
Dependence of Protein Immobilization and Photocurrent Generation in PSI–FTO Electrodes on the Electrodeposition Parameters

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Dependence of the Efficiency of PSI Immobilization on FTO Glass Plates on Deposition Parameters at pH 7 and 8
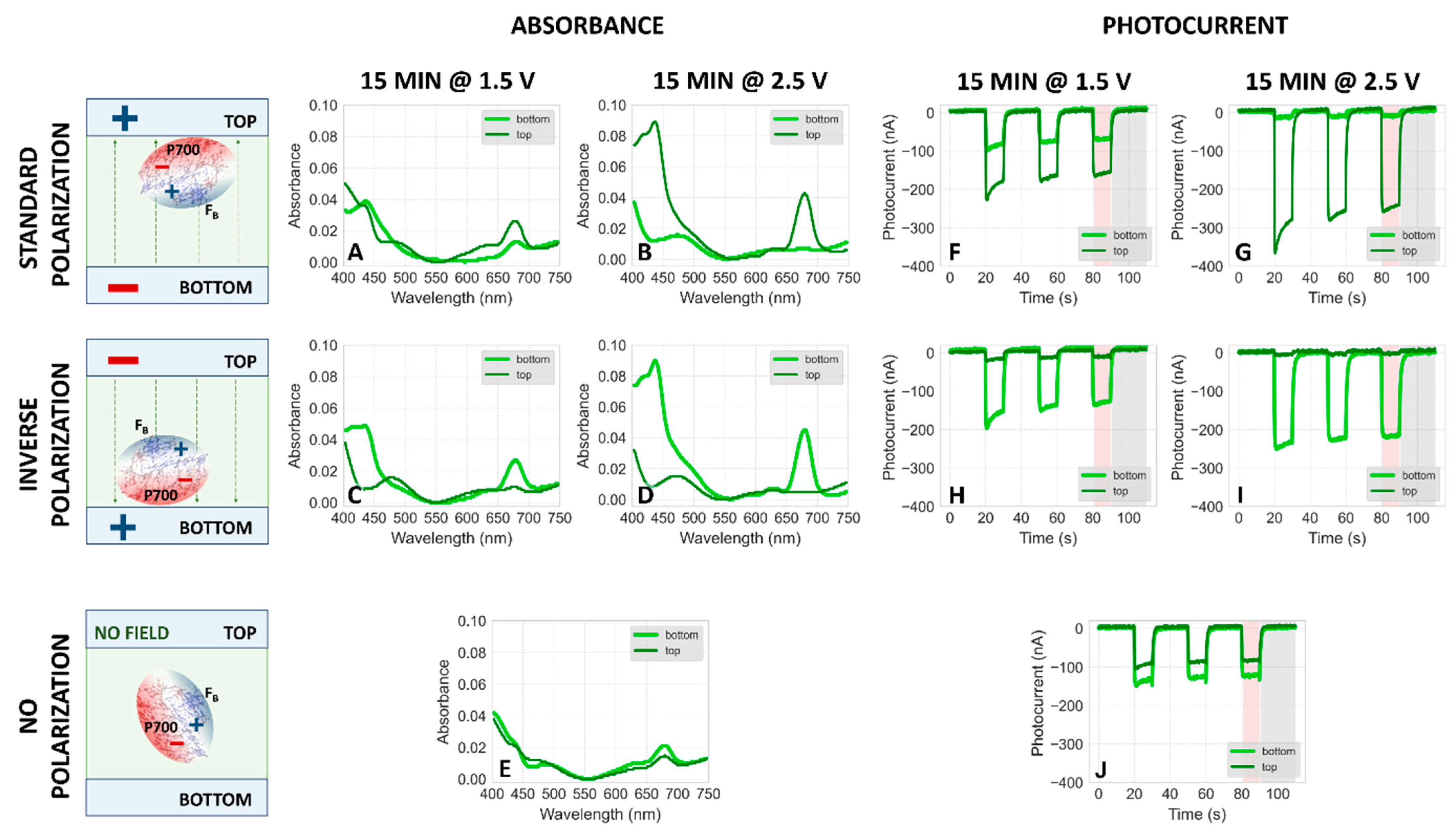
2.1.1. Absorption Spectra of PSI–FTO Plates Fabricated at pH 7
2.1.2. Absorption Spectra of PSI–FTO Plates Fabricated at pH 8
2.1.3. Effect of Deionized Water
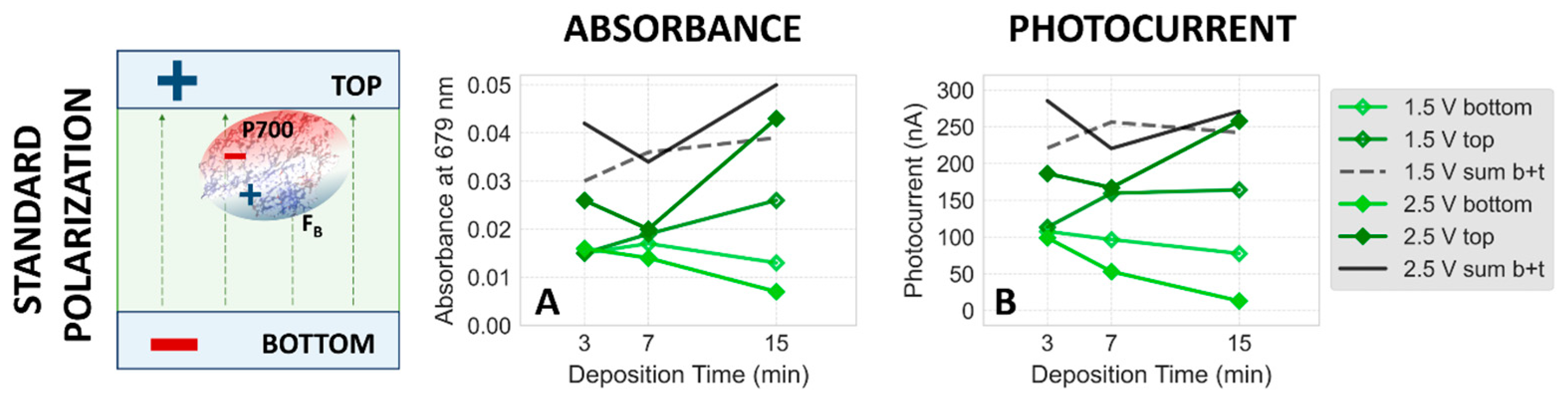
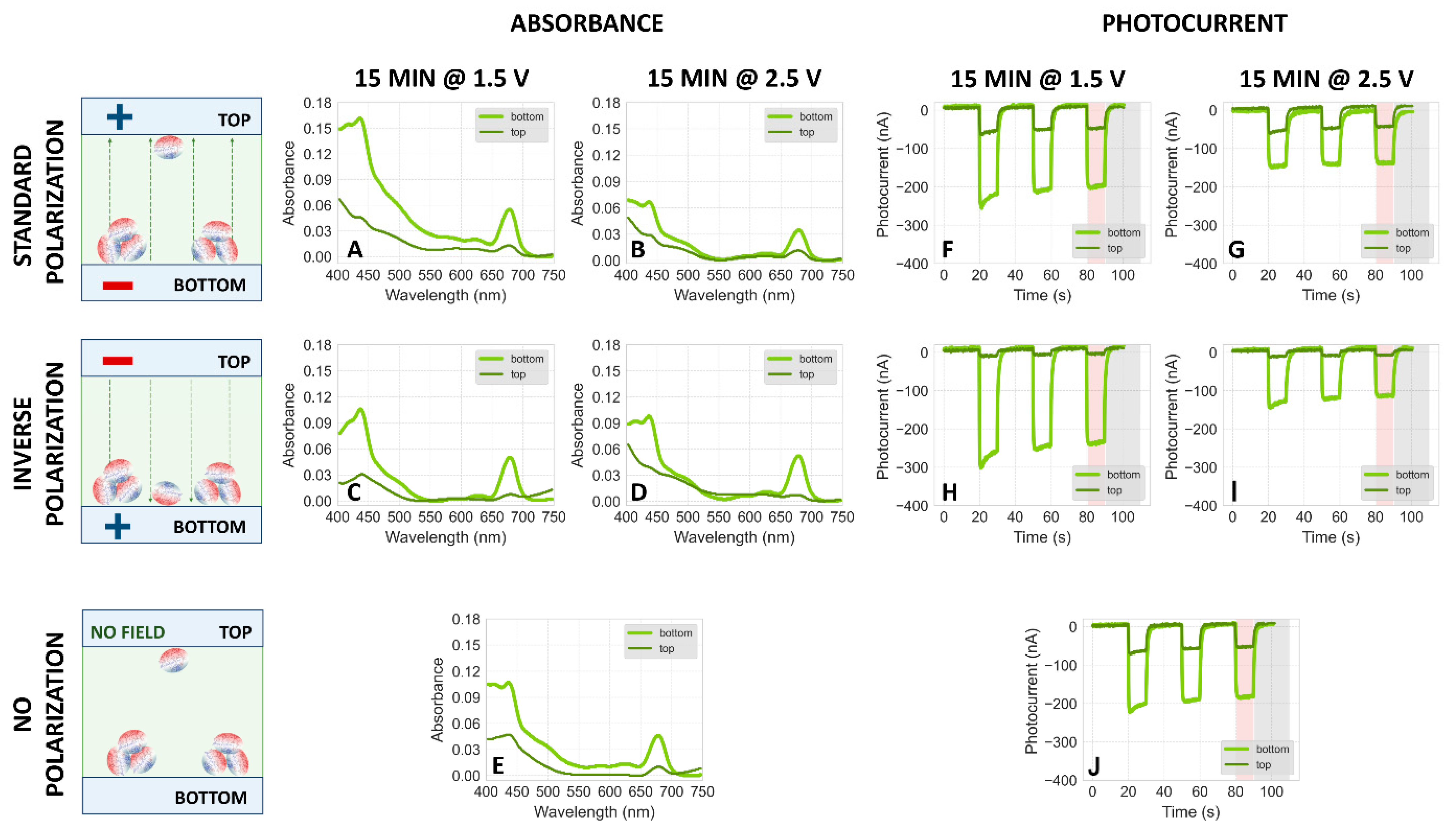
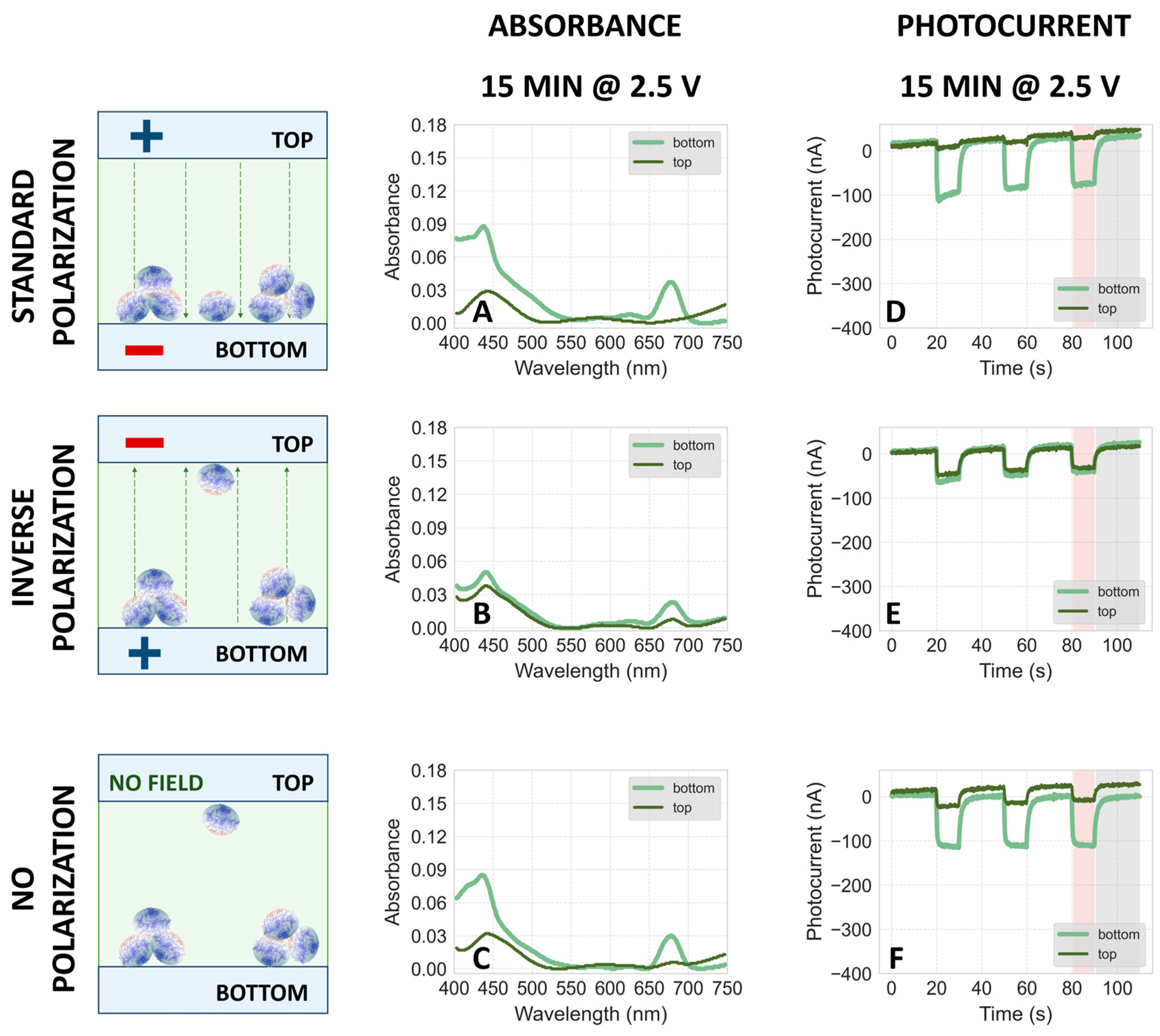
2.1.4. Effect of Higher Voltage
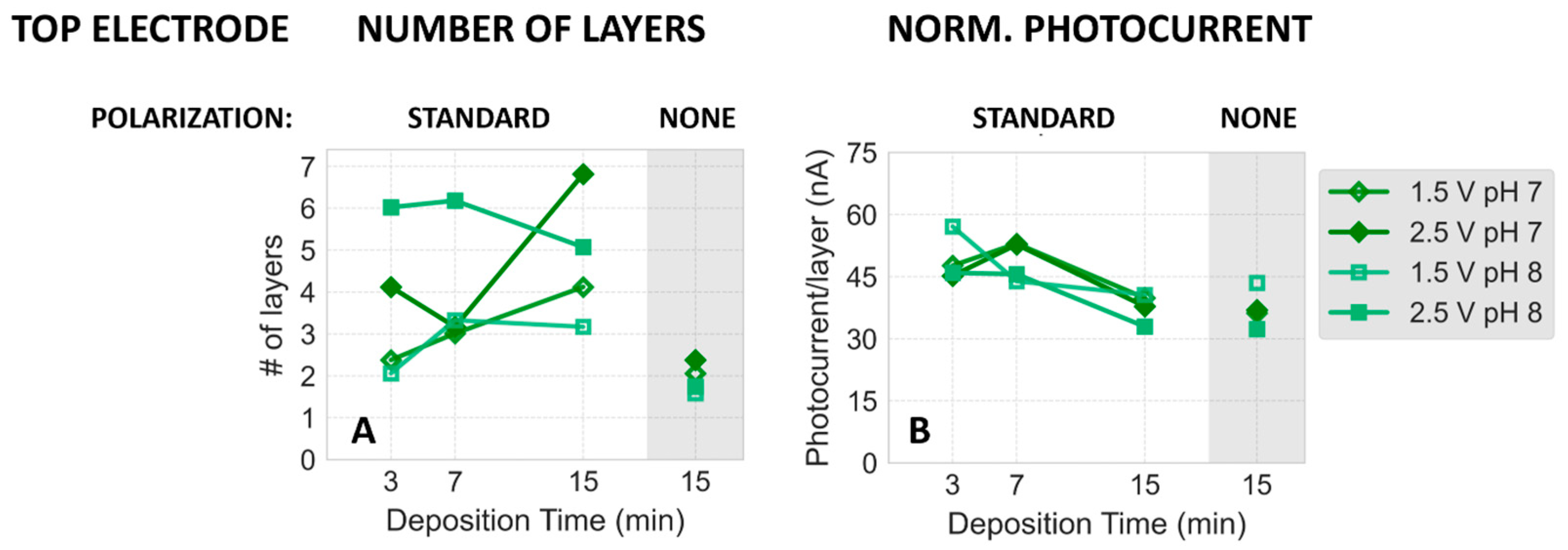
2.2. Dependence of Photocurrent Generated in the PSI–FTO Electrodes on Electrodeposition Parameters at pH 7 and 8
Correlation between Absorption and Photocurrent Signals
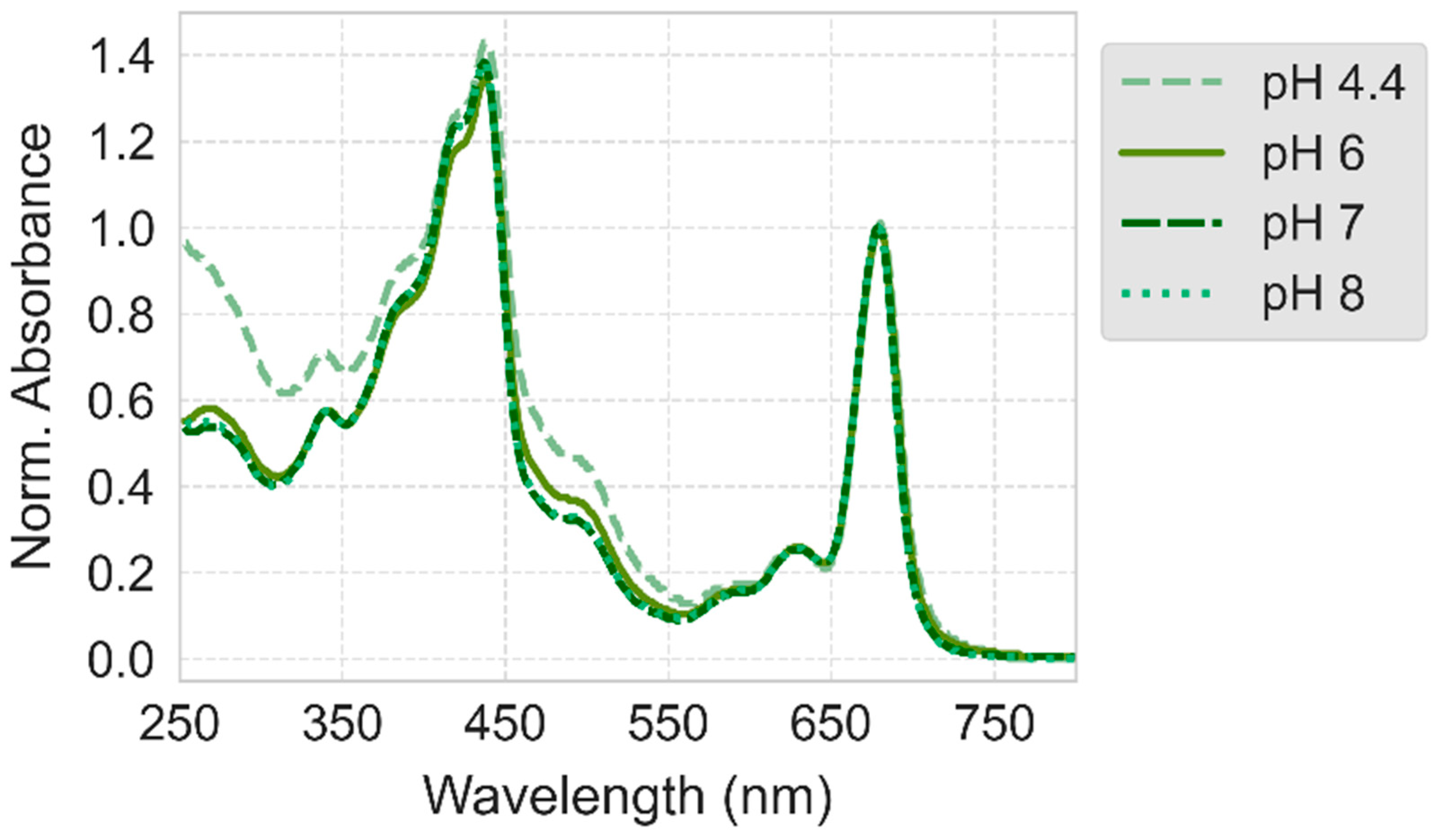
2.3. Absorption and Photocurrent Measurements Following Deposition of PSI Dissolved in Acidic Solutions (pH 4.4, 5, and 6)
2.3.1. Electrodeposition at pH 6
2.3.2. Electrodeposition at pH 4.4 and 5
2.4. Theoretical Predictions of PSI Surface Charges
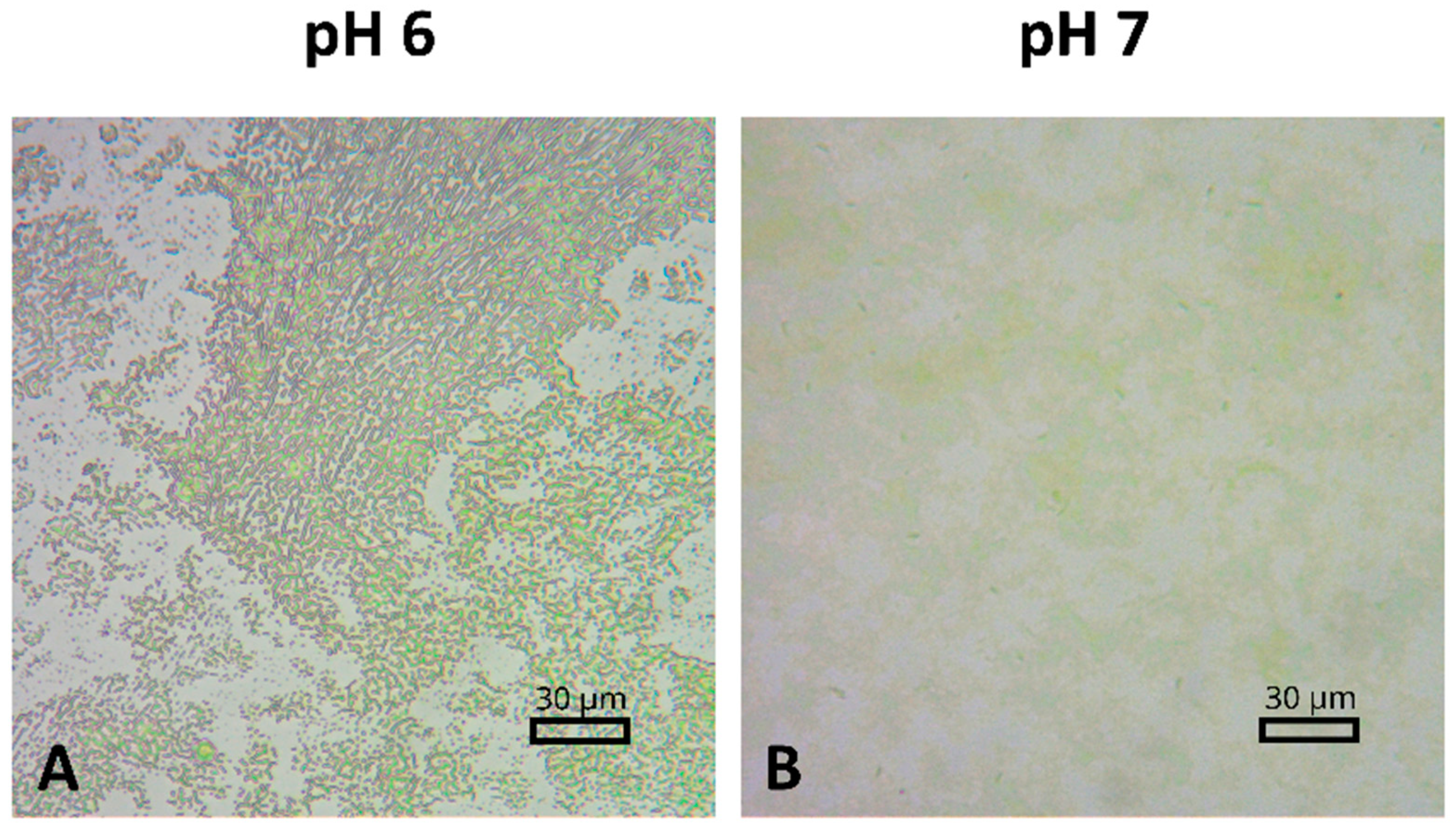
2.5. Characterization of the PSI–FTO Surface by Optical Microscopy
3. Materials and Methods
3.1. Preparation of PSI Particles
3.2. Disaggregation Methods of PSI Aggregates
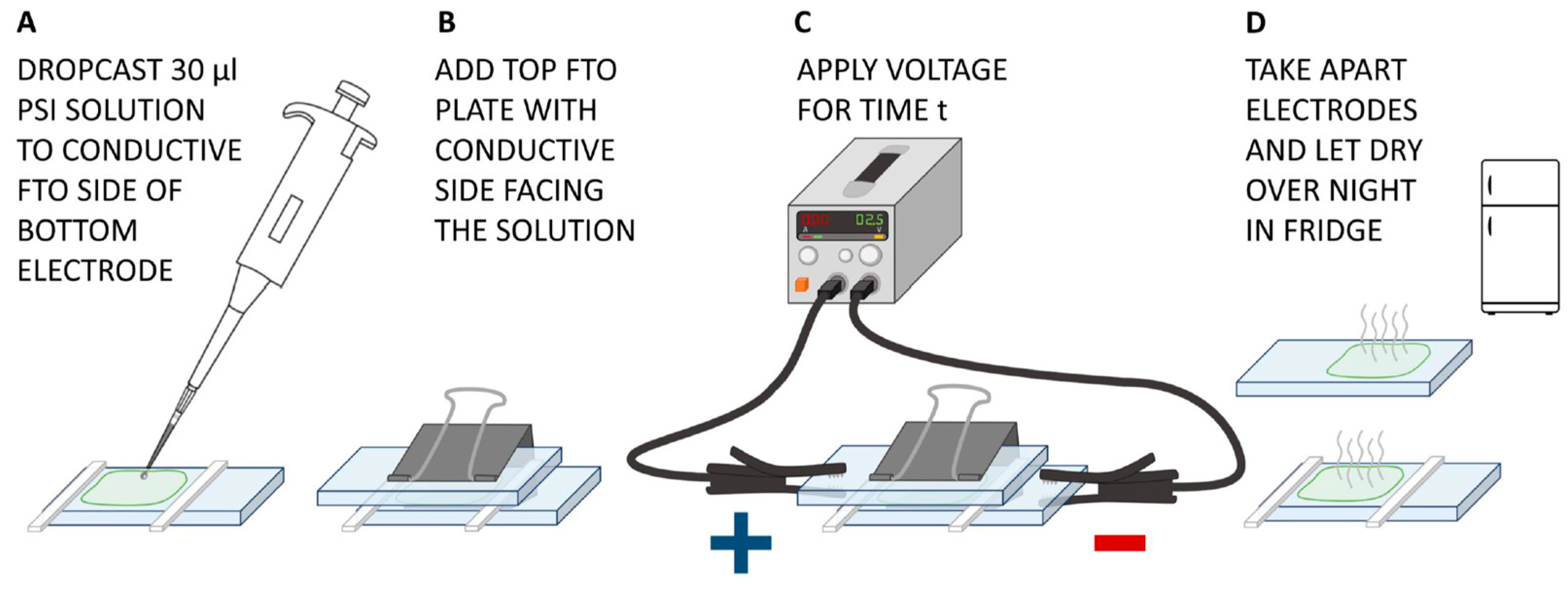
3.3. Preparation of the PSI–FTO Electrodes
3.4. Characterization of the Photoelectrodes
3.4.1. Steady-State Absorption Spectroscopy
3.4.2. Estimation of the Average Number of PSI Monolayers Deposited on FTO
3.4.3. Estimation of Internal Quantum Efficiency
3.4.4. Optical Microscopy
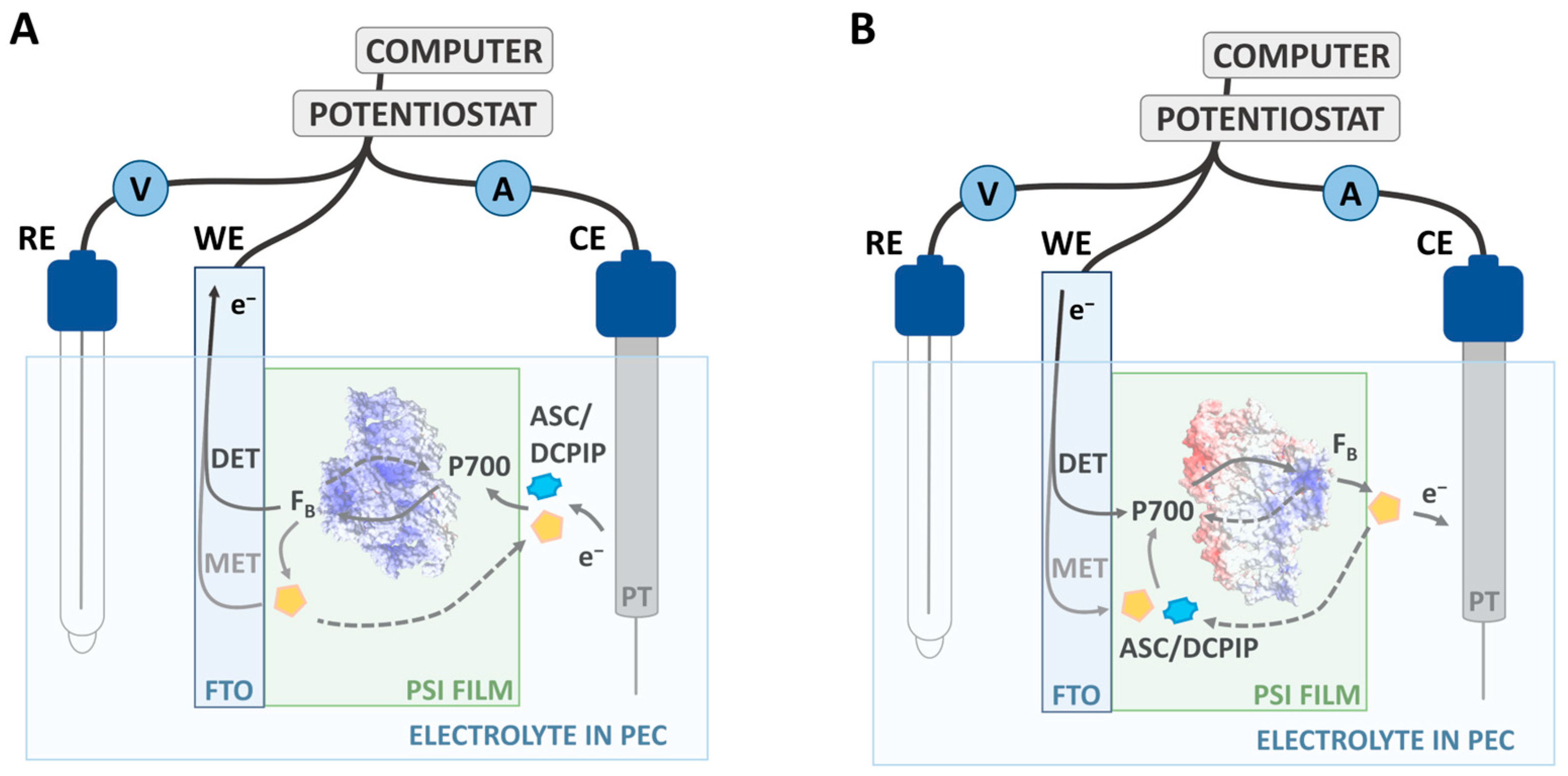
3.4.5. Photochronoamperometric Measurements
3.5. Computation of Surface Charges
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brettel, K. Electron transfer and arrangement of the redox cofactors in photosystem I. Biochim. Biophys. Acta Bioenerg. 1997, 1318, 322–373. [Google Scholar] [CrossRef]
