
Mechanistic Insights into Substrate Recognition of Human Nucleoside Diphosphate Kinase C Based on Nucleotide-Induced Structural Changes

 , , , and
, , , and 
Abstract
:1. Introduction
2. Results and Discussion
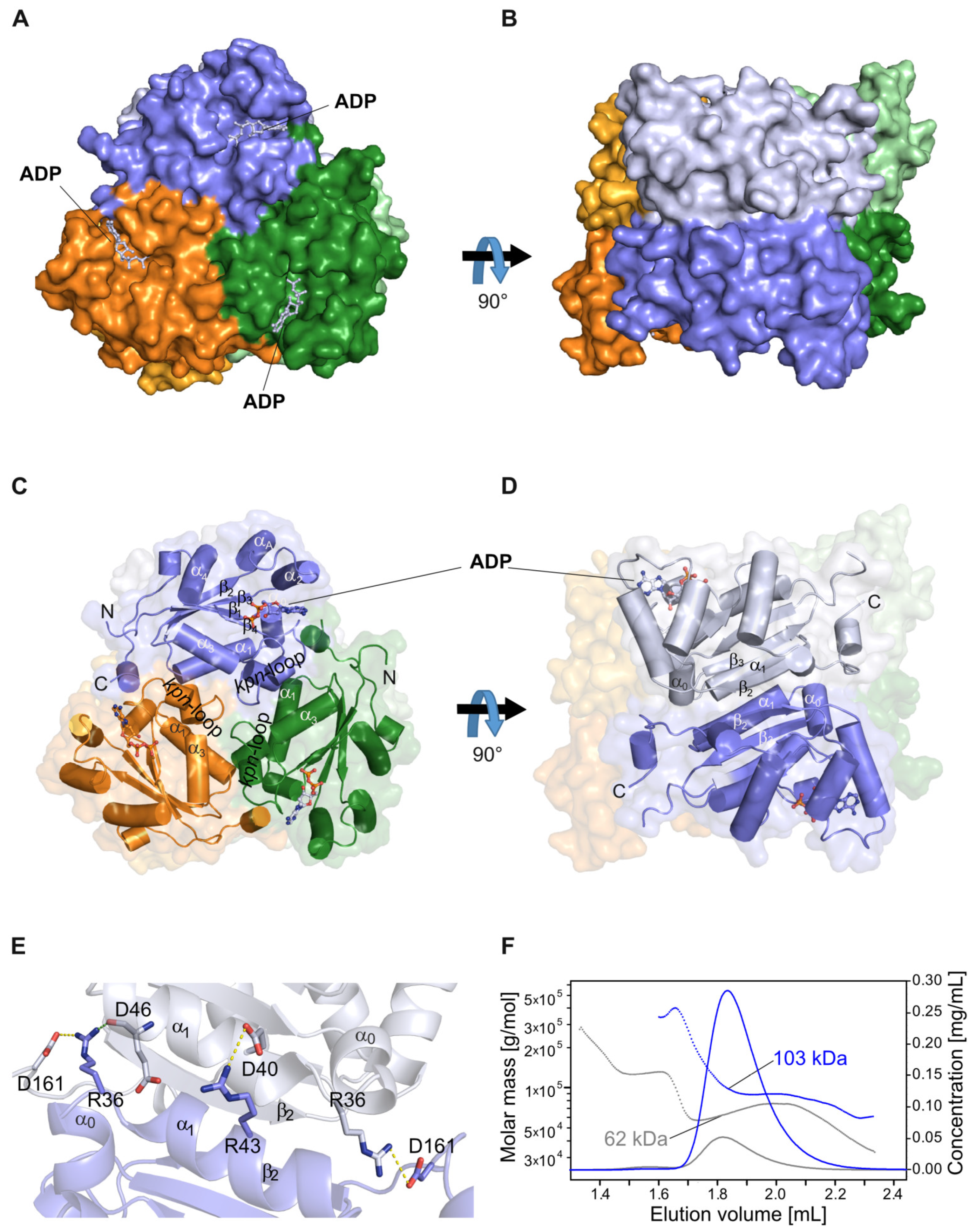
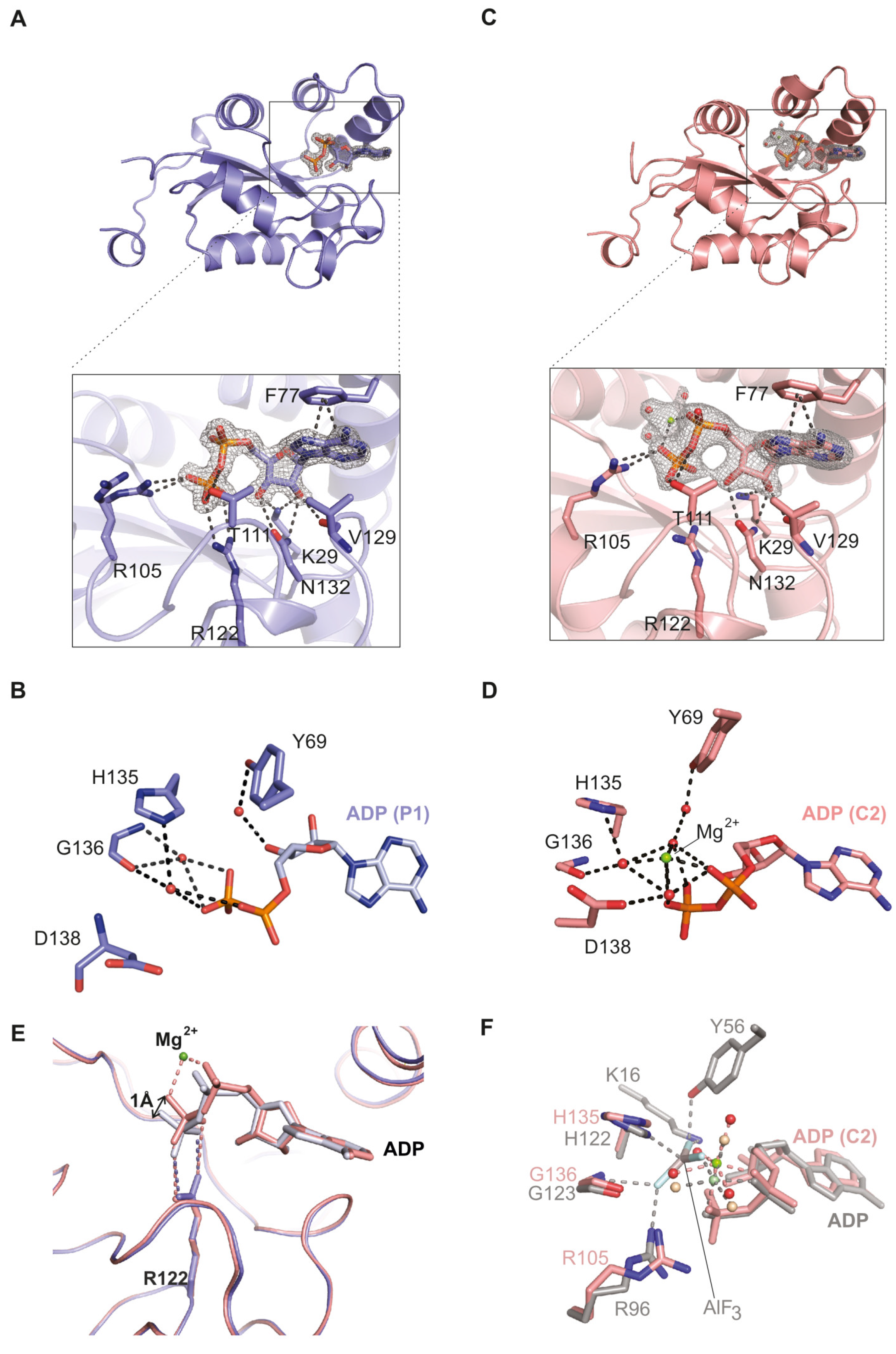
2.1. The NDPK-C ADP Complex in the Absence and Presence of Mg2+
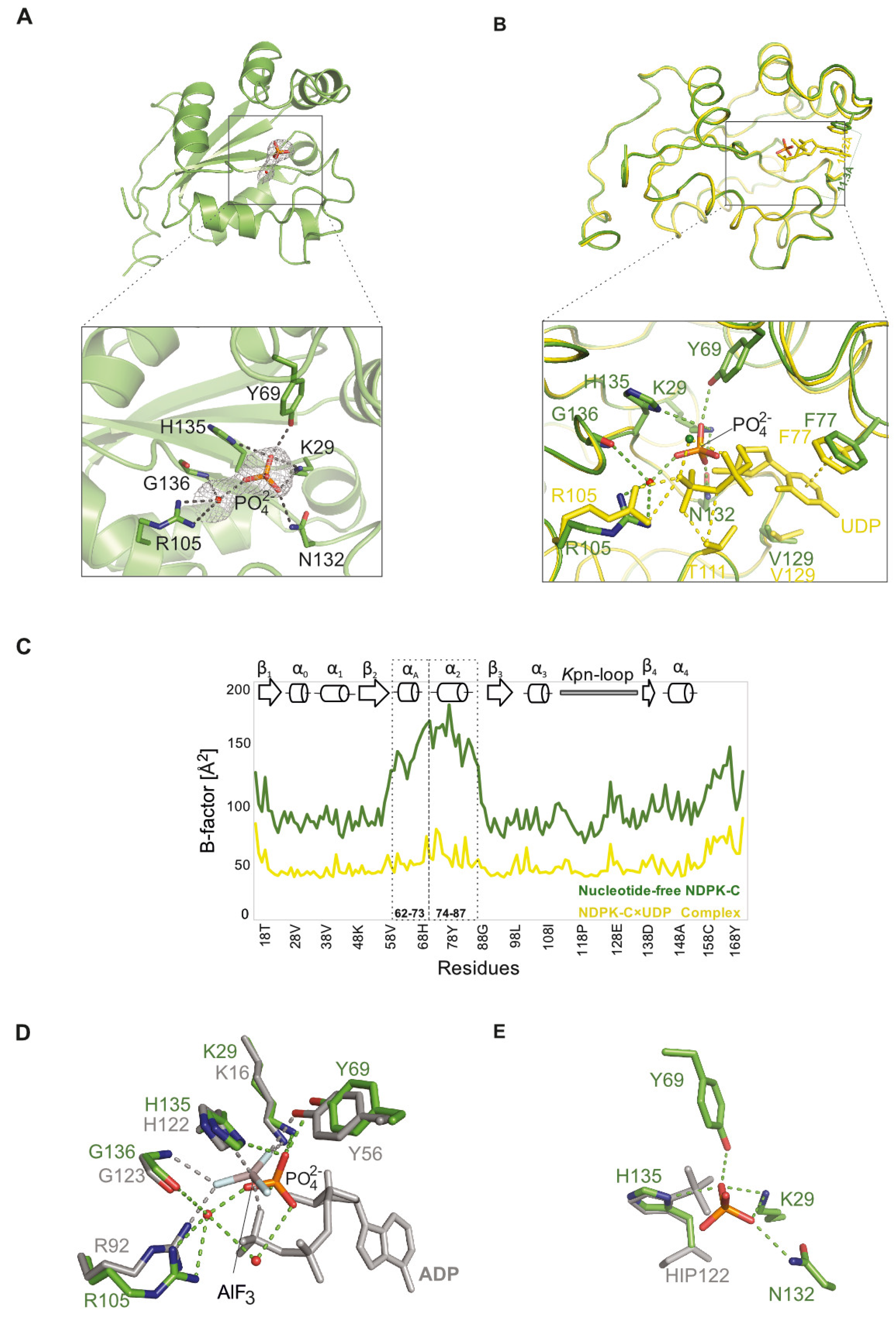
2.2. Comparison of Human NDPK-C Ligated with Different Nucleotides
2.3. Structure of Nucleotide-Free Human NDPK-C
2.4. Differences between the NDPK B and C Isoforms Might Contribute to Isoform-Specific Interactions
3. Materials and Methods
3.1. Cloning, Protein Preparation and Purification
3.2. Protein Characterization and Enzymatic Activity Assays
3.3. Crystallization
3.4. X-ray Data Collection
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boissan, M.; Schlattner, U.; Lacombe, M.L. The NDPK/NME superfamily: State of the art. Lab. Investig. 2018, 98, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Schlattner, U. The Complex Functions of the NME Family—A Matter of Location and Molecular Activity. Int. J. Mol. Sci. 2021, 22, 13083. [Google Scholar] [CrossRef] [PubMed]
- Herbert, E.; Potter, V.R.; Takagi, Y. Nucleotide metabolism: IV. The phosphorylation of 5′-uridine nucleotides by cell fractions from rat liver. J. Biol. Chem. 1955, 213, 923–940. [Google Scholar] [CrossRef]
- Morera, S.; Chiadmi, M.; LeBras, G.; Lascu, I.; Janin, J. Mechanism of phosphate transfer by nucleoside diphosphate kinase: X-ray structures of the phosphohistidine intermediate of the enzymes from Drosophila and Dictyostelium. Biochemistry 1995, 34, 11062–11070. [Google Scholar] [CrossRef]
- Parks, R.; Agarwal, R. Nucleoside diphosphokinases. In The Enzymes; Boyer, P.D., Ed.; Academic Press: Cambridge, MA, USA, 1973; Volume 8, pp. 307–333. [Google Scholar]
- Pedersen, P.L. Coupling of adenosine triphosphate formation in mitochondria to the formation of nucleoside triphosphates: Involvement of nucleoside diphosphokinase. J. Biol. Chem. 1973, 248, 3956–3962. [Google Scholar] [CrossRef]
- Tepper, A.D.; Dammann, H.; Bominaar, A.A.; Veron, M. Investigation of the active site and the conformational stability of nucleoside diphosphate kinase by site-directed mutagenesis. J. Biol. Chem. 1994, 269, 32175–32180. [Google Scholar] [CrossRef]
