
Discriminating Benign from Malignant Lung Diseases Using Plasma Glycosaminoglycans and Cell-Free DNA
 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Results
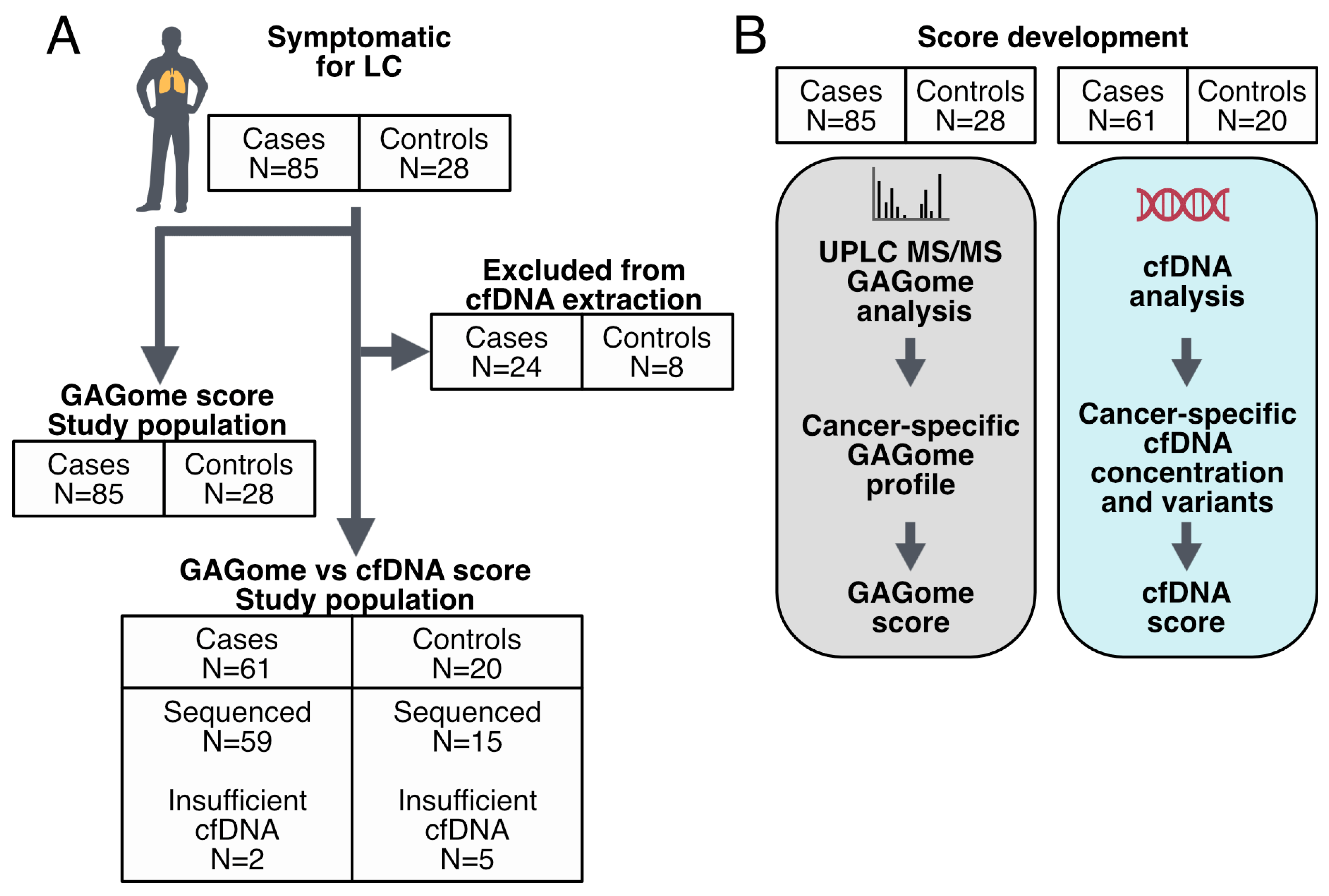
2.1. Cohort Characteristics
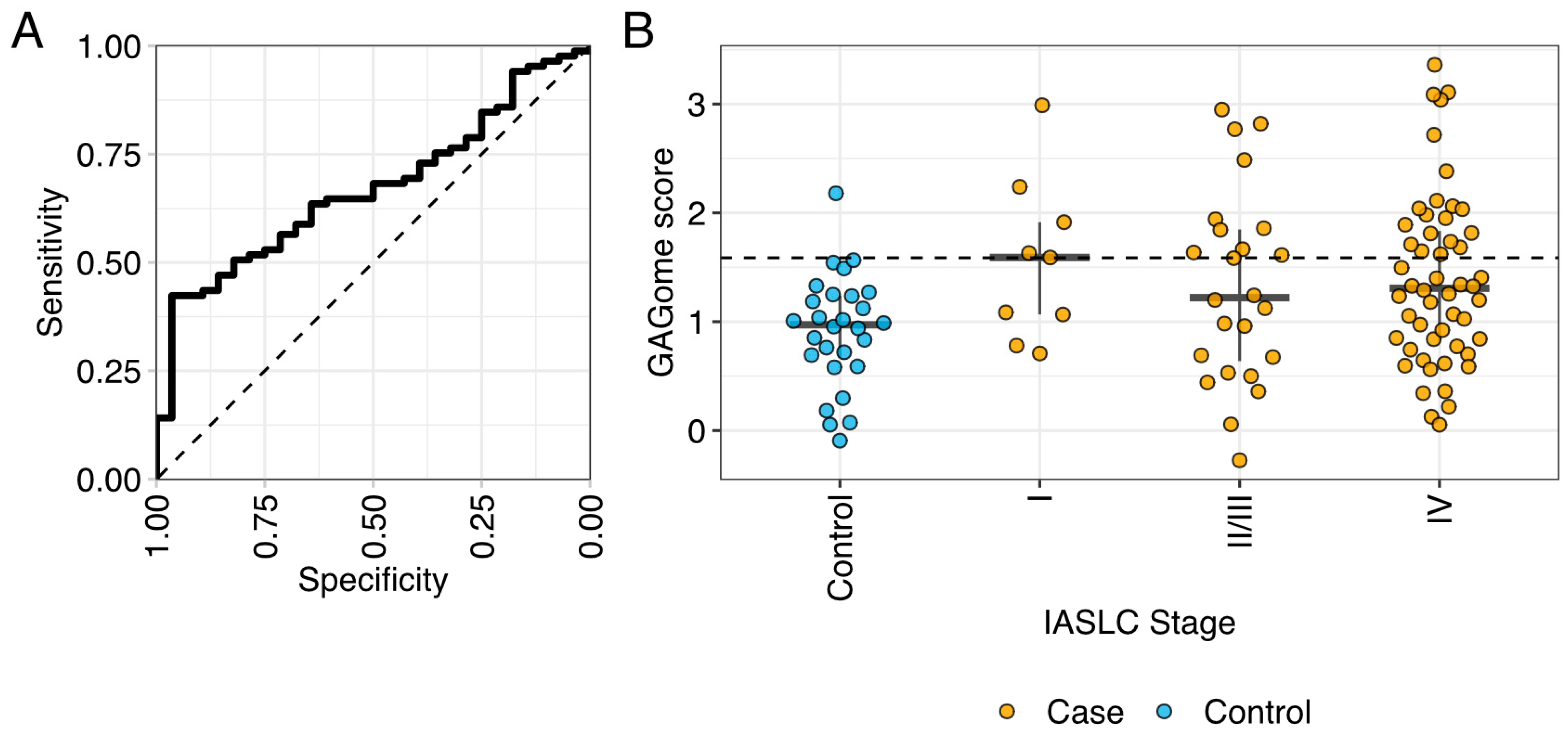
2.2. Correlation between Plasma GAGomes and Lung Cancer Diagnosis
2.3. GAGome and cfDNA Score
3. Discussion
4. Materials and Methods
4.1. Study Design and Cohort Characteristics
4.2. Plasma Collection and Isolation
4.3. GAGome Measurements
4.4. cfDNA Data
4.5. Statistical Analysis
4.5.1. GAGome Analysis
4.5.2. GAGome Score Development
4.5.3. cfDNA Score Development
4.5.4. Combined GAGome and cfDNA Test Pathway
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, Z.; Miao, H.; Zeng, Q.; Xu, S.; Chen, Z.; Liu, K. Circulating cell-free DNA as a diagnostic and prognostic biomarker for non-small-cell lung cancer: A systematic review and meta-analysis. Biomark. Med. 2020, 14, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of Circulating Tumor DNA in Early- and Late-Stage Human Malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Song, Q.; Ren, J.; Zhang, W.; Wang, Y.; Zhou, L.; Wang, D.; Chen, K.; Jiang, L.; Zhang, B.; et al. Simultaneous analysis of mutations and methylations in circulating cell-free DNA for hepatocellular carcinoma detection. Sci. Transl. Med. 2022, 14, eabp8704. [Google Scholar] [CrossRef]
- Visal, T.H.; den Hollander, P.; Cristofanilli, M.; Mani, S.A. Circulating tumour cells in the -omics era: How far are we from achieving the ‘singularity’? Br. J. Cancer 2022, 127, 173. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Sun, J.; Zhao, H.; Jiang, R.; Zheng, J.; Li, Z.; Peng, J.; Shen, H.; Zhang, K.; Zhao, J.; et al. Non-invasive lung cancer diagnosis and prognosis based on multi-analyte liquid biopsy. Mol. Cancer 2021, 20, 23. [Google Scholar] [CrossRef]