- Brettel, K.; Leibl, W. Electron transfer in Photosystem I. Biochim. Biophys. Acta Bioenerg. 2001, 1507, 100–114. [Google Scholar] [CrossRef] [PubMed]
- Ciesielski, P.N.; Scott, A.M.; Faulkner, C.J.; Berron, B.J.; Cliffel, D.E.; Jennings, G.K. Functionalized nanoporous gold leaf electrode films for the immobilization of photosystem I. ACS Nano 2008, 2, 2465–2472. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.; Bruce, B.D. Growing green electricity: Progress and strategies for use of Photosystem I for sustainable photovoltaic energy conversion. Biochim. Biophys. Acta Bioenerg. 2013, 1827, 1553–1566. [Google Scholar] [CrossRef]
- Gordiichuk, P.I.; Wetzelaer, G.A.H.; Rimmerman, D.; Gruszka, A.; de Vries, J.W.; Saller, M.; Gautier, D.A.; Catarci, S.; Pesce, D.; Richter, S.; et al. Solid-State Biophotovoltaic Cells Containing Photosystem I. Adv. Mater. 2014, 26, 4863–4869. [Google Scholar] [CrossRef]
- Musazade, E.; Voloshin, R.; Brady, N.; Mondal, J.; Atashova, S.; Zharmukhamedov, S.K.; Huseynova, I.; Ramakrishna, S.; Najafpour, M.M.; Shen, J.-R.; et al. Biohybrid solar cells: Fundamentals, progress, and challenges. J. Photochem. Photobiol. C Photochem. Rev. 2018, 35, 134–156. [Google Scholar] [CrossRef]
- Teodor, A.H.; Bruce, B.D. Putting Photosystem I to work: Truly green energy. Trends Biotechnol. 2020, 38, 1329–1342. [Google Scholar] [CrossRef]
- Wolfe, K.D.; Dervishogullari, D.; Passantino, J.M.; Stachurski, C.D.; Jennings, G.K.; Cliffel, D.E. Improving the stability of photosystem I–based bioelectrodes for solar energy conversion. Curr. Opin. Electrochem. 2020, 19, 27–34. [Google Scholar] [CrossRef]
- Voloshin, R.A.; Lokteva, E.S.; Allakhverdiev, S.I. Photosystem I in the biohybrid electrodes. Curr. Opin. Green Sustain. Chem. 2023, 41, 100816. [Google Scholar] [CrossRef]
- Mershin, A.; Matsumoto, K.; Kaiser, L.; Yu, D.; Vaughn, M.; Nazeeruddin, M.K.; Bruce, B.D.; Graetzel, M.; Zhang, S. Self-assembled photosystem-I biophotovoltaics on nanostructured TiO2 and ZnO. Sci. Rep. 2021, 2, 234. [Google Scholar] [CrossRef]