- Moréra, S.; Lacombe, M.-L.; Yingwu, X.; LeBras, G.; Janin, J. X-ray structure of human nucleoside diphosphate kinase B complexed with GDP at 2 Å resolution. Structure 1995, 3, 1307–1314. [Google Scholar] [CrossRef] [PubMed]
- Mourad, N.; Parks, R. Erythrocytic nucleoside diphosphokinase: II. Isolation and kinetics. J. Biol. Chem. 1966, 241, 271–278. [Google Scholar] [CrossRef]
- Otero, A.d.S. Transphosphorylation and G protein activation. Biochem. Pharmacol. 1990, 39, 1399–1404. [Google Scholar] [CrossRef]
- Strelkov, S.V.; Perisic, O.; Webb, P.A.; Williams, R.L. The 1.9 Å crystal structure of a nucleoside diphosphate kinase complex with adenosine 3′, 5′-cyclic monophosphate: Evidence for competitive inhibition. J. Mol. Biol. 1995, 249, 665–674. [Google Scholar] [CrossRef]
- Boissan, M.; Montagnac, G.; Shen, Q.; Griparic, L.; Guitton, J.; Romao, M.; Sauvonnet, N.; Lagache, T.; Lascu, I.; Raposo, G. Nucleoside diphosphate kinases fuel dynamin superfamily proteins with GTP for membrane remodeling. Science 2014, 344, 1510–1515. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Gril, B.; Steeg, P.S. Metastasis Suppressors NME1 and NME2 Promote Dynamin 2 Oligomerization and Regulate Tumor Cell Endocytosis, Motility, and Metastasis. Cancer Res. 2019, 79, 4689–4702. [Google Scholar] [CrossRef] [PubMed]
- Annesley, S.J.; Bago, R.; Bosnar, M.H.; Filic, V.; Marinović, M.; Weber, I.; Mehta, A.; Fisher, P.R. Dictyostelium discoideum nucleoside diphosphate kinase C plays a negative regulatory role in phagocytosis, macropinocytosis and exocytosis. PLoS ONE 2011, 6, e26024. [Google Scholar] [CrossRef]
- Cipollini, G.; Berti, A.; Fiore, L.; Rainaldi, G.; Basolo, F.; Bevilacqua, G.; Caligo, M.A. Down-regulation of the nm23. h1 gene inhibits cell proliferation. Int. J. Cancer 1997, 73, 297–302. [Google Scholar] [CrossRef]
- Kang, Y.; Lee, D.-C.; Han, J.; Yoon, S.; Won, M.; Yeom, J.-H.; Seong, M.-J.; Ko, J.-J.; Lee, K.-A.; Lee, K. NM23-H2 involves in negative regulation of Diva and Bcl2L10 in apoptosis signaling. Biochem. Biophys. Res. Commun. 2007, 359, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Fournier, H.-N.; Albigès-Rizo, C.; Block, M.R. New insights into Nm23 control of cell adhesion and migration. J. Bioenerg. Biomembr. 2003, 35, 81–87. [Google Scholar] [CrossRef]
