


The Drug Transporter P-Glycoprotein and Its Impact on Ceramide Metabolism—An Unconventional Ally in Cancer Treatment

 , and
, and
Abstract
:1. Sphingolipid Metabolism at a Glance
1.1. The De Novo Pathway of Ceramide Formation
1.2. Ceramide Metabolism—Intracellular Fate
1.3. Bioactive Sphingolipids
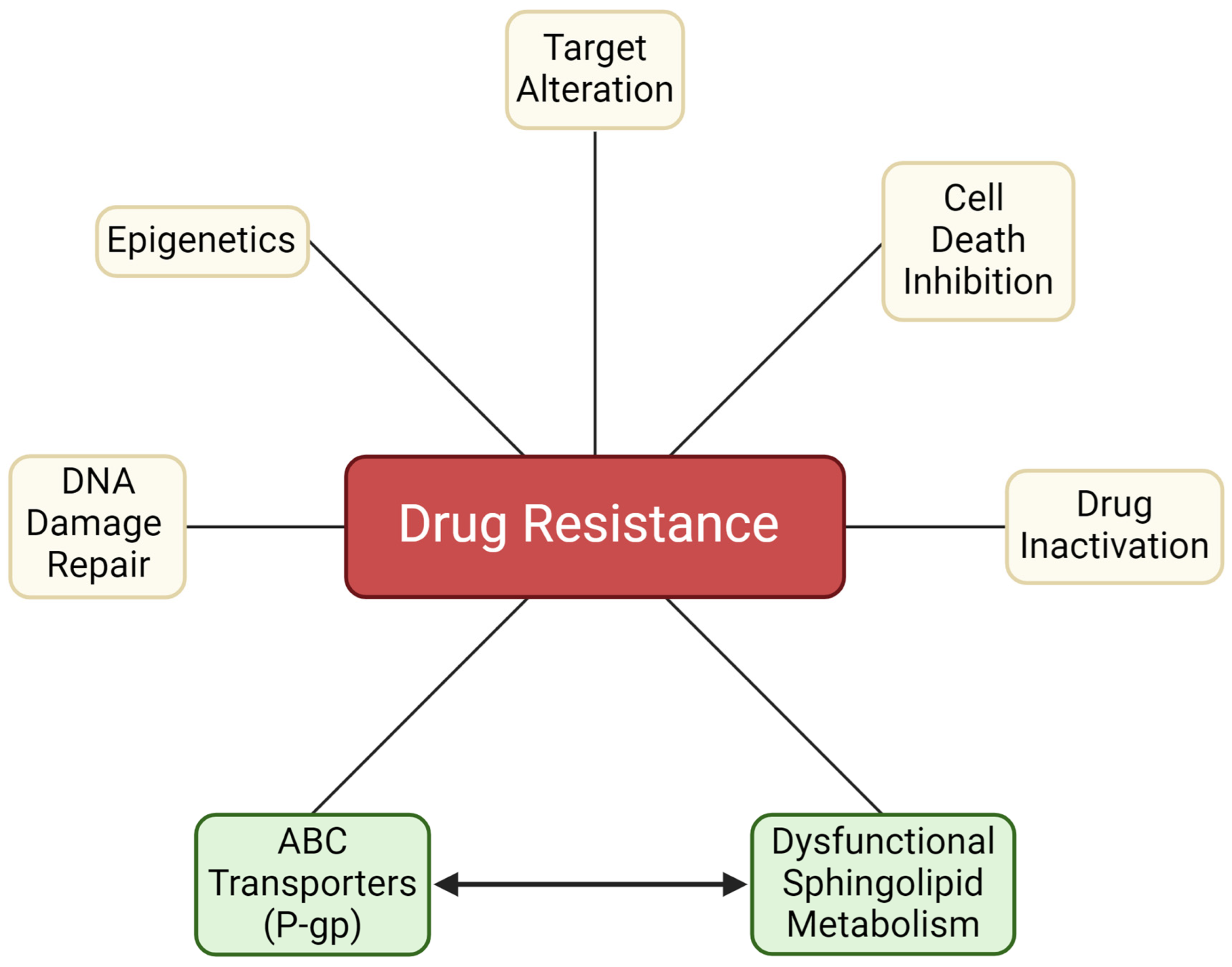
2. An Introduction to Chemotherapy Resistance
2.1. Factors Contributing to Chemotherapy Resistance
2.2. Drug Activation/Inactivation—Drug Metabolism
2.3. Repairing DNA Damage
2.4. Resistance to Apoptosis
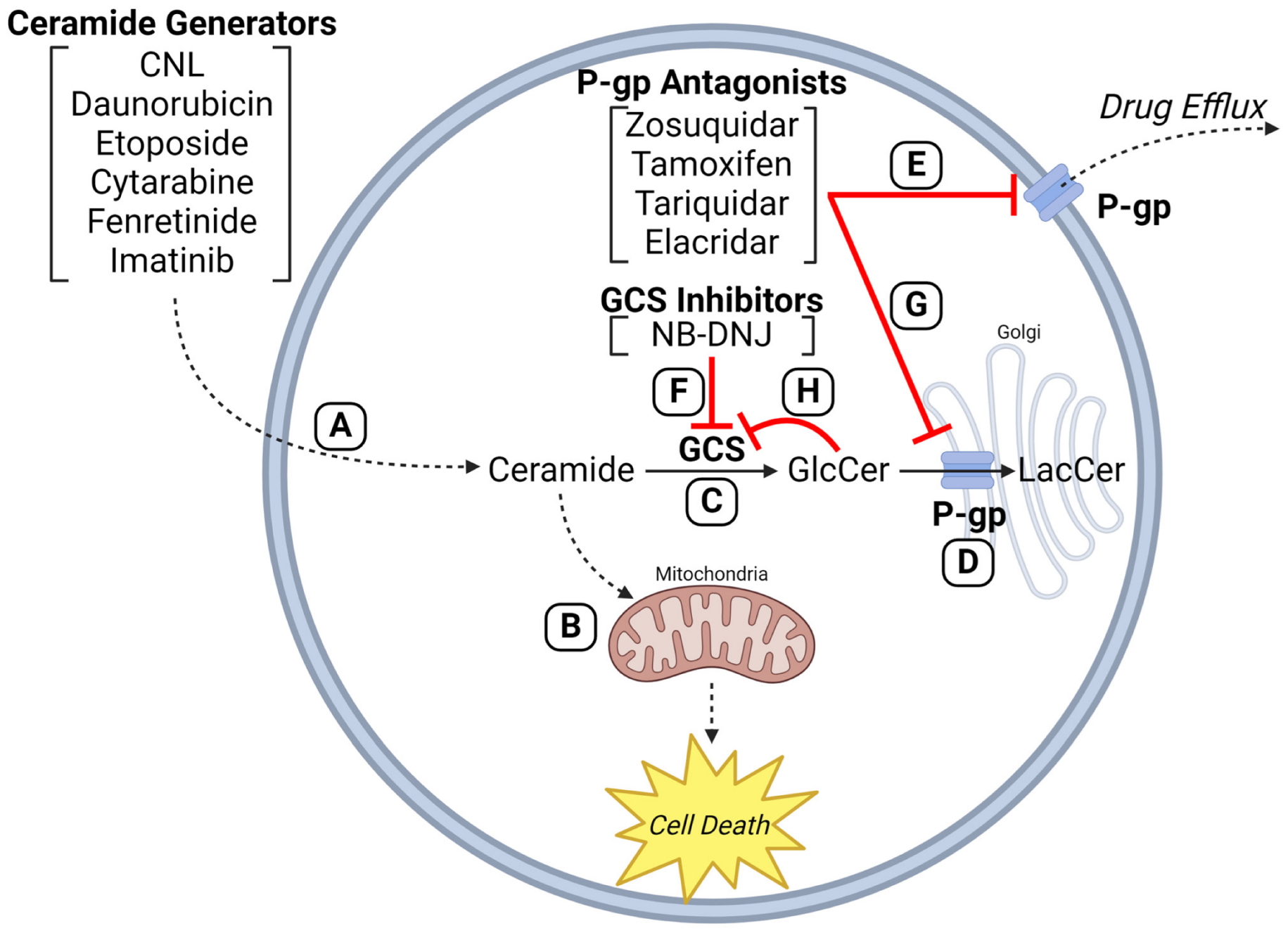
2.5. Interplay between ABC Transporters and Sphingolipid Metabolism—Blunting of the “Ceramide Effect”
3. Chemotherapy Drugs and Sphingolipid Metabolism
3.1. Chemotherapy Selection Pressure Promotes Alterations in Sphingolipid Metabolism
3.2. Ceramide Glycosylation—A Factor in Anthracycline and Vinca Alkaloid Resistance
3.3. Ceramide Glycosylation in Sorafenib, Imatinib, and Platinum Resistance
4. Manipulating GCS Modifies Response to Chemotherapy
5. P-gp, GCS, and Ceramide Glycosylation—Cozy Cooperation
6. P-gp “The Chemotherapy Efflux Pump” versus P-gp “The Ceramide Neutralizer”—Versatility in Drug Resistance
7. P-gp Antagonists and GCS Inhibitors—Duplicitous Roles
8. Do We Target GCS or P-gp to Enhance the Ceramide Effect?
9. Summary and Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Zelnik, I.D.; Rozman, B.; Rosenfeld-Gur, E.; Ben-Dor, S.; Futerman, A.H. A Stroll Down the CerS Lane. Adv. Exp. Med. Biol. 2019, 1159, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Brachtendorf, S.; El-Hindi, K.; Grösch, S. Ceramide synthases in cancer therapy and chemoresistance. Prog. Lipid Res. 2019, 74, 160–185. [Google Scholar] [CrossRef] [PubMed]
- Mullen, T.D.; Hannun, Y.A.; Obeid, L.M. Ceramide synthases at the centre of sphingolipid metabolism and biology. Biochem. J. 2012, 441, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, Y.H.; Jenkins, R.W.; Korman, J.B.; Liu, X.; Obeid, L.M.; Norris, J.S.; Hannun, Y.A. Molecular targeting of acid ceramidase: Implications to cancer therapy. Curr. Drug Targets 2008, 9, 653–661. [Google Scholar] [CrossRef]
- Liu, X.; Elojeimy, S.; Turner, L.S.; Mahdy, A.E.; Zeidan, Y.H.; Bielawska, A.; Bielawski, J.; Dong, J.Y.; El-Zawahry, A.M.; Guo, G.W.; et al. Acid ceramidase inhibition: A novel target for cancer therapy. Front. Biosci. 2008, 13, 2293–2298. [Google Scholar] [CrossRef]
- Dinur, T.; Osiecki, K.M.; Legler, G.; Gatt, S.; Desnick, R.J.; Grabowski, G.A. Human acid beta-glucosidase: Isolation and amino acid sequence of a peptide containing the catalytic site. Proc. Natl. Acad. Sci. USA 1986, 83, 1660–1664. [Google Scholar] [CrossRef]
- Pasmanik-Chor, M.; Laadan, S.; Elroy-Stein, O.; Zimran, A.; Abrahamov, A.; Gatt, S.; Horowitz, M. The glucocerebrosidase D409H mutation in Gaucher disease. Biochem. Mol. Med. 1996, 59, 125–133. [Google Scholar] [CrossRef]
- Bleicher, R.J.; Cabot, M.C. Glucosylceramide synthase and apoptosis. Biochim. Biophys. Acta 2002, 1585, 172–178. [Google Scholar] [CrossRef]
- Lavie, Y.; Cao, H.; Bursten, S.L.; Giuliano, A.E.; Cabot, M.C. Accumulation of glucosylceramides in multidrug-resistant cancer cells. J. Biol. Chem. 1996, 271, 19530–19536. [Google Scholar] [CrossRef]
- Morjani, H.; Aouali, N.; Belhoussine, R.; Veldman, R.J.; Levade, T.; Manfait, M. Elevation of glucosylceramide in multidrug-resistant cancer cells and accumulation in cytoplasmic droplets. Int. J. Cancer 2001, 94, 157–165. [Google Scholar] [CrossRef]
- Gutiérrez-Iglesias, G.; Hurtado, Y.; Palma-Lara, I.; López-Marure, R. Resistance to the antiproliferative effect induced by a short-chain ceramide is associated with an increase of glucosylceramide synthase, P-glycoprotein, and multidrug-resistance gene-1 in cervical cancer cells. Cancer Chemother. Pharmacol. 2014, 74, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Senchenkov, A.; Litvak, D.A.; Cabot, M.C. Targeting ceramide metabolism—A strategy for overcoming drug resistance. J. Natl. Cancer Inst. 2001, 93, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Xie, P.; Shen, Y.F.; Shi, Y.P.; Ge, S.M.; Gu, Z.H.; Wang, J.; Mu, H.J.; Zhang, B.; Qiao, W.Z.; Xie, K.M. Overexpression of glucosylceramide synthase in associated with multidrug resistance of leukemia cells. Leuk. Res. 2008, 32, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Y.; Patwardhan, G.A.; Xie, P.; Gu, X.; Giuliano, A.E.; Cabot, M.C. Glucosylceramide synthase, a factor in modulating drug resistance, is overexpressed in metastatic breast carcinoma. Int. J. Oncol. 2011, 39, 425–431. [Google Scholar] [CrossRef]
- Gouazé-Andersson, V.; Yu, J.Y.; Kreitenberg, A.J.; Bielawska, A.; Giuliano, A.E.; Cabot, M.C. Ceramide and glucosylceramide upregulate expression of the multidrug resistance gene MDR1 in cancer cells. Biochim. Biophys. Acta 2007, 1771, 1407–1417. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Han, T.Y.; Giuliano, A.E.; Cabot, M.C. Ceramide glycosylation potentiates cellular multidrug resistance. FASEB J. 2001, 15, 719–730. [Google Scholar] [CrossRef]
- Baran, Y.; Bielawski, J.; Gunduz, U.; Ogretmen, B. Targeting glucosylceramide synthase sensitizes imatinib-resistant chronic myeloid leukemia cells via endogenous ceramide accumulation. J. Cancer Res. Clin. Oncol. 2011, 137, 1535–1544. [Google Scholar] [CrossRef]
- Camacho, L.; Meca-Cortés, O.; Abad, J.L.; García, S.; Rubio, N.; Díaz, A.; Celià-Terrassa, T.; Cingolani, F.; Bermudo, R.; Fernández, P.L.; et al. Acid ceramidase as a therapeutic target in metastatic prostate cancer. J. Lipid Res. 2013, 54, 1207–1220. [Google Scholar] [CrossRef]
- Liu, X.; Cheng, J.C.; Turner, L.S.; Elojeimy, S.; Beckham, T.H.; Bielawska, A.; Keane, T.E.; Hannun, Y.A.; Norris, J.S. Acid ceramidase upregulation in prostate cancer: Role in tumor development and implications for therapy. Expert Opin. Ther. Targets 2009, 13, 1449–1458. [Google Scholar] [CrossRef]
- Tan, S.F.; Dunton, W.; Liu, X.; Fox, T.E.; Morad, S.A.F.; Desai, D.; Doi, K.; Conaway, M.R.; Amin, S.; Claxton, D.F.; et al. Acid ceramidase promotes drug resistance in acute myeloid leukemia through NF-κB-dependent P-glycoprotein upregulation. J. Lipid Res. 2019, 60, 1078–1086. [Google Scholar] [CrossRef]
- Tan, S.F.; Pearson, J.M.; Feith, D.J.; Loughran, T.P., Jr. The emergence of acid ceramidase as a therapeutic target for acute myeloid leukemia. Expert Opin. Ther. Targets 2017, 21, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Morad, S.A.; Levin, J.C.; Tan, S.F.; Fox, T.E.; Feith, D.J.; Cabot, M.C. Novel off-target effect of tamoxifen—Inhibition of acid ceramidase activity in cancer cells. Biochim. Biophys. Acta 2013, 1831, 1657–1664. [Google Scholar] [CrossRef] [PubMed]