- Yu, D.; Wang, M.; Zhu, G.; Ge, B.; Liu, S.; Huang, F. Enhanced photocurrent production by bio-dyes of photosynthetic macromolecules on designed TiO2 film. Sci. Rep. 2015, 5, 9375. [Google Scholar] [CrossRef] [PubMed]
- Kurashov, V.; Gorka, M.; Milanovsky, G.E.; Johnson, T.W.; Cherepanov, D.A.; Semenov, A.Y.; Golbeck, J.H. Critical evaluation of electron transfer kinetics in P700–FA/FB, P700–FX, and P700–A1 Photosystem I core complexes in liquid and in trehalose glass. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 1288–1301. [Google Scholar] [CrossRef] [PubMed]
- Szewczyk, S.; Białek, R.; Burdziński, G.; Gibasiewicz, K. Photovoltaic activity of electrodes based on intact photosystem I electrodeposited on bare conducting glass. Photosynth. Res. 2020, 144, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Stieger, K.R.; Ciornii, D.; Kolsch, A.; Hejazi, M.; Lokstein, H.; Feifel, S.C.; Zouni, A.; Lisdat, F. Engineering of supramolecular photoactive protein architectures: The defined co-assembly of photosystem I and cytochrome c using a nanoscaled DNA-matrix. Nanoscale 2016, 8, 10695–10705. [Google Scholar] [CrossRef] [PubMed]
- Stieger, K.R.; Feifel, S.C.; Lokstein, H.; Hejazi, M.; Zouni, A.; Lisdat, F. Biohybrid architectures for efficient light-to-current conversion based on photosystem I within scalable 3D mesoporous electrodes. J. Mater. Chem. A 2016, 4, 17009–17017. [Google Scholar] [CrossRef]
- Bottin, H.; Mathis, P. Interaction of plastocyanin with the photosystem I reaction center: A kinetic study by flash absorption spectroscopy. Biochemistry 1985, 24, 6453–6460. [Google Scholar] [CrossRef]
- Hippler, M.; Drepper, F.; Farah, J.; Rochaix, J.D. Fast electron transfer from cytochrome c6 and plastocyanin to photosystem I of Chlamydomonas reinhardtii requires PsaF. Biochemistry 1997, 36, 6343–6349. [Google Scholar] [CrossRef]
- Delosme, R. Electron transfer from cytochrome f to photosystem I in green algae. Photosynth. Res. 1991, 29, 45–54. [Google Scholar] [CrossRef]