- Tomeva, E.; Switzeny, O.J.; Heitzinger, C.; Hippe, B.; Haslberger, A.G. Comprehensive Approach to Distinguish Patients with Solid Tumors from Healthy Controls by Combining Androgen Receptor Mutation p.H875Y with Cell-Free DNA Methylation and Circulating miRNAs. Cancers 2022, 14, 462. [Google Scholar] [CrossRef]
- Lin, A.A.; Nimgaonkar, V.; Issadore, D.; Carpenter, E.L. Extracellular Vesicle–Based Multianalyte Liquid Biopsy as a Diagnostic for Cancer. Annu. Rev. Biomed. Data Sci. 2022, 5, 269–292. [Google Scholar] [CrossRef]
- Li, L.; Ly, M.; Linhardt, R.J. Proteoglycan sequence. Mol. BioSyst. 2012, 8, 1613–1625. [Google Scholar] [CrossRef]
- Bülow, H.E.; Hobert, O. The Molecular Diversity of Glycosaminoglycans Shapes Animal Development. Annu. Rev. Cell Dev. Biol. 2006, 22, 375–407. [Google Scholar] [CrossRef]
- Gray, A.L.; Pun, N.; Ridley, A.J.L.; Dyer, D.P. Role of extracellular matrix proteoglycans in immune cell recruitment. Int. J. Exp. Pathol. 2022, 103, 34–43. [Google Scholar] [CrossRef] [PubMed]
- du Souich, P.; García, A.G.; Vergés, J.; Montell, E. Immunomodulatory and anti-inflammatory effects of chondroitin sulphate. J. Cell. Mol. Med. 2009, 13, 1451–1463. [Google Scholar] [CrossRef]
- Iozzo, R.V.; Schaefer, L. Proteoglycan form and function: A comprehensive nomenclature of proteoglycans. Matrix Biol. 2015, 42, 11–55. [Google Scholar] [CrossRef]
- Gesslbauer, B.; Rek, A.; Falsone, F.; Rajkovic, E.; Kungl, A.J. Proteoglycanomics: Tools to unravel the biological function of glycosaminoglycans. Proteomics 2007, 7, 2870–2880. [Google Scholar] [CrossRef] [PubMed]
- Weyers, A.; Yang, B.; Yoon, D.S.; Park, J.H.; Zhang, F.; Lee, K.B.; Linhardt, R.J. A Structural Analysis of Glycosaminoglycans from Lethal and Nonlethal Breast Cancer Tissues: Toward a Novel Class of Theragnostics for Personalized Medicine in Oncology? OMICS 2012, 16, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Gatto, F.; Bratulic, S.; Jonasch, E.; Limeta, A.; Maccari, F.; Galeotti, F.; Volpi, N.; Lundstam, S.; Nielsen, J.; Stierner, U. Plasma and Urine Free Glycosaminoglycans as Monitoring and Predictive Biomarkers in Metastatic Renal Cell Carcinoma: A Prospective Cohort Study. JCO Precis. Oncol. 2023, 7, e2200361. [Google Scholar] [CrossRef] [PubMed]