- Orr Gandy, K.A.; Obeid, L.M. Targeting the sphingosine kinase/sphingosine 1-phosphate pathway in disease: Review of sphingosine kinase inhibitors. Biochim. Biophys. Acta 2013, 1831, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Hait, N.C.; Oskeritzian, C.A.; Paugh, S.W.; Milstien, S.; Spiegel, S. Sphingosine kinases, sphingosine 1-phosphate, apoptosis and diseases. Biochim. Biophys. Acta 2006, 1758, 2016–2026. [Google Scholar] [CrossRef]
- Maceyka, M.; Rohrbach, T.; Milstien, S.; Spiegel, S. Role of Sphingosine Kinase 1 and Sphingosine-1-Phosphate Axis in Hepatocellular Carcinoma. Handb. Exp. Pharmacol. 2020, 259, 3–17. [Google Scholar] [CrossRef]
- Lu, H.; Zhou, L.; Zuo, H.; Le, W.; Hu, J.; Zhang, T.; Li, M.; Yuan, Y. Overriding sorafenib resistance via blocking lipid metabolism and Ras by sphingomyelin synthase 1 inhibition in hepatocellular carcinoma. Cancer Chemother. Pharmacol. 2021, 87, 217–228. [Google Scholar] [CrossRef]
- Itoh, M.; Kitano, T.; Watanabe, M.; Kondo, T.; Yabu, T.; Taguchi, Y.; Iwai, K.; Tashima, M.; Uchiyama, T.; Okazaki, T. Possible role of ceramide as an indicator of chemoresistance: Decrease of the ceramide content via activation of glucosylceramide synthase and sphingomyelin synthase in chemoresistant leukemia. Clin. Cancer Res. 2003, 9, 415–423. [Google Scholar]
- Rivera, I.G.; Ordoñez, M.; Presa, N.; Gangoiti, P.; Gomez-Larrauri, A.; Trueba, M.; Fox, T.; Kester, M.; Gomez-Muñoz, A. Ceramide 1-phosphate regulates cell migration and invasion of human pancreatic cancer cells. Biochem. Pharmacol. 2016, 102, 107–119. [Google Scholar] [CrossRef]
- Mitra, P.; Maceyka, M.; Payne, S.G.; Lamour, N.; Milstien, S.; Chalfant, C.E.; Spiegel, S. Ceramide kinase regulates growth and survival of A549 human lung adenocarcinoma cells. FEBS Lett. 2007, 581, 735–740. [Google Scholar] [CrossRef]
- Camacho, L.; Ouro, A.; Gomez-Larrauri, A.; Carracedo, A.; Gomez-Muñoz, A. Implication of Ceramide Kinase/C1P in Cancer Development and Progression. Cancers 2022, 14, 227. [Google Scholar] [CrossRef]
- Vu, N.T.; Kim, M.; Stephenson, D.J.; MacKnight, H.P.; Chalfant, C.E. Ceramide Kinase Inhibition Drives Ferroptosis and Sensitivity to Cisplatin in Mutant KRAS Lung Cancer by Dysregulating VDAC-Mediated Mitochondria Function. Mol. Cancer Res. 2022, 20, 1429–1442. [Google Scholar] [CrossRef] [PubMed]
- Morad, S.A.; Cabot, M.C. Ceramide-orchestrated signalling in cancer cells. Nat. Rev. Cancer 2013, 13, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Ung, J.; Tan, S.-F.; Fox, T.E.; Shaw, J.J.P.; Vass, L.R.; Costa-Pinheiro, P.; Garrett-Bakelman, F.E.; Keng, M.K.; Sharma, A.; Claxton, D.F.; et al. Harnessing the power of sphingolipids: Prospects for acute myeloid leukemia. Blood Rev. 2022, 55, 100950. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef]
- Kao, L.P.; Morad, S.A.F.; Davis, T.S.; MacDougall, M.R.; Kassai, M.; Abdelmageed, N.; Fox, T.E.; Kester, M.; Loughran, T.P., Jr.; Abad, J.L.; et al. Chemotherapy selection pressure alters sphingolipid composition and mitochondrial bioenergetics in resistant HL-60 cells. J. Lipid Res. 2019, 60, 1590–1602. [Google Scholar] [CrossRef]
- Fisher-Wellman, K.H.; Hagen, J.T.; Kassai, M.; Kao, L.P.; Nelson, M.A.M.; McLaughlin, K.L.; Coalson, H.S.; Fox, T.E.; Tan, S.F.; Feith, D.J.; et al. Alterations in sphingolipid composition and mitochondrial bioenergetics represent synergistic therapeutic vulnerabilities linked to multidrug resistance in leukemia. FASEB J. 2022, 36, e22094. [Google Scholar] [CrossRef]
- Loh, K.C.; Baldwin, D.; Saba, J.D. Sphingolipid signaling and hematopoietic malignancies: To the rheostat and beyond. Anticancer. Agents Med. Chem. 2011, 11, 782–793. [Google Scholar] [CrossRef]
- García-González, V.; Díaz-Villanueva, J.F.; Galindo-Hernández, O.; Martínez-Navarro, I.; Hurtado-Ureta, G.; Pérez-Arias, A.A. Ceramide Metabolism Balance, a Multifaceted Factor in Critical Steps of Breast Cancer Development. Int. J. Mol. Sci. 2018, 19, 2527. [Google Scholar] [CrossRef]
- LeBlanc, F.R.; Pearson, J.M.; Tan, S.F.; Cheon, H.; Xing, J.C.; Dunton, W.; Feith, D.J.; Loughran, T.P., Jr. Sphingosine kinase-2 is overexpressed in large granular lymphocyte leukaemia and promotes survival through Mcl-1. Br. J. Haematol. 2020, 190, 405–417. [Google Scholar] [CrossRef]
- Dany, M.; Gencer, S.; Nganga, R.; Thomas, R.J.; Oleinik, N.; Baron, K.D.; Szulc, Z.M.; Ruvolo, P.; Kornblau, S.; Andreeff, M.; et al. Targeting FLT3-ITD signaling mediates ceramide-dependent mitophagy and attenuates drug resistance in AML. Blood 2016, 128, 1944–1958. [Google Scholar] [CrossRef]
- Grösch, S.; Schiffmann, S.; Geisslinger, G. Chain length-specific properties of ceramides. Prog. Lipid Res. 2012, 51, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Helke, K.; Angel, P.; Lu, P.; Garrett-Mayer, E.; Ogretmen, B.; Drake, R.; Voelkel-Johnson, C. Ceramide Synthase 6 Deficiency Enhances Inflammation in the DSS model of Colitis. Sci. Rep. 2018, 8, 1627. [Google Scholar] [CrossRef] [PubMed]
- Koch, A.; Grammatikos, G.; Trautmann, S.; Schreiber, Y.; Thomas, D.; Bruns, F.; Pfeilschifter, J.; Badenhoop, K.; Penna-Martinez, M. Vitamin D Supplementation Enhances C18(dihydro)ceramide Levels in Type 2 Diabetes Patients. Int. J. Mol. Sci. 2017, 18, 1532. [Google Scholar] [CrossRef] [PubMed]
- Saleem, M.; Herrmann, N.; Dinoff, A.; Mielke, M.M.; Oh, P.I.; Shammi, P.; Cao, X.; Venkata, S.L.V.; Haughey, N.J.; Lanctôt, K.L. A Lipidomics Approach to Assess the Association Between Plasma Sphingolipids and Verbal Memory Performance in Coronary Artery Disease Patients Undertaking Cardiac Rehabilitation: A C18:0 Signature for Cognitive Response to Exercise. J. Alzheimer’s Dis. 2017, 60, 829–841. [Google Scholar] [CrossRef]
- Tidhar, R.; Ben-Dor, S.; Wang, E.; Kelly, S.; Merrill, A.H., Jr.; Futerman, A.H. Acyl chain specificity of ceramide synthases is determined within a region of 150 residues in the Tram-Lag-CLN8 (TLC) domain. J. Biol. Chem. 2012, 287, 3197–3206. [Google Scholar] [CrossRef]
- Wang, Z.; Wen, L.; Zhu, F.; Wang, Y.; Xie, Q.; Chen, Z.; Li, Y. Overexpression of ceramide synthase 1 increases C18-ceramide and leads to lethal autophagy in human glioma. Oncotarget 2017, 8, 104022–104036. [Google Scholar] [CrossRef]
- Galea, S. An Unhealthy Mismatch. Milbank Q. 2017, 95, 486–489. [Google Scholar] [CrossRef]
- Tosetti, B.; Brodesser, S.; Brunn, A.; Deckert, M.; Blüher, M.; Doehner, W.; Anker, S.D.; Wenzel, D.; Fleischmann, B.; Pongratz, C.; et al. A tissue-specific screen of ceramide expression in aged mice identifies ceramide synthase-1 and ceramide synthase-5 as potential regulators of fiber size and strength in skeletal muscle. Aging Cell 2020, 19, e13049. [Google Scholar] [CrossRef]