- Abu-Taha, I.H.; Heijman, J.; Feng, Y.; Vettel, C.; Dobrev, D.; Wieland, T. Regulation of heterotrimeric G-protein signaling by NDPK/NME proteins and caveolins: An update. Lab. Investig. 2018, 98, 190–197. [Google Scholar] [CrossRef]
- Abu-Taha, I.H.; Heijman, J.; Hippe, H.-J.; Wolf, N.M.; El-Armouche, A.; Nikolaev, V.O.; Schäfer, M.; Würtz, C.M.; Neef, S.; Voigt, N. Nucleoside diphosphate kinase-C suppresses cAMP formation in human heart failure. Circulation 2017, 135, 881–897. [Google Scholar] [CrossRef]
- Sakai, Y.; Hanafusa, H.; Hisamoto, N.; Matsumoto, K. Histidine dephosphorylation of the Gbeta protein GPB-1 promotes axon regeneration in C. elegans. EMBO Rep. 2022, 23, e55076. [Google Scholar] [CrossRef]
- Otero, A.d.S. NM23/nucleoside diphosphate kinase and signal transduction. J. Bioenerg. Biomembr. 2000, 32, 269–275. [Google Scholar] [CrossRef]
- Postel, E.H.; Berberich, S.J.; Rooney, J.W.; Kaetzel, D.M. Human NM23/nucleoside diphosphate kinase regulates gene expression through DNA binding to nuclease-hypersensitive transcriptional elements. J. Bioenerg. Biomembr. 2000, 32, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Postel, E.H. Multiple biochemical activities of NM23/NDP kinase in gene regulation. J. Bioenerg. Biomembr. 2003, 35, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Abu-Taha, I.H.; Vettel, C.; Wieland, T. Targeting altered Nme heterooligomerization in disease? Oncotarget 2018, 9, 1492–1493. [Google Scholar] [CrossRef] [PubMed]
- Georgescauld, F.; Song, Y.; Dautant, A. Structure, Folding and Stability of Nucleoside Diphosphate Kinases. Int. J. Mol. Sci. 2020, 21, 6779. [Google Scholar] [CrossRef] [PubMed]
- Janin, J.; Dumas, C.; Moréra, S.; Xu, Y.; Meyer, P.; Chiadmi, M.; Cherfils, J. Three-dimensional structure of nucleoside diphosphate kinase. J. Bioenerg. Biomembr. 2000, 32, 215–225. [Google Scholar] [CrossRef]
- Adman, E.T.; Sieker, L.C.; Jensen, L.H. The structure of a bacterial ferredoxin. J. Biol. Chem. 1973, 248, 3987–3996. [Google Scholar] [CrossRef]
