, 3289. [Google Scholar] [CrossRef]
- Conte, M.; Chiariello, A.M.; Bianco, S.; Esposito, A.; Abraham, A.; Nicodemi, M. Physics-Based Polymer Models to Probe Chromosome Structure in Single Molecules. In Methods in Molecular Biology; Humana: New York, NY, USA; Springer: Berlin/Heidelberg, Germany, 2023; Volume 2655. [Google Scholar] [CrossRef]
- Defenouillère, Q.; Yao, Y.; Mouaikel, J.; Namane, A.; Galopier, A.; Decourty, L.; Doyen, A.; Malabat, C.; Saveanu, C.; Jacquier, A.; et al. Cdc48-Associated Complex Bound to 60S Particles Is Required for the Clearance of Aberrant Translation Products. Proc. Natl. Acad. Sci. USA 2013, 110, 5046–5051. [Google Scholar] [CrossRef]
- Joazeiro, C.A.P. Mechanisms and Functions of Ribosome-Associated Protein Quality Control. Nat. Rev. Mol. Cell Biol. 2019, 20, 368–383. [Google Scholar] [CrossRef]
- Chu, J.; Hong, N.A.; Masuda, C.A.; Jenkins, B.V.; Nelms, K.A.; Goodnow, C.C.; Glynne, R.J.; Wu, H.; Masliah, E.; Joazeiro, C.A.P.; et al. A Mouse Forward Genetics Screen Identifies LISTERIN as an E3 Ubiquitin Ligase Involved in Neurodegeneration. Proc. Natl. Acad. Sci. USA 2009, 106, 2097–2103. [Google Scholar] [CrossRef]
- Ghosh, A.; Shcherbik, N. Cooperativity between the Ribosome-Associated Chaperone Ssb/RAC and the Ubiquitin Ligase Ltn1 in Ubiquitination of Nascent Polypeptides. Int. J. Mol. Sci. 2020, 21, 6815. [Google Scholar] [CrossRef]
- Farhang, S.; Sabaie, H.; Gharesouran, J.; Asadi, M.R.; Arsang-Jang, S.; Ghafouri-Fard, S.; Taheri, M.; Rezazadeh, M. Expression Analysis of Ermin and Listerin E3 Ubiquitin Protein Ligase 1 Genes in the Periphery of Patients with Schizophrenia. J. Mol. Neurosci. 2022, 72, 246–254. [Google Scholar] [CrossRef]
- Bengtson, M.H.; Joazeiro, C.A.P. Role of a Ribosome-Associated E3 Ubiquitin Ligase in Protein Quality Control. Nature 2010, 467, 470–473. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Brown, A.; Santhanam, B.; Hegde, R.S. Structure and Assembly Pathway of the Ribosome Quality Control Complex. Mol. Cell 2015, 57, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, Y.; Ikeuchi, K.; Saeki, Y.; Iwasaki, S.; Schmidt, C.; Udagawa, T.; Sato, F.; Tsuchiya, H.; Becker, T.; Tanaka, K.; et al. Ubiquitination of Stalled Ribosome Triggers Ribosome-Associated Quality Control. Nat. Commun. 2017, 8, 159. [Google Scholar] [CrossRef] [PubMed]
- Belaghzal, H.; Dekker, J.; Gibcus, J.H. Hi-C 2.0: An Optimized Hi-C Procedure for High-Resolution Genome-Wide Mapping of Chromosome Conformation. Methods 2017, 123, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Reiff, S.B.; Schroeder, A.J.; Kırlı, K.; Cosolo, A.; Bakker, C.; Lee, S.; Veit, A.D.; Balashov, A.K.; Vitzthum, C.; Ronchetti, W.; et al. The 4D Nucleome Data Portal as a Resource for Searching and Visualizing Curated Nucleomics Data. Nat. Commun. 2022, 13, 2365. [Google Scholar] [CrossRef]
- Dekker, J.; Belmont, A.S.; Guttman, M.; Leshyk, V.O.; Lis, J.T.; Lomvardas, S.; Mirny, L.A.; O’Shea, C.C.; Park, P.J.; Ren, B.; et al. The 4D Nucleome Project. Nature 2017, 549, 219–226. [Google Scholar] [CrossRef]
- De Gennes, P.G. Scaling Concepts in Polymer Physics; Cornell University Press: Ithaca, NY, USA, 1979; ISBN 080141203X. [Google Scholar]
- Shrinivas, K.; Sabari, B.R.; Coffey, E.L.; Klein, I.A.; Boija, A.; Zamudio, A.V.; Schuijers, J.; Hannett, N.M.; Sharp, P.A.; Young, R.A.; et al. Enhancer Features That Drive Formation of Transcriptional Condensates. Mol. Cell 2019, 75, 549–561.e7. [Google Scholar] [CrossRef]