- Siddique, M.M.; Bikman, B.T.; Wang, L.; Ying, L.; Reinhardt, E.; Shui, G.; Wenk, M.R.; Summers, S.A. Ablation of dihydroceramide desaturase confers resistance to etoposide-induced apoptosis in vitro. PLoS ONE 2012, 7, e44042. [Google Scholar] [CrossRef]
- Rudd, A.K.; Devaraj, N.K. Traceless synthesis of ceramides in living cells reveals saturation-dependent apoptotic effects. Proc. Natl. Acad. Sci. USA 2018, 115, 7485–7490. [Google Scholar] [CrossRef]
- Schellenberg, B.; Wang, P.; Keeble, J.A.; Rodriguez-Enriquez, R.; Walker, S.; Owens, T.W.; Foster, F.; Tanianis-Hughes, J.; Brennan, K.; Streuli, C.H.; et al. Bax Exists in a Dynamic Equilibrium between the Cytosol and Mitochondria to Control Apoptotic Priming. Mol. Cell 2013, 49, 959–971. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Li, Z.; Liu, Y.; Ding, X.; Wang, Y.; Fan, S. The ceramide synthase (CERS/LASS) family: Functions involved in cancer progression. Cell Oncol. 2023, 46, 825–845. [Google Scholar] [CrossRef] [PubMed]
- Aldoghachi, A.F.; Baharudin, A.; Ahmad, U.; Chan, S.C.; Ong, T.A.; Yunus, R.; Razack, A.H.; Yusoff, K.; Veerakumarasivam, A. Evaluation of CERS2 Gene as a Potential Biomarker for Bladder Cancer. Dis. Markers 2019, 2019, 3875147. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Sakamoto, W.; Canals, D.; Ishibashi, M.; Matsuda, M.; Nishida, K.; Toyoshima, M.; Shigeta, S.; Taniguchi, M.; Senkal, C.E.; et al. Ceramide synthase 2-C(24:1)-ceramide axis limits the metastatic potential of ovarian cancer cells. FASEB J. 2021, 35, e21287. [Google Scholar] [CrossRef] [PubMed]
- Holliday, M.W., Jr.; Cox, S.B.; Kang, M.H.; Maurer, B.J. C22:0- and C24:0-dihydroceramides confer mixed cytotoxicity in T-cell acute lymphoblastic leukemia cell lines. PLoS ONE 2013, 8, e74768. [Google Scholar] [CrossRef]
- Trayssac, M.; Clarke, C.J.; Stith, J.L.; Snider, J.M.; Newen, N.; Gault, C.R.; Hannun, Y.A.; Obeid, L.M. Targeting sphingosine kinase 1 (SK1) enhances oncogene-induced senescence through ceramide synthase 2 (CerS2)-mediated generation of very-long-chain ceramides. Cell Death Dis. 2021, 12, 27. [Google Scholar] [CrossRef]
- Dugan, V.G.; Blanton, L.; Elal, A.I.A.; Alabi, N.; Barnes, J.; Brammer, L.; Burns, E.; Cummings, C.N.; Davis, T.; Flannery, B.; et al. Update: Influenza Activity—United States, October 1–November 25, 2017. MMWR Morb. Mortal. Wkly. Rep. 2017, 66, 1318–1326. [Google Scholar] [CrossRef]
- Hussain, M.; Galvin, H.D.; Haw, T.Y.; Nutsford, A.N.; Husain, M. Drug resistance in influenza A virus: The epidemiology and management. Infect. Drug Resist. 2017, 10, 121–134. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Lavi, O.; Hall, M.D.; Gillet, J.-P. Toward a Better Understanding of the Complexity of Cancer Drug Resistance. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 85–102. [Google Scholar] [CrossRef]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [PubMed]
- Keating, P.; Cambrosio, A.; Nelson, N.; Mogoutov, A.; Cointet, J.-P. Therapy’s Shadow: A Short History of the Study of Resistance to Cancer Chemotherapy. Front. Pharmacol. 2013, 4, 58. [Google Scholar] [CrossRef] [PubMed]
- Lippert, T.H.; Ruoff, H.J.; Volm, M. Intrinsic and acquired drug resistance in malignant tumors: The main reason for therapeutic failure. Arzneimittelforschung 2008, 58, 261–264. [Google Scholar] [CrossRef] [PubMed]
- Niero, E.L.; Rocha-Sales, B.; Lauand, C.; Cortez, B.A.; de Souza, M.M.; Rezende-Teixeira, P.; Urabayashi, M.S.; Martens, A.A.; Neves, J.H.; Machado-Santelli, G.M. The multiple facets of drug resistance: One history, different approaches. J. Exp. Clin. Cancer Res. 2014, 33, 37. [Google Scholar] [CrossRef]
- Frei, E., III; Karon, M.; Levin, R.H.; Freireich, E.J.; Taylor, R.J.; Hananian, J.; Selawry, O.; HOLLAND, J.F.; HOOGSTRATEN, B.; WOLMAN, I.J.; et al. The Effectiveness of Combinations of Antileukemic Agents in Inducing and Maintaining Remission in Children with Acute Leukemia. Blood 1965, 26, 642–656. [Google Scholar] [CrossRef]
- Kreutzfeldt, J.; Rozeboom, B.; Dey, N.; De, P. The trastuzumab era: Current and upcoming targeted HER2+ breast cancer therapies. Am. J. Cancer Res. 2020, 10, 1045–1067. [Google Scholar]
- Winder, M.; Virós, A. Mechanisms of Drug Resistance in Melanoma. Handb. Exp. Pharmacol. 2018, 249, 91–108. [Google Scholar] [CrossRef]
- Asano, T. Drug Resistance in Cancer Therapy and the Role of Epigenetics. J. Nippon. Med. Sch. 2020, 87, 244–251. [Google Scholar] [CrossRef]
- Abraham, A.; Devasia, A.J.; Varatharajan, S.; Karathedath, S.; Balasubramanian, P.; Mathews, V. Effect of cytosine arabinoside metabolizing enzyme expression on drug toxicity in acute myeloid leukemia. Ann. Hematol. 2015, 94, 883–885. [Google Scholar] [CrossRef]
- Candelaria, M.; Corrales-Alfaro, C.; Gutiérrez-Hernández, O.; Díaz-Chavez, J.; Labardini-Méndez, J.; Vidal-Millán, S.; Herrera, L.A. Expression Levels of Human Equilibrative Nucleoside Transporter 1 and Deoxycytidine Kinase Enzyme as Prognostic Factors in Patients with Acute Myeloid Leukemia Treated with Cytarabine. Chemotherapy 2016, 61, 313–318. [Google Scholar] [CrossRef]
- Galmarini, C.M.; Thomas, X.; Graham, K.; El Jafaari, A.; Cros, E.; Jordheim, L.; Mackey, J.R.; Dumontet, C. Deoxycytidine kinase and cN-II nucleotidase expression in blast cells predict survival in acute myeloid leukaemia patients treated with cytarabine. Br. J. Haematol. 2003, 122, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Meijer, C.; Mulder, N.H.; Timmer-Bosscha, H.; Sluiter, W.J.; Meersma, G.J.; de Vries, E.G.E. Relationship of Cellular Glutathione to the Cytotoxicity and Resistance of Seven Platinum Compounds. Cancer Res. 1992, 52, 6885–6889. [Google Scholar] [PubMed]
- Pathania, S.; Bhatia, R.; Baldi, A.; Singh, R.; Rawal, R.K. Drug metabolizing enzymes and their inhibitors’ role in cancer resistance. Biomed. Pharmacother. 2018, 105, 53–65. [Google Scholar] [CrossRef]
- Woods, D.; Turchi, J.J. Chemotherapy induced DNA damage response: Convergence of drugs and pathways. Cancer Biol. Ther. 2013, 14, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Pilié, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef]
- Bonanno, L.; Favaretto, A.; Rosell, R. Platinum drugs and DNA repair mechanisms in lung cancer. Anticancer. Res. 2014, 34, 493–501. [Google Scholar]
- Damia, G.; Broggini, M. Platinum Resistance in Ovarian Cancer: Role of DNA Repair. Cancers 2019, 11, 119. [Google Scholar] [CrossRef]