- Ramesh, V.M.; Guergova-Kuras, M.; Joliot, P.; Webber, A.N. Electron Transfer from Plastocyanin to the Photosystem I Reaction Center in Mutants with Increased Potential of the Primary Donor in Chlamydomonas reinhardtii. Biochemistry 2002, 41, 14652–14658. [Google Scholar] [CrossRef]
- Setif, P.; Bottin, H. Laser flash absorption spectroscopy study of ferredoxin reduction by photosystem I in Synechocystis sp. PCC 6803: Evidence for submicrosecond and microsecond kinetics. Biochemistry 1994, 33, 8495–8504. [Google Scholar] [CrossRef]
- Setif, P.; Bottin, H. Laser flash absorption spectroscopy study of ferredoxin reduction by photosystem I: Spectral and kinetic evidence for the existence of several photosystem I-ferredoxin complexes. Biochemistry 1995, 34, 9059–9070. [Google Scholar] [CrossRef] [PubMed]
- Fischer, N.; Setif, P.; Rochaix, J.D. Targeted mutations in the psaC gene of Chlamydomonas reinhardtii: Preferential reduction of FB at low temperature is not accompanied by altered electron flow from photosystem I to ferredoxin. Biochemistry 1997, 36, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Fischer, N.; Hippler, M.; Setif, P.; Jacquot, J.P.; Rochaix, J.D. The PsaC subunit of photosystem I provides an essential lysine residue for fast electron transfer to ferredoxin. EMBO J. 1998, 17, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Setif, P. Ferredoxin and flavodoxin reduction by photosystem I. Biochim. Biophys. Acta Bioenerg. 2001, 1507, 161–179. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Wang, P.; Ruff, A.; Hartmann, V.; Zacarias, S.; Pereira, I.A.C.; Nowaczyk, M.; Rogner, M.; Conzuelo, F.; Schuhmann, W. A photosystem I monolayer with anisotropic electron flow enables Z-scheme like photosynthetic water splitting. Energy Environ. Sci. 2019, 12, 3133–3143. [Google Scholar] [CrossRef]
- Wang, P.; Frank, A.; Zhao, F.; Nowaczyk, M.M.; Conzuelo, F.; Schuhmann, W. A biomimetic assembly of folded photosystem I monolayers for an improved light utilization in biophotovoltaic devices. Bioelectrochemistry 2023, 149, 108288. [Google Scholar] [CrossRef]