- Gatto, F.; Dabestani, S.; Bratulic, S.; Limeta, A.; Maccari, F.; Galeotti, F.; Volpi, N.; Stierner, U.; Nielsen, J.; Lundstam, S. Plasma and Urine Free Glycosaminoglycans as Monitoring Biomarkers in Nonmetastatic Renal Cell Carcinoma—A Prospective Cohort Study. Eur. Urol. Open Sci. 2022, 42, 30–39. [Google Scholar] [CrossRef]
- Al-Nakouzi, N.; Wang, C.K.; Oo, H.Z.; Nelepcu, I.; Lallous, N.; Spliid, C.B.; Khazamipour, N.; Lo, J.; Truong, S.; Collins, C.; et al. Reformation of the chondroitin sulfate glycocalyx enables progression of AR-independent prostate cancer. Nat. Commun. 2022, 13, 4760. [Google Scholar] [CrossRef]
- Theocharis, A.D.; Vynios, D.H.; Papageorgakopoulou, N.; Skandalis, S.S.; Theocharis, D.A. Altered content composition and structure of glycosaminoglycans and proteoglycans in gastric carcinoma. Int. J. Biochem. Cell Biol. 2003, 35, 376–390. [Google Scholar] [CrossRef]
- Balbisi, M.; Sugár, S.; Schlosser, G.; Szeitz, B.; Fillinger, J.; Moldvay, J.; Drahos, L.; Szász, A.M.; Tóth, G.; Turiák, L. Inter- and intratumoral proteomics and glycosaminoglycan characterization of ALK rearranged lung adenocarcinoma tissues: A pilot study. Sci. Rep. 2023, 13, 6268. [Google Scholar] [CrossRef]
- Li, G.; Li, L.; Joo, E.J.; Son, J.W.; Kim, Y.J.; Kang, J.K.; Lee, K.B.; Zhang, F.; Linhardt, R.J. Glycosaminoglycans and glycolipids as potential biomarkers in lung cancer. Glycoconj. J. 2017, 34, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Pál, D.; Tóth, G.; Sugár, S.; Fügedi, K.D.; Szabó, D.; Kovalszky, I.; Papp, D.; Schlosser, G.; Tóth, C.; Tornóczky, T.; et al. Compositional Analysis of Glycosaminoglycans in Different Lung Cancer Types—A Pilot Study. Int. J. Mol. Sci. 2023, 24, 7050. [Google Scholar] [CrossRef] [PubMed]
- Mattox, A.K.; Douville, C.; Wang, Y.; Popoli, M.; Ptak, J.; Silliman, N.; Dobbyn, L.; Schaefer, J.; Lu, S.; Pearlman, A.H.; et al. The Origin of Highly Elevated Cell-Free DNA in Healthy Individuals and Patients with Pancreatic, Colorectal, Lung, or Ovarian Cancer. Cancer Discov. 2023, 13, 2166–2179. [Google Scholar] [CrossRef]