- Timucin, A.C.; Basaga, H.; Kutuk, O. Selective targeting of antiapoptotic BCL-2 proteins in cancer. Med. Res. Rev. 2019, 39, 146–175. [Google Scholar] [CrossRef]
- Punnoose, E.A.; Leverson, J.D.; Peale, F.; Boghaert, E.R.; Belmont, L.D.; Tan, N.; Young, A.; Mitten, M.; Ingalla, E.; Darbonne, W.C.; et al. Expression Profile of BCL-2, BCL-XL, and MCL-1 Predicts Pharmacological Response to the BCL-2 Selective Antagonist Venetoclax in Multiple Myeloma Models. Mol. Cancer Ther. 2016, 15, 1132–1144. [Google Scholar] [CrossRef]
- Mohammad, R.M.; Muqbil, I.; Lowe, L.; Yedjou, C.; Hsu, H.Y.; Lin, L.T.; Siegelin, M.D.; Fimognari, C.; Kumar, N.B.; Dou, Q.P.; et al. Broad targeting of resistance to apoptosis in cancer. Semin. Cancer Biol. 2015, 35, S78–S103. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Fairbrother, W.J.; Leverson, J.D.; Souers, A.J. From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors. Nat. Rev. Drug Discov. 2017, 16, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Kartal-Yandim, M.; Adan-Gokbulut, A.; Baran, Y. Molecular mechanisms of drug resistance and its reversal in cancer. Crit. Rev. Biotechnol. 2016, 36, 716–726. [Google Scholar] [CrossRef] [PubMed]
- Zahreddine, H.; Borden, K. Mechanisms and insights into drug resistance in cancer. Front. Pharmacol. 2013, 4, 28. [Google Scholar] [CrossRef] [PubMed]
- Bradley, G.; Ling, V. P-glycoprotein, multidrug resistance and tumor progression. Cancer Metastasis Rev. 1994, 13, 223–233. [Google Scholar] [CrossRef]
- Gottesman, M.M. Mechanisms of cancer drug resistance. Annu. Rev. Med. 2002, 53, 615–627. [Google Scholar] [CrossRef]
- Sun, Y.L.; Patel, A.; Kumar, P.; Chen, Z.S. Role of ABC transporters in cancer chemotherapy. Chin. J. Cancer 2012, 31, 51–57. [Google Scholar] [CrossRef]
- Sandor, V.; Fojo, T.; Bates, S.E. Future perspectives for the development of P-glycoprotein modulators. Drug Resist. Updat. 1998, 1, 190–200. [Google Scholar] [CrossRef]
- Rudas, M.; Filipits, M.; Taucher, S.; Stranzl, T.; Steger, G.G.; Jakesz, R.; Pirker, R.; Pohl, G. Expression of MRP1, LRP and Pgp in breast carcinoma patients treated with preoperative chemotherapy. Breast Cancer Res. Treat. 2003, 81, 149–157. [Google Scholar] [CrossRef]
- Pilotto Heming, C.; Muriithi, W.; Wanjiku Macharia, L.; Niemeyer Filho, P.; Moura-Neto, V.; Aran, V. P-glycoprotein and cancer: What do we currently know? Heliyon 2022, 8, e11171. [Google Scholar] [CrossRef]
- Callaghan, R.; Luk, F.; Bebawy, M. Inhibition of the Multidrug Resistance P-Glycoprotein: Time for a Change of Strategy? Drug Metab. Dispos. 2014, 42, 623–631. [Google Scholar] [CrossRef]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Leopoldo, M.; Nardulli, P.; Contino, M.; Leonetti, F.; Luurtsema, G.; Colabufo, N.A. An updated patent review on P-glycoprotein inhibitors (2011–2018). Expert. Opin. Ther. Pat. 2019, 29, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Amawi, H.; Sim, H.M.; Tiwari, A.K.; Ambudkar, S.V.; Shukla, S. ABC Transporter-Mediated Multidrug-Resistant Cancer. Adv. Exp. Med. Biol. 2019, 1141, 549–580. [Google Scholar] [CrossRef] [PubMed]
- Budani, M.; Auray-Blais, C.; Lingwood, C. ATP-binding cassette transporters mediate differential biosynthesis of glycosphingolipid species. J. Lipid Res. 2021, 62, 100128. [Google Scholar] [CrossRef]
- Kamani, M.; Budani, M.; Binnington, B.; Lingwood, C. ABC Transporters Can Regulate Glycosphingolipid Biosynthesis within the Golgi. Ann. Pharma Res. 2022, 10, 762–777. [Google Scholar]
- Lai, J.-I.; Tseng, Y.-J.; Chen, M.-H.; Huang, C.-Y.F.; Chang, P.M.-H. Clinical Perspective of FDA Approved Drugs With P-Glycoprotein Inhibition Activities for Potential Cancer Therapeutics. Front. Oncol. 2020, 10, 561936. [Google Scholar] [CrossRef]
- Companioni, O.; Mir, C.; Garcia-Mayea, Y.; Lleonart, M.E. Targeting Sphingolipids for Cancer Therapy. Front. Oncol. 2021, 11, 745092. [Google Scholar] [CrossRef]
- Bose, R.; Verheij, M.; Haimovitz-Friedman, A.; Scotto, K.; Fuks, Z.; Kolesnick, R. Ceramide synthase mediates daunorubicin-induced apoptosis: An alternative mechanism for generating death signals. Cell 1995, 82, 405–414. [Google Scholar] [CrossRef]
- Jaffrézou, J.P.; Levade, T.; Bettaïeb, A.; Andrieu, N.; Bezombes, C.; Maestre, N.; Vermeersch, S.; Rousse, A.; Laurent, G. Daunorubicin-induced apoptosis: Triggering of ceramide generation through sphingomyelin hydrolysis. EMBO J. 1996, 15, 2417–2424. [Google Scholar] [CrossRef]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Tumor suppressive functions of ceramide: Evidence and mechanisms. Apoptosis 2015, 20, 689–711. [Google Scholar] [CrossRef]
- Gouazé, V.; Yu, J.Y.; Bleicher, R.J.; Han, T.Y.; Liu, Y.Y.; Wang, H.; Gottesman, M.M.; Bitterman, A.; Giuliano, A.E.; Cabot, M.C. Overexpression of glucosylceramide synthase and P-glycoprotein in cancer cells selected for resistance to natural product chemotherapy. Mol. Cancer Ther. 2004, 3, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Hajj, C.; Becker-Flegler, K.A.; Haimovitz-Friedman, A. Novel mechanisms of action of classical chemotherapeutic agents on sphingolipid pathways. Biol. Chem. 2015, 396, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Y.; Han, T.Y.; Giuliano, A.E.; Cabot, M.C. Expression of glucosylceramide synthase, converting ceramide to glucosylceramide, confers adriamycin resistance in human breast cancer cells. J. Biol. Chem. 1999, 274, 1140–1146. [Google Scholar] [CrossRef] [PubMed]
- Lavie, Y.; Cao, H.; Volner, A.; Lucci, A.; Han, T.Y.; Geffen, V.; Giuliano, A.E.; Cabot, M.C. Agents that reverse multidrug resistance, tamoxifen, verapamil, and cyclosporin A, block glycosphingolipid metabolism by inhibiting ceramide glycosylation in human cancer cells. J. Biol. Chem. 1997, 272, 1682–1687. [Google Scholar] [CrossRef]
- Aoyama, Y.; Sobue, S.; Mizutani, N.; Inoue, C.; Kawamoto, Y.; Nishizawa, Y.; Ichihara, M.; Kyogashima, M.; Suzuki, M.; Nozawa, Y.; et al. Modulation of the sphingolipid rheostat is involved in paclitaxel resistance of the human prostate cancer cell line PC3-PR. Biochem. Biophys. Res. Commun. 2017, 486, 551–557. [Google Scholar] [CrossRef]
- Sietsma, H.; Veldman, R.J.; Kok, J.W. The involvement of sphingolipids in multidrug resistance. J. Membr. Biol. 2001, 181, 153–162. [Google Scholar] [CrossRef]
- Kester, M.; Bassler, J.; Fox, T.E.; Carter, C.J.; Davidson, J.A.; Parette, M.R. Preclinical development of a C6-ceramide NanoLiposome, a novel sphingolipid therapeutic. Biol. Chem. 2015, 396, 737–747. [Google Scholar] [CrossRef]