- Ciesielski, P.N.; Hijazi, F.M.; Scott, A.M.; Faulkner, C.J.; Beard, L.; Emmett, K.; Rosenthal, S.J.; Cliffel, D.; Jennings, G.K. Photosystem I—Based biohybrid photoelectrochemical cells. Bioresour. Technol. 2010, 101, 3047–3053. [Google Scholar] [CrossRef]
- Ciesielski, P.N.; Faulkner, C.J.; Irwin, M.T.; Gregory, J.M.; Tolk, N.H.; Cliffel, D.E.; Jennings, G.K. Enhanced Photocurrent Production by Photosystem I Multilayer Assemblies. Adv. Funct. Mater. 2010, 20, 4048–4054. [Google Scholar] [CrossRef]
- Manocchi, A.K.; Baker, D.R.; Pendley, S.S.; Nguyen, K.; Hurley, M.M.; Bruce, B.D.; Sumner, J.J.; Lundgren, C.A. Photocurrent generation from surface assembled photosystem I on alkanethiol modified electrodes. Langmuir 2013, 29, 2412–2419. [Google Scholar] [CrossRef]
- Izzo, M.; Jacquet, M.; Fujiwara, T.; Harputlu, E.; Mazur, R.; Wróbel, P.; Góral, T.; Unlu, C.G.; Ocakoglu, K.; Miyagishima, S.; et al. Development of a novel nanoarchitecture of the robust photosystem I from a volcanic microalga Cyanidioschyzon merolae on single graphene for improved photocurrent generation. Int. J. Mol. Sci. 2021, 22, 8369. [Google Scholar] [CrossRef]
- Badura, A.; Guschin, D.; Kothe, T.; Kopczak, M.J.; Schuhmann, W.; Roegner, M. Photocurrent generation by photosystem 1 integrated in crosslinked redox hydrogels. Energy Environ. Sci. 2011, 4, 2435. [Google Scholar] [CrossRef]
- Gizzie, E.A.; LeBlanc, G.; Jennings, G.K.; Cliffel, D.E. Electrochemical preparation of Photosystem I-polyaniline composite films for biohybrid solar energy conversion. ACS Appl. Mater. Interfaces 2015, 7, 9328–9335. [Google Scholar] [CrossRef] [PubMed]
- Ciornii, D.; Kölsch, A.; Zouni, A.; Lisdat, F. Exploiting new ways for a more efficient orientation and wiring of PSI to electrodes: A fullerene C70 approach. Electrochim. Acta 2019, 299, 531–539. [Google Scholar] [CrossRef]
- Morlock, S.; Subramanian, S.K.; Zouni, A.; Lisdat, F. Bio-inorganic hybrid structures for direct electron transfer to photosystem I in photobioelectrodes. Biosens. Bioelectron. 2022, 214, 114495. [Google Scholar] [CrossRef] [PubMed]