- Tafuri, F.; Chiariello, A.M. The Effect of Configurational Complexity in Hetero-Polymers on the Coil-Globule Phase Transition. Eur. Phys. J. Plus 2023, 138, 150. [Google Scholar] [CrossRef]
- Esposito, A.; Bianco, S.; Chiariello, A.M.; Abraham, A.; Fiorillo, L.; Conte, M.; Campanile, R.; Nicodemi, M. Polymer Physics Reveals a Combinatorial Code Linking 3D Chromatin Architecture to 1D Chromatin States. Cell Rep. 2022, 38, 110601. [Google Scholar] [CrossRef]
- Thompson, A.P.; Aktulga, H.M.; Berger, R.; Bolintineanu, D.S.; Brown, W.M.; Crozier, P.S.; in ’t Veld, P.J.; Kohlmeyer, A.; Moore, S.G.; Nguyen, T.D.; et al. LAMMPS—A Flexible Simulation Tool for Particle-Based Materials Modeling at the Atomic, Meso, and Continuum Scales. Comput. Phys. Commun. 2022, 271, 108171. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Conte, M.; Esposito, A.; Fiorillo, L.; Campanile, R.; Annunziatella, C.; Corrado, A.; Chiariello, M.G.; Bianco, S.; Chiariello, A.M. Efficient Computational Implementation of Polymer Physics Models to Explore Chromatin Structure. Int. J. Parallel Emergent Distrib. Syst. 2022, 37, 91–102. [Google Scholar] [CrossRef]
- Kremer, K.; Grest, G.S. Dynamics of Entangled Linear Polymer Melts: A Molecular-Dynamics Simulation. J. Chem. Phys. 1990, 92, 5057–5086. [Google Scholar] [CrossRef]
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids (Oxford Science Publications); Oxford University Press: Oxford, UK, 1989. [Google Scholar]
- Conte, M.; Fiorillo, L.; Annunziatella, C.; Esposito, A.; Musella, F.; Abraham, A.; Bianco, S.; Chiariello, A.M. Dynamic and Equilibrium Properties of Finite-Size Polymer Models of Chromosome Folding. Phys. Rev. E 2021, 104, 054402. [Google Scholar] [CrossRef] [PubMed]
- Chiariello, A.M.; Corberi, F.; Salerno, M. The Interplay between Phase Separation and Gene-Enhancer Communication: A Theoretical Study. Biophys. J. 2020, 119, 873–883. [Google Scholar] [CrossRef] [PubMed]
- Baum, M.; Erdel, F.; Wachsmuth, M.; Rippe, K. Retrieving the Intracellular Topology from Multi-Scale Protein Mobility Mapping in Living Cells. Nat. Commun. 2014, 5, 4494. [Google Scholar] [CrossRef]
- Conte, M.; Chiariello, A.M.; Abraham, A.; Bianco, S.; Esposito, A.; Nicodemi, M.; Matteuzzi, T.; Vercellone, F. Polymer Models of Chromatin Imaging Data in Single Cells. Algorithms 2022, 15, 330. [Google Scholar] [CrossRef]
- Trussart, M.; Serra, F.; Baù, D.; Junier, I.; Serrano, L.; Marti-Renom, M.A. Assessing the Limits of Restraint-Based 3D Modeling of Genomes and Genomic Domains. Nucleic Acids Res. 2015, 43, 3465–3477. [Google Scholar] [CrossRef]
- Nagano, T.; Lubling, Y.; Stevens, T.J.; Schoenfelder, S.; Yaffe, E.; Dean, W.; Laue, E.D.; Tanay, A.; Fraser, P. Single-Cell Hi-C Reveals Cell-to-Cell Variability in Chromosome Structure. Nature 2013, 502, 59–64. [Google Scholar] [CrossRef]
- Stevens, T.J.; Lando, D.; Basu, S.; Atkinson, L.P.; Cao, Y.; Lee, S.F.; Leeb, M.; Wohlfahrt, K.J.; Boucher, W.; O’Shaughnessy-Kirwan, A.; et al. 3D Structures of Individual Mammalian Genomes Studied by Single-Cell Hi-C. Nature 2017, 544, 59–64. [Google Scholar] [CrossRef]