- Galmarini, C.M.; Thomas, X.; Calvo, F.; Rousselot, P.; Rabilloud, M.; El Jaffari, A.; Cros, E.; Dumontet, C. In vivo mechanisms of resistance to cytarabine in acute myeloid leukaemia. Br. J. Haematol. 2002, 117, 860–868. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Han, T.Y.; Giuliano, A.E.; Hansen, N.; Cabot, M.C. Uncoupling ceramide glycosylation by transfection of glucosylceramide synthase antisense reverses adriamycin resistance. J. Biol. Chem. 2000, 275, 7138–7143. [Google Scholar] [CrossRef]
- Patwardhan, G.A.; Zhang, Q.J.; Yin, D.; Gupta, V.; Bao, J.; Senkal, C.E.; Ogretmen, B.; Cabot, M.C.; Shah, G.V.; Sylvester, P.W.; et al. A new mixed-backbone oligonucleotide against glucosylceramide synthase sensitizes multidrug-resistant tumors to apoptosis. PLoS ONE 2009, 4, e6938. [Google Scholar] [CrossRef]
- Stefanovic, M.; Tutusaus, A.; Martinez-Nieto, G.A.; Bárcena, C.; de Gregorio, E.; Moutinho, C.; Barbero-Camps, E.; Villanueva, A.; Colell, A.; Marí, M.; et al. Targeting glucosylceramide synthase upregulation reverts sorafenib resistance in experimental hepatocellular carcinoma. Oncotarget 2016, 7, 8253–8267. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.F.; Liu, X.; Fox, T.E.; Barth, B.M.; Sharma, A.; Turner, S.D.; Awwad, A.; Dewey, A.; Doi, K.; Spitzer, B.; et al. Acid ceramidase is upregulated in AML and represents a novel therapeutic target. Oncotarget 2016, 7, 83208–83222. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, Y.; Lankadasari, M.B.; Harikumar, K.B. Acid Ceramidase: A Novel Therapeutic Target in Cancer. Curr. Top. Med. Chem. 2019, 19, 1512–1520. [Google Scholar] [CrossRef] [PubMed]
- Geffken, K.; Spiegel, S. Sphingosine kinase 1 in breast cancer. Adv. Biol. Regul. 2018, 67, 59–65. [Google Scholar] [CrossRef]
- Pyne, S.; Bittman, R.; Pyne, N.J. Sphingosine kinase inhibitors and cancer: Seeking the golden sword of Hercules. Cancer Res. 2011, 71, 6576–6582. [Google Scholar] [CrossRef]
- Pyne, N.J.; El Buri, A.; Adams, D.R.; Pyne, S. Sphingosine 1-phosphate and cancer. Adv. Biol. Regul. 2018, 68, 97–106. [Google Scholar] [CrossRef]
- Kok, J.W.; Sietsma, H. Sphingolipid metabolism enzymes as targets for anticancer therapy. Curr. Drug Targets 2004, 5, 375–382. [Google Scholar] [CrossRef]
- Ung, J.; Tan, S.-F.; Fox, T.E.; Shaw, J.J.P.; Taori, M.; Horton, B.J.; Golla, U.; Sharma, A.; Szulc, Z.M.; Wang, H.-G.; et al. Acid Ceramidase Inhibitor LCL-805 Antagonizes Akt Signaling and Promotes Iron-Dependent Cell Death in Acute Myeloid Leukemia. Cancers 2023, 15, 5866. [Google Scholar] [CrossRef]
- Ryland, L.K.; Fox, T.E.; Liu, X.; Loughran, T.P.; Kester, M. Dysregulation of sphingolipid metabolism in cancer. Cancer Biol. Ther. 2011, 11, 138–149. [Google Scholar] [CrossRef]
- Zheng, W.; Kollmeyer, J.; Symolon, H.; Momin, A.; Munter, E.; Wang, E.; Kelly, S.; Allegood, J.C.; Liu, Y.; Peng, Q.; et al. Ceramides and other bioactive sphingolipid backbones in health and disease: Lipidomic analysis, metabolism and roles in membrane structure, dynamics, signaling and autophagy. Biochim. Biophys. Acta 2006, 1758, 1864–1884. [Google Scholar] [CrossRef]
- Ichikawa, S.; Hirabayashi, Y. Glucosylceramide synthase and glycosphingolipid synthesis. Trends Cell Biol. 1998, 8, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Messner, M.C.; Cabot, M.C. Glucosylceramide in humans. Adv. Exp. Med. Biol. 2010, 688, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Lucci, A.; Cho, W.I.; Han, T.Y.; Giuliano, A.E.; Morton, D.L.; Cabot, M.C. Glucosylceramide: A marker for multiple-drug resistant cancers. Anticancer. Res. 1998, 18, 475–480. [Google Scholar]
- Veldman, R.J.; Klappe, K.; Hinrichs, J.; Hummel, I.; van der Schaaf, G.; Sietsma, H.; Kok, J.W. Altered sphingolipid metabolism in multidrug-resistant ovarian cancer cells is due to uncoupling of glycolipid biosynthesis in the Golgi apparatus. FASEB J. 2002, 16, 1111–1113. [Google Scholar] [CrossRef] [PubMed]
- Che, J.; Huang, Y.; Xu, C.; Zhang, P. Increased ceramide production sensitizes breast cancer cell response to chemotherapy. Cancer Chemother. Pharmacol. 2017, 79, 933–941. [Google Scholar] [CrossRef]
- Fisher-Wellman, K.; Hagen, J.T.; Neufer, P.D.; Kassai, M.; Cabot, M.C. On the nature of ceramide-mitochondria interactions—Dissection using comprehensive mitochondrial phenotyping. Cell. Signal. 2021, 78, 109838. [Google Scholar] [CrossRef]
- Roh, J.L.; Kim, E.H.; Park, J.Y.; Kim, J.W. Inhibition of Glucosylceramide Synthase Sensitizes Head and Neck Cancer to Cisplatin. Mol. Cancer Ther. 2015, 14, 1907–1915. [Google Scholar] [CrossRef]
- Dupre, T.V.; Doll, M.A.; Shah, P.P.; Sharp, C.N.; Siow, D.; Megyesi, J.; Shayman, J.; Bielawska, A.; Bielawski, J.; Beverly, L.J.; et al. Inhibiting glucosylceramide synthase exacerbates cisplatin-induced acute kidney injury. J. Lipid Res. 2017, 58, 1439–1452. [Google Scholar] [CrossRef]
- Rebillard, A.; Tekpli, X.; Meurette, O.; Sergent, O.; LeMoigne-Muller, G.; Vernhet, L.; Gorria, M.; Chevanne, M.; Christmann, M.; Kaina, B.; et al. Cisplatin-induced apoptosis involves membrane fluidification via inhibition of NHE1 in human colon cancer cells. Cancer Res. 2007, 67, 7865–7874. [Google Scholar] [CrossRef]
- Madigan, J.P.; Robey, R.W.; Poprawski, J.E.; Huang, H.; Clarke, C.J.; Gottesman, M.M.; Cabot, M.C.; Rosenberg, D.W. A role for ceramide glycosylation in resistance to oxaliplatin in colorectal cancer. Exp. Cell Res. 2020, 388, 111860. [Google Scholar] [CrossRef]
- Salustiano, E.J.; da Costa, K.M.; Freire-de-Lima, L.; Mendonça-Previato, L.; Previato, J.O. Inhibition of glycosphingolipid biosynthesis reverts multidrug resistance by differentially modulating ABC transporters in chronic myeloid leukemias. J. Biol. Chem. 2020, 295, 6457–6471. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Y.; Cabot, M.C. Development of a mammalian Tet-on expression cell line: Glucosylceramide synthase regulates TNF-alpha-induced apoptosis. Methods Mol. Biol. 2004, 249, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Y.; Han, T.Y.; Giuliano, A.E.; Ichikawa, S.; Hirabayashi, Y.; Cabot, M.C. Glycosylation of ceramide potentiates cellular resistance to tumor necrosis factor-α-induced apoptosis. Exp. Cell Res. 1999, 252, 464–470. [Google Scholar] [CrossRef]
- Peña, L.A.; Fuks, Z.; Koksnick, R. Stress-induced apoptosis and the sphingomyelin pathway. Biochem. Pharmacol. 1997, 53, 615–621. [Google Scholar] [CrossRef]
- Deng, W.; Li, R.; Guerrera, M.; Liu, Y.; Ladisch, S. Transfection of glucosylceramide synthase antisense inhibits mouse melanoma formation. Glycobiology 2002, 12, 145–152. [Google Scholar] [CrossRef]
- Di Sano, F.; Di Bartolomeo, S.; Fazi, B.; Fiorentini, C.; Matarrese, P.; Spinedi, A.; Piacentini, M. Antisense to glucosylceramide synthase in human neuroepithelioma affects cell growth but not apoptosis. Cell Death Differ. 2002, 9, 693–695. [Google Scholar] [CrossRef]