- Morlock, S.; Subramanian, S.K.; Zouni, A.; Lisdat, F. Closing the green gap of photosystem I with synthetic fluorophores for enhanced photocurrent generation in photobiocathodes. Chem. Sci. 2023, 14, 1696–1708. [Google Scholar] [CrossRef]
- Ocakoglu, K.; Krupnik, T.; van den Bosch, B.; Harputlu, E.; Gullo, M.P.; Olmos, J.D.J.; Yildirimcan, S.; Gupta, R.K.; Yakuphanoglu, F.; Barbieri, A.; et al. Photosystem I-based biophotovoltaics on nanostructured hematite. Adv. Funct. Mater. 2014, 24, 7467–7477. [Google Scholar] [CrossRef]
- Zhang, J.Z.; Sokol, K.P.; Paul, N.; Romero, E.; van Grondelle, R.; Reisner, E. Competing charge transfer pathways at the photosystem II-electrode interface. Nat. Chem. Biol. 2016, 12, 1046–1052. [Google Scholar] [CrossRef]
- Mukherjee, D.; May, M.; Vaughn, M.; Bruce, B.D.; Khomami, B. Controlling the Morphology of Photosystem I Assembly on Thiol-Activated Au Substrates. Langmuir 2010, 26, 16048–16054. [Google Scholar] [CrossRef]
- Mukherjee, D.; May, M.; Khomami, B. Detergent–protein interactions in aqueous buffer suspensions of Photosystem I (PS I). J. Colloid Interface Sci. 2011, 358, 477–484. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, X.; Wang, M.; Liu, J.; Cao, M.; Lu, J.; Cui, Z. Characterization of photosystem I from spinach: Effect of solution pH. Photosynth. Res. 2012, 112, 63–70. [Google Scholar] [CrossRef]
- Tamura, N.; Itoh, S.; Yamamoto, Y.; Nishimura, M. Electrostatic interaction between plastocyanin and P700 in the electron transfer reaction of photosystem I-enriched particles. Plant Cell Physiol. 1981, 22, 603–612. [Google Scholar]
- Szewczyk, S.; Giera, W.; D’Haene, S.; van Grondelle, R.; Gibasiewicz, K. Comparison of excitation energy transfer in cyanobacterial photosystem I in solution and immobilized on conducting glass. Photosynth. Res. 2017, 132, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Haniewicz, P.; Abram, M.; Nosek, L.; Kirkpatrick, J.; El-Mohsnawy, E.; Olmos, J.D.J.; Kouril, R.; Kargul, J.M. Molecular mechanisms of photoadaptation of photosystem I supercomplex from an evolutionary cyanobacterial/algal intermediate. Plant Physiol. 2018, 176, 1433–1451. [Google Scholar] [CrossRef] [PubMed]