- Demmerle, J.; Innocent, C.; North, A.J.; Ball, G.; Müller, M.; Miron, E.; Matsuda, A.; Dobbie, I.M.; Markaki, Y.; Schermelleh, L. Strategic and Practical Guidelines for Successful Structured Illumination Microscopy. Nat. Protoc. 2017, 12, 988–1010. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, M.G.L.; Shao, L.; Carlton, P.M.; Wang, C.J.R.; Golubovskaya, I.N.; Cande, W.Z.; Agard, D.A.; Sedat, J.W. Three-Dimensional Resolution Doubling in Wide-Field Fluorescence Microscopy by Structured Illumination. Biophys. J. 2008, 94, 4957–4970. [Google Scholar] [CrossRef] [PubMed]
- Markaki, Y.; Smeets, D.; Fiedler, S.; Schmid, V.J.; Schermelleh, L.; Cremer, T.; Cremer, M. The Potential of 3D-FISH and Super-resolution Structured Illumination Microscopy for Studies of 3D Nuclear Architecture. BioEssays 2012, 34, 412–426. [Google Scholar] [CrossRef] [PubMed]
- Bishop, M.; Michels, J.P.J. Polymer Shapes in Three Dimensions. J. Chem. Phys. 1986, 85, 5961–5962. [Google Scholar] [CrossRef]
- Coniglio, A.; Nicodemi, M. A Statistical Mechanics Approach to the Inherent States of Granular Media. Phys. A Stat. Mech. Its Appl. 2001, 296, 451–459. [Google Scholar] [CrossRef]
- Nicodemi, M. Force Correlations and Arch Formation in Granular Assemblies. Phys. Rev. Lett. 1998, 80, 1340–1343. [Google Scholar] [CrossRef]
- Guha, S.; Mitra, M.K. Multivalent Binding Proteins Can Drive Collapse and Reswelling of Chromatin in Confinement. Soft Matter 2023, 19, 153–163. [Google Scholar] [CrossRef]
- Brangwynne, C.P.; Tompa, P.; Pappu, R.V. Polymer Physics of Intracellular Phase Transitions. Nat. Phys. 2015, 11, 899–904. [Google Scholar] [CrossRef]
- Choi, J.-M.; Holehouse, A.S.; Pappu, R.V. Physical Principles Underlying the Complex Biology of Intracellular Phase Transitions. Annu. Rev. Biophys. 2020, 49, 107–133. [Google Scholar] [CrossRef]
- Scolari, V.F.; Cosentino Lagomarsino, M. Combined Collapse by Bridging and Self-Adhesion in a Prototypical Polymer Model Inspired by the Bacterial Nucleoid. Soft Matter 2015, 11, 1677–1687. [Google Scholar] [CrossRef]
- Scolari, V.F.; Mercy, G.; Koszul, R.; Lesne, A.; Mozziconacci, J. Kinetic Signature of Cooperativity in the Irreversible Collapse of a Polymer. Phys. Rev. Lett. 2018, 121, 057801. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, W.M.; Frenkel, D. Phase Transitions in Biological Systems with Many Components. Biophys. J. 2017, 112, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.-K.; Hwang, D.-E.; Choi, J.-M. Current Understanding of Molecular Phase Separation in Chromosomes. Int. J. Mol. Sci. 2021, 22, 10736. [Google Scholar] [CrossRef] [PubMed]
- Ancona, M.; Brackley, C.A. Simulating the Chromatin-Mediated Phase Separation of Model Proteins with Multiple Domains. Biophys. J. 2022, 121, 2600–2612. [Google Scholar] [CrossRef] [PubMed]
- Micheletti, C.; Marenduzzo, D.; Orlandini, E. Polymers with Spatial or Topological Constraints: Theoretical and Computational Results. Phys. Rep. 2011, 504, 1–73. [Google Scholar] [CrossRef]
- Oliveira, L.P.; Jensen, H.J.; Nicodemi, M.; Sibani, P. Record Dynamics and the Observed Temperature Plateau in the Magnetic Creep-Rate of Type-II Superconductors. Phys. Rev. B—Condens. Matter Mater. Phys. 2005, 71, 104526. [Google Scholar] [CrossRef]
- Nicodemi, M.; Fierro, A.; Coniglio, A. Segregation in Hard-Sphere Mixtures under Gravity. An Extension of Edwards Approach with Two Thermodynamical Parameters. Europhys. Lett. 2002, 60, 684–690. [Google Scholar] [CrossRef]
- Arenzon, J.J.; Nicodemi, M.; Sellitto, M. Equilibrium Properties of the Ising Frustrated Lattice Gas. J. Phys. I 1996, 6, 1143–1152. [Google Scholar] [CrossRef]