- Hummel, I.; Klappe, K.; Kok, J.W. Up-regulation of lactosylceramide synthase in MDR1 overexpressing human liver tumour cells. FEBS Lett. 2005, 579, 3381–3384. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, X.; Li, J.; Sun, Y.; Gao, P.; Zhang, C.; Zhang, H.; Zhou, G. MDR1 (multidrug resistence 1) can regulate GCS (glucosylceramide synthase) in breast cancer cells. J. Surg. Oncol. 2011, 104, 466–471. [Google Scholar] [CrossRef]
- Paul, P.; Kamisaka, Y.; Marks, D.L.; Pagano, R.E. Purification and characterization of UDP-glucose:ceramide glucosyltransferase from rat liver Golgi membranes. J. Biol. Chem. 1996, 271, 2287–2293. [Google Scholar] [CrossRef]
- Marks, D.L.; Wu, K.; Paul, P.; Kamisaka, Y.; Watanabe, R.; Pagano, R.E. Oligomerization and topology of the Golgi membrane protein glucosylceramide synthase. J. Biol. Chem. 1999, 274, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Jeckel, D.; Karrenbauer, A.; Burger, K.N.; van Meer, G.; Wieland, F. Glucosylceramide is synthesized at the cytosolic surface of various Golgi subfractions. J. Cell Biol. 1992, 117, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Nemoto-Sasaki, Y.; Matsumoto, N.; Hama, K.; Tanikawa, T.; Oka, S.; Saeki, T.; Kumasaka, T.; Koizumi, T.; Arai, S.; et al. Complex formation of sphingomyelin synthase 1 with glucosylceramide synthase increases sphingomyelin and decreases glucosylceramide levels. J. Biol. Chem. 2018, 293, 17505–17522. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, P.M.; Roninson, I.B. Induction of multidrug resistance in human cells by transient exposure to different chemotherapeutic drugs. J. Natl. Cancer Inst. 1993, 85, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Kohno, K.; Sato, S.; Takano, H.; Matsuo, K.; Kuwano, M. The direct activation of human multidrug resistance gene (MDR1) by anticancer agents. Biochem. Biophys. Res. Commun. 1989, 165, 1415–1421. [Google Scholar] [CrossRef]
- Klappe, K.; Hinrichs, J.W.; Kroesen, B.J.; Sietsma, H.; Kok, J.W. MRP1 and glucosylceramide are coordinately over expressed and enriched in rafts during multidrug resistance acquisition in colon cancer cells. Int. J. Cancer 2004, 110, 511–522. [Google Scholar] [CrossRef]
- Kok, J.W.; Veldman, R.J.; Klappe, K.; Koning, H.; Filipeanu, C.M.; Müller, M. Differential expression of sphingolipids in MRP1 overexpressing HT29 cells. Int. J. Cancer 2000, 87, 172–178. [Google Scholar] [CrossRef]
- Lançon, A.; Hanet, N.; Jannin, B.; Delmas, D.; Heydel, J.M.; Lizard, G.; Chagnon, M.C.; Artur, Y.; Latruffe, N. Resveratrol in human hepatoma HepG2 cells: Metabolism and inducibility of detoxifying enzymes. Drug Metab. Dispos. 2007, 35, 699–703. [Google Scholar] [CrossRef]
- van Helvoort, A.; Smith, A.J.; Sprong, H.; Fritzsche, I.; Schinkel, A.H.; Borst, P.; van Meer, G. MDR1 P-glycoprotein is a lipid translocase of broad specificity, while MDR3 P-glycoprotein specifically translocates phosphatidylcholine. Cell 1996, 87, 507–517. [Google Scholar] [CrossRef]
- Lee, W.K.; Kolesnick, R.N. Sphingolipid abnormalities in cancer multidrug resistance: Chicken or egg? Cell. Signal. 2017, 38, 134–145. [Google Scholar] [CrossRef]
- Song, M.; Zang, W.; Zhang, B.; Cao, J.; Yang, G. GCS overexpression is associated with multidrug resistance of human HCT-8 colon cancer cells. J. Exp. Clin. Cancer Res. 2012, 31, 23. [Google Scholar] [CrossRef] [PubMed]
- Wegner, M.S.; Gruber, L.; Mattjus, P.; Geisslinger, G.; Grösch, S. The UDP-glucose ceramide glycosyltransferase (UGCG) and the link to multidrug resistance protein 1 (MDR1). BMC Cancer 2018, 18, 153. [Google Scholar] [CrossRef] [PubMed]
- Gouazé, V.; Liu, Y.Y.; Prickett, C.S.; Yu, J.Y.; Giuliano, A.E.; Cabot, M.C. Glucosylceramide synthase blockade down-regulates P-glycoprotein and resensitizes multidrug-resistant breast cancer cells to anticancer drugs. Cancer Res. 2005, 65, 3861–3867. [Google Scholar] [CrossRef] [PubMed]
- van Helvoort, A.; Giudici, M.L.; Thielemans, M.; van Meer, G. Transport of sphingomyelin to the cell surface is inhibited by brefeldin A and in mitosis, where C6-NBD-sphingomyelin is translocated across the plasma membrane by a multidrug transporter activity. J. Cell Sci. 1997, 110 Pt 1, 75–83. [Google Scholar] [CrossRef]
- Smyth, M.J.; Krasovskis, E.; Sutton, V.R.; Johnstone, R.W. The drug efflux protein, P-glycoprotein, additionally protects drug-resistant tumor cells from multiple forms of caspase-dependent apoptosis. Proc. Natl. Acad. Sci. USA 1998, 95, 7024–7029. [Google Scholar] [CrossRef]
- Shabbits, J.A.; Mayer, L.D. P-glycoprotein modulates ceramide-mediated sensitivity of human breast cancer cells to tubulin-binding anticancer drugs. Mol. Cancer Ther. 2002, 1, 205–213. [Google Scholar]
- Charles, A.G.; Han, T.Y.; Liu, Y.Y.; Hansen, N.; Giuliano, A.E.; Cabot, M.C. Taxol-induced ceramide generation and apoptosis in human breast cancer cells. Cancer Chemother. Pharmacol. 2001, 47, 444–450. [Google Scholar] [CrossRef]
- Morad, S.A.F.; Davis, T.S.; MacDougall, M.R.; Tan, S.F.; Feith, D.J.; Desai, D.H.; Amin, S.G.; Kester, M.; Loughran, T.P., Jr.; Cabot, M.C. Role of P-glycoprotein inhibitors in ceramide-based therapeutics for treatment of cancer. Biochem. Pharmacol. 2017, 130, 21–33. [Google Scholar] [CrossRef]
- Sietsma, H.; Dijkhuis, A.J.; Kamps, W.; Kok, J.W. Sphingolipids in neuroblastoma: Their role in drug resistance mechanisms. Neurochem. Res. 2002, 27, 665–674. [Google Scholar] [CrossRef]
- D’Angelo, G.; Polishchuk, E.; Di Tullio, G.; Santoro, M.; Di Campli, A.; Godi, A.; West, G.; Bielawski, J.; Chuang, C.C.; van der Spoel, A.C.; et al. Glycosphingolipid synthesis requires FAPP2 transfer of glucosylceramide. Nature 2007, 449, 62–67. [Google Scholar] [CrossRef]
- D’Angelo, G.; Uemura, T.; Chuang, C.C.; Polishchuk, E.; Santoro, M.; Ohvo-Rekilä, H.; Sato, T.; Di Tullio, G.; Varriale, A.; D’Auria, S.; et al. Vesicular and non-vesicular transport feed distinct glycosylation pathways in the Golgi. Nature 2013, 501, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Turzanski, J.; Grundy, M.; Shang, S.; Russell, N.; Pallis, M. P-glycoprotein is implicated in the inhibition of ceramide-induced apoptosis in TF-1 acute myeloid leukemia cells by modulation of the glucosylceramide synthase pathway. Exp. Hematol. 2005, 33, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.V.; Gouazé-Andersson, V.; Cabot, M.C. Expression of P-glycoprotein in HeLa cells confers resistance to ceramide cytotoxicity. Int. J. Oncol. 2010, 37, 1591–1597. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.V.; Gouazé-Andersson, V.; Karimi, R.; Messner, M.C.; Cabot, M.C. P-glycoprotein antagonists confer synergistic sensitivity to short-chain ceramide in human multidrug-resistant cancer cells. Exp. Cell Res. 2011, 317, 1736–1745. [Google Scholar] [CrossRef]