- Lascu, I.; Giartosio, A.; Ransac, S.; Erent, M. Quaternary structure of nucleoside diphosphate kinases. J. Bioenerg. Biomembr. 2000, 32, 227–236. [Google Scholar] [CrossRef]
- Dumas, C.; Lascu, I.; Morera, S.; Glaser, P.; Fourme, R.; Wallet, V.; Lacombe, M.; Veron, M.; Janin, J. X-ray structure of nucleoside diphosphate kinase. EMBO J. 1992, 11, 3203–3208. [Google Scholar] [CrossRef]
- Lacombe, M.-L.L.; Munier, A.; Mehus, J.G.; Lambeth, D.O. The human Nm23/nucleoside diphosphate kinases. J. Bioenerg. Biomembr. 2000, 32, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Desvignes, T.; Pontarotti, P.; Fauvel, C.; Bobe, J. Nme protein family evolutionary history, a vertebrate perspective. BMC Evol. Biol. 2009, 9, 256. [Google Scholar] [CrossRef]
- Lombardi, D.; Lacombe, M.L.; Paggi, M.G. nm23: Unraveling its biological function in cell differentiation. J. Cell. Physiol. 2000, 182, 144–149. [Google Scholar] [CrossRef]
- Gilles, A.-M.; Presecan, E.; Vonica, A.; Lascu, I. Nucleoside diphosphate kinase from human erythrocytes. Structural characterization of the two polypeptide chains responsible for heterogeneity of the hexameric enzyme. J. Biol. Chem. 1991, 266, 8784–8789. [Google Scholar] [CrossRef] [PubMed]
- Urano, T.; Takamiya, K.; Furukawa, K.; Shiku, H. Molecular cloning and functional expression of the second mouse nm23/NDP kinase gene, nm23-M2. FEBS Lett. 1992, 309, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Potel, C.M.; Fasci, D.; Heck, A.J. Mix and match of the tumor metastasis suppressor Nm23 protein isoforms in vitro and in vivo. FEBS J. 2018, 285, 2856–2868. [Google Scholar] [CrossRef]
- Milon, L.; Meyer, P.; Chiadmi, M.; Munier, A.; Johansson, M.; Karlsson, A.; Lascu, I.; Capeau, J.; Janin, J.; Lacombe, M.-L. The human nm23-H4 gene product is a mitochondrial nucleoside diphosphate kinase. J. Biol. Chem. 2000, 275, 14264–14272. [Google Scholar] [CrossRef]
- Schlattner, U.; Tokarska-Schlattner, M.; Ramirez, S.; Tyurina, Y.Y.; Amoscato, A.A.; Mohammadyani, D.; Huang, Z.; Jiang, J.; Yanamala, N.; Seffouh, A. Dual Function of Mitochondrial Nm23-H4 Protein in Phosphotransfer and Intermembrane Lipid Transfer: A CARDIOLIPIN-DEPENDENT SWITCH*♦. J. Biol. Chem. 2013, 288, 111–121. [Google Scholar] [CrossRef]