- Netzer-El, S.Y.; Caspy, I.; Nelson, N. Crystal Structure of Photosystem I Monomer from Synechocystis PCC 6803. Front. Plant Sci. 2019, 9, 1865. [Google Scholar] [CrossRef] [PubMed]
- Goyal, A.; Szewczyk, S.; Burdziński, G.; Abram, M.; Kargul, J.; Gibasiewicz, K. Competition between intra-protein charge recombination and electron transfer outside photosystem I complexes used for photovoltaic applications. Photochem. Photobiol. Sci. 2022, 21, 319–336. [Google Scholar] [CrossRef]
- Szewczyk, S.; Goyal, A.; Abram, M.; Burdziński, G.; Kargul, J.; Gibasiewicz, K. Electron transfer in a bio-photoelectrode based on photosystem I multilayer immobilized on the conducting glass. Int. J. Mol. Sci. 2022, 23, 4774. [Google Scholar] [CrossRef]
- Kruip, J.; Chitnis, P.R.; Lagoutte, B.; Roegner, M.; Boekema, E.J. Structural organization of the major subunits in cyanobacterial photosystem 1. J. Biol. Chem. 1997, 272, 17061–17069. [Google Scholar] [CrossRef]
- Schubert, W.D.; Klukas, O.; Krauß, N.; Saenger, W.; Fromme, P.; Witt, H.T. Photosystem I of Synechococcus elongatus at 4 Å resolution: Comprehensive structure analysis. J. Mol. Biol. 1997, 272, 741–769. [Google Scholar] [CrossRef]
- Müh, F.; Zouni, A. Extinction coefficients and critical solubilisation concentrations of photosystems I and II from Thermosynechococcus elongatus. Biochim. Biophys. Acta Bioenerg. 2005, 1708, 219–228. [Google Scholar] [CrossRef]
- Jordan, P.; Fromme, P.; Witt, H.T.; Klukas, O.; Saenger, W.; Krauß, N. Three-dimensional structure of cyanobacterial photosystem I at 2.5 Å resolution. Nature 2001, 411, 909–917. [Google Scholar] [CrossRef]