- Fu, D.; Arias, I.M. Intracellular trafficking of P-glycoprotein. Int. J. Biochem. Cell Biol. 2012, 44, 461–464. [Google Scholar] [CrossRef]
- Fisher-Wellman, K.H.; Kassai, M.; Hagen, J.T.; Neufer, P.D.; Kester, M.; Loughran, T.P.; Chalfant, C.E.; Feith, D.J.; Tan, S.-F.; Fox, T.E.; et al. Simultaneous Inhibition of Ceramide Hydrolysis and Glycosylation Synergizes to Corrupt Mitochondrial Respiration and Signal Caspase Driven Cell Death in Drug-Resistant Acute Myeloid Leukemia. Cancers 2023, 15, 1883. [Google Scholar] [CrossRef]
- Plo, I.; Lehne, G.; Beckstrøm, K.J.; Maestre, N.; Bettaïeb, A.; Laurent, G.; Lautier, D. Influence of ceramide metabolism on P-glycoprotein function in immature acute myeloid leukemia KG1a cells. Mol. Pharmacol. 2002, 62, 304–312. [Google Scholar] [CrossRef]
- Chen, B.; Yin, L.; Cheng, J.; Ding, J.; Gao, C.; Sun, Y.; Zhao, G.; Wang, J.; Bao, W.; Xia, G.; et al. Effect of D, L-threo-1-phenyl-2-decanoylamino-3-morpholino-1-propanol and tetrandrine on the reversion of multidrug resistance in K562/A02 cells. Hematology 2011, 16, 24–30. [Google Scholar] [CrossRef]
- Lee, L.; Abe, A.; Shayman, J.A. Improved inhibitors of glucosylceramide synthase. J. Biol. Chem. 1999, 274, 14662–14669. [Google Scholar] [CrossRef]
- Sietsma, H.; Veldman, R.J.; Kolk, D.; Ausema, B.; Nijhof, W.; Kamps, W.; Vellenga, E.; Kok, J.W. 1-phenyl-2-decanoylamino-3-morpholino-1-propanol chemosensitizes neuroblastoma cells for taxol and vincristine. Clin. Cancer Res. 2000, 6, 942–948. [Google Scholar]
- Chai, L.; McLaren, R.P.; Byrne, A.; Chuang, W.L.; Huang, Y.; Dufault, M.R.; Pacheco, J.; Madhiwalla, S.; Zhang, X.; Zhang, M.; et al. The chemosensitizing activity of inhibitors of glucosylceramide synthase is mediated primarily through modulation of P-gp function. Int. J. Oncol. 2011, 38, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Norris-Cervetto, E.; Callaghan, R.; Platt, F.M.; Dwek, R.A.; Butters, T.D. Inhibition of glucosylceramide synthase does not reverse drug resistance in cancer cells. J. Biol. Chem. 2004, 279, 40412–40418. [Google Scholar] [CrossRef] [PubMed]
- Bieberich, E.; Freischütz, B.; Suzuki, M.; Yu, R.K. Differential Effects of Glycolipid Biosynthesis Inhibitors on Ceramide-Induced Cell Death in Neuroblastoma Cells. J. Neurochem. 1999, 72, 1040–1049. [Google Scholar] [CrossRef] [PubMed]
- Piulats, J.M.; Vidal, A.; García-Rodríguez, F.J.; Muñoz, C.; Nadal, M.; Moutinho, C.; Martínez-Iniesta, M.; Mora, J.; Figueras, A.; Guinó, E.; et al. Orthoxenografts of Testicular Germ Cell Tumors Demonstrate Genomic Changes Associated with Cisplatin Resistance and Identify PDMP as a Resensitizing Agent. Clin. Cancer Res. 2018, 24, 3755–3766. [Google Scholar] [CrossRef]
- Huang, C.; Tu, Y.; Freter, C.E. Fludarabine-resistance associates with ceramide metabolism and leukemia stem cell development in chronic lymphocytic leukemia. Oncotarget 2018, 9, 33124–33137. [Google Scholar] [CrossRef]
- Tanaka, K.; Takada, H.; Isonishi, S.; Aoki, D.; Mikami, M.; Kiguchi, K.; Iwamori, M. Possible involvement of glycolipids in anticancer drug resistance of human ovarian serous carcinoma-derived cells. J. Biochem. 2012, 152, 587–594. [Google Scholar] [CrossRef]
- Wang, T.; Wei, J.; Wang, N.; Ma, J.L.; Hui, P.P. The glucosylceramide synthase inhibitor PDMP sensitizes pancreatic cancer cells to MEK/ERK inhibitor AZD-6244. Biochem. Biophys. Res. Commun. 2015, 456, 821–826. [Google Scholar] [CrossRef]
- Jiang, Y.; DiVittore, N.A.; Kaiser, J.M.; Shanmugavelandy, S.S.; Fritz, J.L.; Heakal, Y.; Tagaram, H.R.; Cheng, H.; Cabot, M.C.; Staveley-O’Carroll, K.F.; et al. Combinatorial therapies improve the therapeutic efficacy of nanoliposomal ceramide for pancreatic cancer. Cancer Biol. Ther. 2011, 12, 574–585. [Google Scholar] [CrossRef]
- Chiu, W.H.; Su, W.C.; Li, C.L.; Chen, C.L.; Lin, C.F. An increase in glucosylceramide synthase induces Bcl-xL-mediated cell survival in vinorelbine-resistant lung adenocarcinoma cells. Oncotarget 2015, 6, 20513–20524. [Google Scholar] [CrossRef]
- Casson, L.; Howell, L.; Mathews, L.A.; Ferrer, M.; Southall, N.; Guha, R.; Keller, J.M.; Thomas, C.; Siskind, L.J.; Beverly, L.J. Inhibition of ceramide metabolism sensitizes human leukemia cells to inhibition of BCL2-like proteins. PLoS ONE 2013, 8, e54525. [Google Scholar] [CrossRef]
- Xu, W.; Xu, B.; Yao, Y.; Yu, X.; Shen, J. The novel HDAC inhibitor AR-42-induced anti-colon cancer cell activity is associated with ceramide production. Biochem. Biophys. Res. Commun. 2015, 463, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Fan, G.; Wang, X. Pre-clinical assessment of A-674563 as an anti-melanoma agent. Biochem. Biophys. Res. Commun. 2016, 477, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.C.; Tsai, C.C.; Chen, C.L.; Chen, T.Y.; Chen, Y.P.; Lin, Y.S.; Lu, P.J.; Lin, C.M.; Wang, S.H.; Tsao, C.W.; et al. Glucosylceramide synthase inhibitor PDMP sensitizes chronic myeloid leukemia T315I mutant to Bcr-Abl inhibitor and cooperatively induces glycogen synthase kinase-3-regulated apoptosis. FASEB J. 2011, 25, 3661–3673. [Google Scholar] [CrossRef] [PubMed]
- Li, D.Q.; Wang, Z.B.; Bai, J.; Zhao, J.; Wang, Y.; Hu, K.; Du, Y.H. Reversal of multidrug resistance in drug-resistant human gastric cancer cell line SGC7901/VCR by antiprogestin drug mifepristone. World J. Gastroenterol. 2004, 10, 1722–1725. [Google Scholar] [CrossRef]
- Lecureur, V.; Fardel, O.; Guillouzo, A. The antiprogestatin drug RU 486 potentiates doxorubicin cytotoxicity in multidrug resistant cells through inhibition of P-glycoprotein function. FEBS Lett. 1994, 355, 187–191. [Google Scholar] [CrossRef]
- Al-Abd, A.M.; Mahmoud, A.M.; El-Sherbiny, G.A.; El-Moselhy, M.A.; Nofal, S.M.; El-Latif, H.A.; El-Eraky, W.I.; El-Shemy, H.A. Resveratrol enhances the cytotoxic profile of docetaxel and doxorubicin in solid tumour cell lines in vitro. Cell Prolif. 2011, 44, 591–601. [Google Scholar] [CrossRef]
- Jain, V.; Harper, S.L.; Versace, A.M.; Fingerman, D.; Brown, G.S.; Bhardwaj, M.; Crissey, M.A.S.; Goldman, A.R.; Ruthel, G.; Liu, Q.; et al. Targeting UGCG Overcomes Resistance to Lysosomal Autophagy Inhibition. Cancer Discov. 2023, 13, 454–473. [Google Scholar] [CrossRef]
- Vogler, M.; Dickens, D.; Dyer, M.J.; Owen, A.; Pirmohamed, M.; Cohen, G.M. The B-cell lymphoma 2 (BCL2)-inhibitors, ABT-737 and ABT-263, are substrates for P-glycoprotein. Biochem. Biophys. Res. Commun. 2011, 408, 344–349. [Google Scholar] [CrossRef]