- Chen, C.W.; Su, C.; Huang, C.Y.; Huang, X.R.; Cuili, X.; Chao, T.; Fan, C.H.; Ting, C.W.; Tsai, Y.W.; Yang, K.C.; et al. NME3 is a gatekeeper for DRP1-dependent mitophagy in hypoxia. Nat. Commun. 2024, 15, 2264. [Google Scholar] [CrossRef]
- Chen, C.W.; Wang, H.L.; Huang, C.W.; Huang, C.Y.; Lim, W.K.; Tu, I.C.; Koorapati, A.; Hsieh, S.T.; Kan, H.W.; Tzeng, S.R.; et al. Two separate functions of NME3 critical for cell survival underlie a neurodegenerative disorder. Proc. Natl. Acad. Sci. USA 2019, 116, 566–574. [Google Scholar] [CrossRef]
- Negroni, A.; Venturelli, D.; Tanno, B.; Amendola, R.; Ransac, S.; Cesi, V.; Calabretta, B.; Raschella, G. Neuroblastoma specific effects of DR-nm23 and its mutant forms on differentiation and apoptosis. Cell Death Differ. 2000, 7, 843–850. [Google Scholar] [CrossRef]
- Zala, D.; Schlattner, U.; Desvignes, T.; Bobe, J.; Roux, A.; Chavrier, P.; Boissan, M. The advantage of channeling nucleotides for very processive functions. F1000Research 2017, 6, 724. [Google Scholar] [CrossRef]
- Erent, M.; Gonin, P.; Cherfils, J.; Tissier, P.; Raschellà, G.; Giartosio, A.; Agou, F.; Sarger, C.; Lacombe, M.L.; Konrad, M. Structural and catalytic properties and homology modelling of the human nucleoside diphosphate kinase C, product of the DRnm23 gene. Eur. J. Biochem. 2001, 268, 1972–1981. [Google Scholar] [CrossRef] [PubMed]
- Anciaux, K.; Van Dommelen, K.; Willems, R.; Roymans, D.; Slegers, H. Inhibition of nucleoside diphosphate kinase (NDPK/nm23) by cAMP analogues. FEBS Lett. 1997, 400, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Wittinghofer, A. Signaling mechanistics: Aluminum fluoride for molecule of the year. Curr. Biol. 1997, 7, R682–R685. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.-W.; Moréra, S.; Janin, J.; Cherfils, J. AlF3 mimics the transition state of protein phosphorylation in the crystal structure of nucleoside diphosphate kinase and MgADP. Proc. Natl. Acad. Sci. USA 1997, 94, 3579–3583. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.L.; Oren, D.A.; Muñoz-Dorado, J.; Inouye, S.; Inouye, M.; Arnold, E. Crystal Structure of Myxococcus xanthus Nucleoside Diphosphate Kinase and its Interaction with a Nucleotide Substrate at 2· 0 Å Resolution. J. Mol. Biol. 1993, 234, 1230–1247. [Google Scholar] [CrossRef]