- Bratulic, S.; Limeta, A.; Dabestani, S.; Birgisson, H.; Enblad, G.; Stålberg, K.; Hesselager, G.; Häggman, M.; Höglund, M.; Simonson, O.E.; et al. Noninvasive detection of any-stage cancer using free glycosaminoglycans. Proc. Natl. Acad. Sci. USA 2022, 119, e2115328119. [Google Scholar] [CrossRef]
- Wang, F.; Li, X.; Li, M.; Liu, W.; Lu, L.; Li, Y.; Chen, X.; Yang, S.; Liu, T.; Cheng, W.; et al. Ultra-short cell-free DNA fragments enhance cancer early detection in a multi-analyte blood test combining mutation, protein and fragmentomics. Clin. Chem. Lab. Med. 2023, 62, 168–177. [Google Scholar] [CrossRef] [PubMed]
- D’Ambrosi, S.; Giannoukakos, S.; Antunes-Ferreira, M.; Pedraz-Valdunciel, C.; Bracht, J.W.; Potie, N.; Gimenez-Capitan, A.; Hackenberg, M.; Fernandez Hilario, A.; Molina-Vila, M.A.; et al. Combinatorial Blood Platelets-Derived circRNA and mRNA Signature for Early-Stage Lung Cancer Detection. Int. J. Mol. Sci. 2023, 24, 4881. [Google Scholar] [CrossRef]
- Bratulic, S.; Limeta, A.; Maccari, F.; Galeotti, F.; Volpi, N.; Levin, M.; Nielsen, J.; Gatto, F. Analysis of normal levels of free glycosaminoglycans in urine and plasma in adults. J. Biol. Chem. 2022, 298, 101575. [Google Scholar] [CrossRef]
- da Silva, M.N.R.; Mendes, A.; Martins, J.R.M.; Tobias-Machado, M.; Pinhal, M.A.d.S. Prospective Evaluation of Chondroitin Sulfate, Heparan Sulfate and Hyaluronic Acid in Prostate Cancer. Int. Braz. J. Urol. 2018, 44, 1139–1146. [Google Scholar] [CrossRef]
- Saito, A.; Munakata, H. Analysis of plasma proteins that bind to glycosaminoglycans. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2007, 1770, 241–246. [Google Scholar] [CrossRef]
- Prabhakar, V.; Sasisekharan, R. The biosynthesis and catabolism of galactosaminoglycans. Adv. Pharmacol. 2006, 53, 69–115. [Google Scholar]