- Cataudella, V.; Franzese, G.; Nicodemi, M.; Scala, A.; Coniglio, A. Critical Clusters and Efficient Dynamics for Frustrated Spin Models. Phys. Rev. Lett. 1994, 72, 1541. [Google Scholar] [CrossRef]
- Borrelli, A.; De Falco, I.; Della Cioppa, A.; Nicodemi, M.; Trautteur, G. Performance of Genetic Programming to Extract the Trend in Noisy Data Series. Phys. A Stat. Mech. Its Appl. 2006, 370, 104–108. [Google Scholar] [CrossRef]
- Caglioti, E.; Coniglio, A.; Herrmann, H.J.; Loreto, V.; Nicodemi, M. Segregation of Granular Mixtures in the Presence of Compaction. Europhys. Lett. 1998, 43, 591. [Google Scholar] [CrossRef]
- Tarzia, M.; de Candia, A.; Fierro, A.; Nicodemi, M.; Coniglio, A. Glass Transition in Granular Media. Europhys. Lett. 2004, 66, 531–537. [Google Scholar] [CrossRef]
- Nicodemi, M.; Coniglio, A. Macroscopic Glassy Relaxations and Microscopic Motions in a Frustrated Lattice Gas. Phys. Rev. E 1998, 57, R39–R42. [Google Scholar] [CrossRef]
- Weischenfeldt, J.; Symmons, O.; Spitz, F.; Korbel, J.O. Phenotypic Impact of Genomic Structural Variation: Insights from and for Human Disease. Nat. Rev. Genet. 2013, 14, 125–138. [Google Scholar] [CrossRef]
- Collins, R.L.; Brand, H.; Karczewski, K.J.; Zhao, X.; Alföldi, J.; Francioli, L.C.; Khera, A.V.; Lowther, C.; Gauthier, L.D.; Wang, H.; et al. A Structural Variation Reference for Medical and Population Genetics. Nature 2020, 581, 444–451. [Google Scholar] [CrossRef]
- Iannone, F.; Ambrosino, F.; Bracco, G.; De Rosa, M.; Funel, A.; Guarnieri, G.; Migliori, S.; Palombi, F.; Ponti, G.; Santomauro, G.; et al. CRESCO ENEA HPC Clusters: A Working Example of a Multifabric GPFS Spectrum Scale Layout. In Proceedings of the 2019 International Conference on High Performance Computing & Simulation (HPCS), Dublin, Ireland, 15–19 July 2019; IEEE: Piscataway, NJ, USA, 2019; pp. 1051–1052. [Google Scholar]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conte, M.; Abraham, A.; Esposito, A.; Yang, L.; Gibcus, J.H.; Parsi, K.M.; Vercellone, F.; Fontana, A.; Di Pierno, F.; Dekker, J.; et al. Polymer Physics Models Reveal Structural Folding Features of Single-Molecule Gene Chromatin Conformations. Int. J. Mol. Sci. 2024, 25, 10215. https://doi.org/10.3390/ijms251810215
Conte M, Abraham A, Esposito A, Yang L, Gibcus JH, Parsi KM, Vercellone F, Fontana A, Di Pierno F, Dekker J, et al. Polymer Physics Models Reveal Structural Folding Features of Single-Molecule Gene Chromatin Conformations. International Journal of Molecular Sciences. 2024; 25(18):10215. https://doi.org/10.3390/ijms251810215
Chicago/Turabian StyleConte, Mattia, Alex Abraham, Andrea Esposito, Liyan Yang, Johan H. Gibcus, Krishna M. Parsi, Francesca Vercellone, Andrea Fontana, Florinda Di Pierno, Job Dekker, and et al. 2024. "Polymer Physics Models Reveal Structural Folding Features of Single-Molecule Gene Chromatin Conformations" International Journal of Molecular Sciences 25, no. 18: 10215. https://doi.org/10.3390/ijms251810215
APA StyleConte, M., Abraham, A., Esposito, A., Yang, L., Gibcus, J. H., Parsi, K. M., Vercellone, F., Fontana, A., Di Pierno, F., Dekker, J., & Nicodemi, M. (2024). Polymer Physics Models Reveal Structural Folding Features of Single-Molecule Gene Chromatin Conformations. International Journal of Molecular Sciences, 25(18), 10215. https://doi.org/10.3390/ijms251810215