- Romani, A.; Scarpa, A. Regulation of cell magnesium. Arch. Biochem. Biophys. 1992, 298, 1–12. [Google Scholar] [CrossRef]
- Fuhs, S.R.; Meisenhelder, J.; Aslanian, A.; Ma, L.; Zagorska, A.; Stankova, M.; Binnie, A.; Al-Obeidi, F.; Mauger, J.; Lemke, G.; et al. Monoclonal 1- and 3-Phosphohistidine Antibodies: New Tools to Study Histidine Phosphorylation. Cell 2015, 162, 198–210. [Google Scholar] [CrossRef]
- Dautant, A.; Henri, J.; Wales, T.E.; Meyer, P.; Engen, J.R.; Georgescauld, F. Remodeling of the binding site of nucleoside diphosphate kinase revealed by X-ray structure and H/D exchange. Biochemistry 2019, 58, 1440–1449. [Google Scholar] [CrossRef]
- Attwood, P.V.; Wieland, T. Nucleoside diphosphate kinase as protein histidine kinase. Naunyn Schmiedebergs Arch. Pharmacol. 2015, 388, 153–160. [Google Scholar] [CrossRef]
- Nurizzo, D.; Mairs, T.; Guijarro, M.; Rey, V.; Meyer, J.; Fajardo, P.; Chavanne, J.; Biasci, J.C.; McSweeney, S.; Mitchell, E. The ID23-1 structural biology beamline at the ESRF. J. Synchrotron Radiat. 2006, 13, 227–238. [Google Scholar] [CrossRef]
- Nanao, M.; Basu, S.; Zander, U.; Giraud, T.; Surr, J.; Guijarro, M.; Lentini, M.; Felisaz, F.; Sinoir, J.; Morawe, C.; et al. ID23-2: An automated and high-performance microfocus beamline for macromolecular crystallography at the ESRF. J. Synchrotron Radiat. 2022, 29, 581–590. [Google Scholar] [CrossRef] [PubMed]
- von Stetten, D.; Carpentier, P.; Flot, D.; Beteva, A.; Caserotto, H.; Dobias, F.; Guijarro, M.; Giraud, T.; Lentini, M.; McSweeney, S.; et al. ID30A-3 (MASSIF-3)—A beamline for macromolecular crystallography at the ESRF with a small intense beam. J. Synchrotron Radiat. 2020, 27, 844–851. [Google Scholar] [CrossRef]
- Powell, H.R.; Battye, T.G.G.; Kontogiannis, L.; Johnson, O.; Leslie, A.G.W. Integrating macromolecular X-ray diffraction data with the graphical user interface iMosflm. Nat. Protoc. 2017, 12, 1310–1325. [Google Scholar] [CrossRef]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. Xds. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Winn, M.D.; Murshudov, G.N.; Papiz, M.Z. Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 2003, 374, 300–321. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997, 53, 240–255. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB Entry | 8QW3 | 8QVZ | 8QW0 | 8QW1 | 8QW2 | 8QVY |