- Cohen, J.F.; Korevaar, D.; Altman, D.G.; Bruns, D.; Gatsonis, C.; Hooft, L.; Irwig, L.; Levine, D.; Reitsma, J.B.; de Vet, H.C.W.; et al. STARD 2015 guidelines for reporting diagnostic accuracy studies: Explanation and elaboration. BMJ Open 2016, 6, e012799. [Google Scholar] [CrossRef] [PubMed]
- Detterbeck, F.C.; Boffa, D.J.; Kim, A.W.; Tanoue, L.T. The Eighth Edition Lung Cancer Stage Classification. Chest 2017, 151, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Travis, W.D.; Brambilla, E.; Müller-Hermelink, H.K.; Harris, C.C. Pathology and Genetics of Tumours of the Lung, Pleura, Thymus and Heart; IARC Press: Lyon, France, 2004. [Google Scholar]
- Tamburro, D.; Bratulic, S.; Abou Shameh, S.; Soni, N.K.; Bacconi, A.; Maccari, F.; Galeotti, F.; Mattsson, K.; Volpi, N.; Nielsen, J.; et al. Analytical performance of a standardized kit for mass spectrometry-based measurements of human glycosaminoglycans. J. Chromatogr. B 2021, 1177, 122761. [Google Scholar] [CrossRef]
- Volpi, N.; Galeotti, F.; Yang, B.; Linhardt, R.J. Analysis of glycosaminoglycan-derived, precolumn, 2-aminoacridone–labeled disaccharides with LC-fluorescence and LC-MS detection. Nat. Protoc. 2014, 9, 541–558. [Google Scholar] [CrossRef]
- Qvick, A.; Stenmark, B.; Carlsson, J.; Isaksson, J.; Karlsson, C.; Helenius, G. Liquid biopsy as an option for predictive testing and prognosis in patients with lung cancer. Mol. Med. 2021, 27, 68. [Google Scholar] [CrossRef]
- Kruschke, J.K. Bayesian estimation supersedes the t test. J. Exp. Psychol. Gen. 2013, 142, 573–603. [Google Scholar] [CrossRef]
- Vehtari, A.; Gelman, A.; Simpson, D.; Carpenter, B.; Bürkner, P.-C. Rank-Normalization, Folding, and Localization: An Improved for Assessing Convergence of MCMC (with Discussion). Bayesian Anal. 2021, 16, 667–718. [Google Scholar] [CrossRef]
- Bürkner, P.-C. brms: An R Package for Bayesian Multilevel Models Using Stan. J. Stat. Softw. 2017, 80, 1–28. [Google Scholar] [CrossRef]