- Passantino, J.M.; Wolfe, K.D.; Simon, K.T.; Cliffel, D.E.; Jennings, G.K. Photosystem I Enhances the Efficiency of a Natural, Gel-Based Dye-Sensitized Solar Cell. ACS Appl. Bio Mater. 2020, 3, 4465–4473. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Duvaud, S.; Gabella, C.; Lisacek, F.; Stockinger, H.; Ioannidis, V.; Durinx, C. Expasy, the Swiss Bioinformatics Resource Portal, as designed by its users. Nucleic Acids Res. 2021, 49, W216–W227. [Google Scholar] [CrossRef] [PubMed]
- Guex, N.; Peitsch, M.C. Swiss-model and the Swiss-PDB viewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef] [PubMed]
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K.; et al. Improvements to APBS biomolecular solvation software suite. Protein Sci. 2017, 27, 112–128. [Google Scholar] [CrossRef]
- Nnyigide, O.S.; Nnyigide, T.O.; Lee, S.-G.; Hyun, K. Protein repair and analysis server: A web server to repair PDB structures, add missing heavy atoms and hydrogen atoms, and assign secondary structures by amide interactions. J. Chem. Inf. Model. 2022, 62, 4232–4246. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.C.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF chimerax: Structure visualization for researchers, educators, and developers. Protein Sci. 2020, 30, 70–82. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- The Pandas Development Team. pandas-dev/pandas: Pandas (v2.2.2). Zenodo 2024. [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kehler, T.; Szewczyk, S.; Gibasiewicz, K. Dependence of Protein Immobilization and Photocurrent Generation in PSI–FTO Electrodes on the Electrodeposition Parameters. Int. J. Mol. Sci. 2024, 25, 9772. https://doi.org/10.3390/ijms25189772
Kehler T, Szewczyk S, Gibasiewicz K. Dependence of Protein Immobilization and Photocurrent Generation in PSI–FTO Electrodes on the Electrodeposition Parameters. International Journal of Molecular Sciences. 2024; 25(18):9772. https://doi.org/10.3390/ijms25189772
Chicago/Turabian StyleKehler, Theresa, Sebastian Szewczyk, and Krzysztof Gibasiewicz. 2024. "Dependence of Protein Immobilization and Photocurrent Generation in PSI–FTO Electrodes on the Electrodeposition Parameters" International Journal of Molecular Sciences 25, no. 18: 9772. https://doi.org/10.3390/ijms25189772