|---|---|---|---|---|---|---|
| Ligands | ADP | cAMP | GDP | ADP, Mg2+ | UDP, Mg2+ | None |
| Data Collection | ||||||
| X-ray source | ESRF ID23-1 | ESRF ID30A-3 | ESRF ID30A-3 | ESRF ID23-2 | ESRF ID23-1 | ESRF ID30A-3 |
| Wavelength (Å) | 1.005 | 0.9677 | 0.9677 | 0.8731 | 0.6888 | 0.9677 |
| Detector | PILATUS 6M-F | EIGER X 4M | EIGER X 4M | PILATUS3 X 2M | EIGER2 X CdTe 16M | EIGER X 4M |
| Space group | P1 | P1 | P21 | C2 | C2 | C2 |
| Unit cell dimensions | ||||||
| a (Å) | 52.32 | 52.02 | 104.3 | 106.5 | 106.5 | 106.2 |
| b (Å) | 67.87 | 67.41 | 78.64 | 116.3 | 116.0 | 115.5 |
| c (Å) | 68.42 | 68.15 | 112.4 | 84.07 | 83.84 | 82.61 |
| α (°) | 108.5 | 108.7 | 90 | 90 | 90 | 90 |
| β (°) | 112.3 | 112.4 | 97.56 | 93.05 | 93.28 | 94.32 |
| γ (°) | 101.1 | 100.9 | 90 | 90 | 90 | 90 |
| Vm (Å3/Da) | 1.89 | 1.86 | 2.17 | 2.47 | 2.46 | 2.40 |
| Solvent content (%) | 35.1 | 34.0 | 43.5 | 50.3 | 50.0 | 48.9 |
| Resolution range (Å) | 60.1–1.25 (1.28–1.25) | 45.0–1.77 (1.81–1.77) | 46.6–2.17 (2.23–2.17) | 42.0–2.10 (2.15–2.10) | 47.7–1.87 (1.94–1.87) | 47.3–2.64 (2.71–2.64) |
| Rmerge (%) | 4.9 (94.7) | 12.7 (101.4) | 15.2 (138.8) | 18.4 (150.9) | 4.2 (118.7) | 12.7 (239.0) |
| <I/σ(I)> | 8.6 (0.7) | 6.2 (1.2) | 9.6 (1.0) | 10.0 (1.7) | 15.0 (1.1) | 9.45 (0.9) |
| CC1/2 (%) | 97.0 (40.1) | 99.3 (61.2) | 99.8 (54.9) | 99.5 (46.6) | 99.7 (69.7) | 99.2 (48.0) |
| Data completeness (%) | 94.2 (89.0) | 97.3 (88.4) | 99.5 (94.2) | 95.8 (73.1) | 99.9 (99.5) | 96.4 (98.2) |
| Average redundancy | 2.8 (1.8) | 4.6 (4.4) | 10.3 (7.2) | 6.6 (5.9) | 5.2 (4.9) | 7.3 (7.4) |
| Wilson B (Å2) | 12.8 | 20.8 | 46.2 | 41.6 | 44.0 | 79.1 |
| Refinement | ||||||
| Max. resolution (Å) | 1.25 | 1.77 | 2.17 | 2.10 | 1.87 | 2.64 |
| Total nr. reflections | 198,505 | 71,473 | 94,493 | 57,087 | 83,594 | 28,215 |
| Test set | 2119 | 2091 | 1996 | 2382 | 2091 | 1263 |
| Rwork (%) | 14.7 | 16.5 | 18.9 | 17.2 | 17.0 | 19.7 |