- Makowski, D.; Ben-Shachar, M.S.; Lüdecke, D. bayestestR: Describing Effects and their Uncertainty, Existence and Significance within the Bayesian Framework. J. Open Source Softw. 2019, 4, 1541. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case (n = 85) | Control (n = 28) | Case vs. control, p-Value | Overall (n = 113) | |
|---|---|---|---|---|
| Age | ||||
| Mean (SD) | 70.4 (8.5) | 67.0 (14.4) | 0.246 | 69.6 (10.3) |
| Gender | ||||
| Female | 45 (52.9%) | 17 (60.7%) | 0.473 | 63 (55.3%) |
| Male | 40 (47.1%) | 11 (39.3%) | 51 (44.7%) | |
| Smoking | ||||
| Smoker | 30 (35.3%) | 5 (17.9%) | 0.047 | 35 (31.0%) |
| Ex-Smoker | 43 (50.6%) | 12 (42.9%) | 55 (48.7%) | |
| Never-Smoker | 12 (14.1%) | 9 (32.1%) | 21 (18.6%) | |
| Missing | 0 (0%) | 2 (7.1%) | 2 (1.8%) | |
| Tumor size (cm) | ||||
| Mean (SD) | 4.5 (2.4) | NA (NA) | 4.5 (2.4) | |
| Median [Min, Max] | 4.0 [0.9, 12.5] | NA [NA, NA] | 4.0 [0.9, 12.5] | |
| Missing | 4 (4.7%) | 28 (100%) | 32 (28.3%) | |
| IASLC stage | ||||
| I | 9 (10.6%) | 0 (0%) | 9 (8.0%) | |
| II | 6 (7.1%) | 0 (0%) | 6 (5.3%) | |
| III | 18 (21.2%) | 0 (0%) | 18 (15.9%) | |
| IV | 52 (61.2%) | 0 (0%) | 52 (46.0%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qvick, A.; Bratulic, S.; Carlsson, J.; Stenmark, B.; Karlsson, C.; Nielsen, J.; Gatto, F.; Helenius, G. Discriminating Benign from Malignant Lung Diseases Using Plasma Glycosaminoglycans and Cell-Free DNA. Int. J. Mol. Sci. 2024, 25, 9777. https://doi.org/10.3390/ijms25189777
Qvick A, Bratulic S, Carlsson J, Stenmark B, Karlsson C, Nielsen J, Gatto F, Helenius G. Discriminating Benign from Malignant Lung Diseases Using Plasma Glycosaminoglycans and Cell-Free DNA. International Journal of Molecular Sciences. 2024; 25(18):9777. https://doi.org/10.3390/ijms25189777
Chicago/Turabian StyleQvick, Alvida, Sinisa Bratulic, Jessica Carlsson, Bianca Stenmark, Christina Karlsson, Jens Nielsen, Francesco Gatto, and Gisela Helenius. 2024. "Discriminating Benign from Malignant Lung Diseases Using Plasma Glycosaminoglycans and Cell-Free DNA" International Journal of Molecular Sciences 25, no. 18: 9777. https://doi.org/10.3390/ijms25189777