| Rfree (%) | 17.5 | 21.0 | 22.9 | 20.5 | 20.3 | 24.7 |
| Protein atoms | 7425 | 7481 | 14,483 | 7284 | 7300 | 7073 |
| Other non-solvent atoms | 177 | 147 | 346 | 168 | 156 | 30 |
| Solvent atoms | 542 | 408 | 303 | 399 | 313 | 0 |
| RMSD bond lengths (Å) | 0.0116 | 0.0106 | 0.0112 | 0.0091 | 0.0092 | 0.0073 |
| RMSD bond angles (°) | 1.671 | 1.664 | 1.812 | 1.641 | 1.618 | 1.470 |
| Ramachandran | ||||||
| favored (%) | 98 | 98 | 98 | 98 | 98 | 97 |
| allowed (%) | 2 | 2 | 2 | 2 | 2 | 3 |
| outliers (%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Average B (Å2) | ||||||
| protein | 21.3 | 26.8 | 50.8 | 41.4 | 61.0 | 103.3 |
| other non-solvent | 27.9 | 30.1 | 66.9 | 51.0 | 77.4 | 133.1 |
| solvent | 30.9 | 30.1 | 41.1 | 41.8 | 57.3 | - |
| PDB Entry | 8QW3 | 8QVZ | 8QW0 | 8QW1 | 8QW2 |
|---|---|---|---|---|---|
| Ligands | ADP | cAMP | GDP | ADP, Mg2+ | UDP, Mg2+ |
| Nucleotide concentration | 1 mM | 5 mM | 3 mM | 1 mM | 2 mM |
| Space group | P1 | P1 | P21 | C2 | C2 |
| Crystallization condition | 6% PEG 3350 | 7% PEG 8000 | |||
| 150 mM Li2SO4 | 27% Glycerol | ||||
| 100 mM Tri-sodium citrate pH 5.4 | 40 mM KH2PO4 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amjadi, R.; Werten, S.; Lomada, S.K.; Baldin, C.; Scheffzek, K.; Dunzendorfer-Matt, T.; Wieland, T. Mechanistic Insights into Substrate Recognition of Human Nucleoside Diphosphate Kinase C Based on Nucleotide-Induced Structural Changes. Int. J. Mol. Sci. 2024, 25, 9768. https://doi.org/10.3390/ijms25189768
Amjadi R, Werten S, Lomada SK, Baldin C, Scheffzek K, Dunzendorfer-Matt T, Wieland T. Mechanistic Insights into Substrate Recognition of Human Nucleoside Diphosphate Kinase C Based on Nucleotide-Induced Structural Changes. International Journal of Molecular Sciences. 2024; 25(18):9768. https://doi.org/10.3390/ijms25189768
Chicago/Turabian StyleAmjadi, Rezan, Sebastiaan Werten, Santosh Kumar Lomada, Clara Baldin, Klaus Scheffzek, Theresia Dunzendorfer-Matt, and Thomas Wieland. 2024. "Mechanistic Insights into Substrate Recognition of Human Nucleoside Diphosphate Kinase C Based on Nucleotide-Induced Structural Changes" International Journal of Molecular Sciences 25, no. 18: 9768. https://doi.org/10.3390/ijms25189768