- Ammar, M.; Louati, N.; Frikha, I.; Medhaffar, M.; Ghozzi, H.; Elloumi, M.; Menif, H.; Zeghal, K.; Ben Mahmoud, L. Overexpression of P-glycoprotein and resistance to Imatinib in chronic myeloid leukemia patients. J. Clin. Lab. Anal. 2020, 34, e23374. [Google Scholar] [CrossRef]
- Yamakawa, Y.; Hamada, A.; Uchida, T.; Sato, D.; Yuki, M.; Hayashi, M.; Kawaguchi, T.; Saito, H. Distinct interaction of nilotinib and imatinib with P-Glycoprotein in intracellular accumulation and cytotoxicity in CML Cell Line K562 cells. Biol. Pharm. Bull. 2014, 37, 1330–1335. [Google Scholar] [CrossRef]
- Baran, Y.; Salas, A.; Senkal, C.E.; Gunduz, U.; Bielawski, J.; Obeid, L.M.; Ogretmen, B. Alterations of ceramide/sphingosine 1-phosphate rheostat involved in the regulation of resistance to imatinib-induced apoptosis in K562 human chronic myeloid leukemia cells. J. Biol. Chem. 2007, 282, 10922–10934. [Google Scholar] [CrossRef] [PubMed]
- Lucci, A.; Han, T.Y.; Liu, Y.Y.; Giuliano, A.E.; Cabot, M.C. Modification of ceramide metabolism increases cancer cell sensitivity to cytotoxics. Int. J. Oncol. 1999, 15, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Kartal, M.; Saydam, G.; Sahin, F.; Baran, Y. Resveratrol triggers apoptosis through regulating ceramide metabolizing genes in human K562 chronic myeloid leukemia cells. Nutr. Cancer 2011, 63, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Scarlatti, F.; Sala, G.; Somenzi, G.; Signorelli, P.; Sacchi, N.; Ghidoni, R. Resveratrol induces growth inhibition and apoptosis in metastatic breast cancer cells via de novo ceramide signaling. FASEB J. 2003, 17, 2339–2341. [Google Scholar] [CrossRef]
- Li, G.; Liu, D.; Kimchi, E.T.; Kaifi, J.T.; Qi, X.; Manjunath, Y.; Liu, X.; Deering, T.; Avella, D.M.; Fox, T.; et al. Nanoliposome C6-Ceramide Increases the Anti-tumor Immune Response and Slows Growth of Liver Tumors in Mice. Gastroenterology 2018, 154, 1024–1036.e1029. [Google Scholar] [CrossRef]
- Morad, S.A.; Madigan, J.P.; Levin, J.C.; Abdelmageed, N.; Karimi, R.; Rosenberg, D.W.; Kester, M.; Shanmugavelandy, S.S.; Cabot, M.C. Tamoxifen magnifies therapeutic impact of ceramide in human colorectal cancer cells independent of p53. Biochem. Pharmacol. 2013, 85, 1057–1065. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Action | Agents | Model | Reference |
|---|---|---|---|
| A. Increase Adriamycin sensitivity | Tamoxifen; PPMP | MCF-7/AdrR cells (Adriamycin-resistant, human ovarian cancer cell line) | [104] |
| B. Increase Taxol, vincristine sensitivity | PDMP | MCF-7/AdrR | [156] |
| C. Increase C6-ceramide sensitivity | Tamoxifen; tariquidar; zosuquidar; cyclosporin A | HL-60/VCR (vincristine-resistant human acute myeloid leukemia cell line); KG-1a (human acute myeloid cell line); LoVo (human colon cancer cell line) | [158] |
| D. Enhance C8-ceramide sensitivity | Elacridar; cyclosporin A | KG-1a; TF-1 (human acute myeloid leukemia cell lines) | [162] |
| E. Increase C6-ceramide sensitivity | Tamoxifen | P-gp-overexpressing HeLa cells | [163] |
| F. Increase daunorubicin and vincristine sensitivity; increase rhodamine retention | PDMP | KG-1a/200 (daunorubicin- and vincristine-resistant); K562/138 (vincristine-resistant chronic myeloid leukemia cell line); K562/MDR-1 (stably transfected to express MDR-1) | [167] |
| G. Reverse daunorubicin resistance; enhance daunorubicin accumulation | PDMP; tetrandrine | K562/A02 (Adriamycin-resistant) | [168] |
| H. Enhanced C6-, C8-ceramide cytotoxicity | Tamoxifen; cyclosporin A; VX-710 (Biricodar); P4 (analog of PPPP) | 2780AD (doxorubicin-resistant human ovarian cancer cell line); NCI/ADR-RES (doxorubicin-resistant, human ovarian cancer cell line) | [164] |
| I. Increase sensitivity to Taxol and vincristine; decrease efflux | PDMP; PSC833 (Valspodar); MK571 | Neuro-2a; C1300 (murine neuroblastoma cell lines) | [170] |
| J. Sensitize drug-resistant cells to chemotherapy (Vincas; Adriamycin; Taxotere) | Genz-123346; PDMP; verapamil | KBV-1 (human cervical carcinoma cell line); HCT-15 (human colorectal cancer cell line) | [171] |
| K. Reverse Imatinib resistance | PDMP | K562/IMA (imatinib-resistant chronic myeloid leukemia cell line) | [17] |
| L. Reverse Cisplatin resistance | PDMP | Orthoxenografts, testicular germ cell tumors | [174] |
| M. Reverse Fludarabine resistance | PDMP | Fludarabine-resistant MEC-2 cells (human B cell leukemia cell line) | [175] |
| N. Enhance sensitivity to Taxol and cisplatin | PDMP | Paclitaxel-resistant PX2 and KF28; cisplatin-resistant C13 and KFr13 (human ovarian cancer cell lines) | [176] |
| O. Enhance selumetinib (AZD-6244) sensitivity | PDMP | PANC-1; AsPC-1; MIA PaCa-2 (human pancreatic cancer cell lines) | [177] |
| P. Increase sensitivity to C6-ceramide nanoliposome (CNL) | PDMP | PANC-1 | [178] |
| Q. Enhance vinorelbine sensitivity | PDMP | A549; CL1-5 (human lung adenocarcinoma cell lines) | [179] |
| R. Sensitization to ABT-263 (Bcl-2 inhibitor) | PDMP | U937; K562; HL-60; RPMI-8226 (various human leukemia cell lines) | [180] |
| S. Enhance AR-42 (HDAC inhibitor) sensitivity | PDMP | SW-620 (human colon cancer cell line) | [181] |
| T. Potentiate A-674563 (Akt1 inhibitor) cytotoxicity | PDMP | A375 (human melanoma cell line) | [182] |
| U. Sensitize to GNF-2 (Bcr-Abl inhibitor); imatinib; nilotinib | PDMP | K562; Ba/F3-p210T315I (Bcr-Abl mutant murine chronic myeloid leukemia cell line) | [183] |
| V. Enhance vincristine and Adriamycin sensitivity; decrease drug efflux | Mifepristone | SGC7901/VCR (vincristine-resistant human gastric cancer cell line); rat hepatoma cells; K562-R7 (multidrug-resistant) | [184,185] |
| W. Enhance sensitivity to Taxol and Adriamycin | Resveratrol | MCF-7; HepG2, HeLa (various human solid tumor cell lines) | [186] |
| X. Enhance vincristine, daunorubicin, cisplatin sensitivity | Ethylenedioxy-P4 | Lucena-1 (vincristine-resistant K562); FEPS (daunorubicin-resistant K562) | [131] |
| Y. Enhance SACLAC (acid ceramidase inhibitor) sensitivity | PDMP | HL-60/VCR (vincristine-resistant); HL-60/DNR (daunorubicin-resistant) | [166] |
| Z. Enhance hydroxychloroquine or DC661 sensitivity (lysosomal autophagy inhibitors) | Genz-123346 | A375-P (human melanoma cell line); DLD-1 (human colorectal adenocarcinoma cell line); B16-F10 (murine melanoma cell line); MIA PaCa-2; A549; PDXWM4380 and PDX WM4552 (patient-derived cell lines) | [187] |
| AA. Reverse sorafenib resistance | PDMP | Hep3B; HepG2 (hepatocellular carcinoma cell lines) | [111] |
| AB. Sensitize to cisplatin | PPMP | HN9-cisR (cisplatin-resistant human head and neck cancer cell line) | [127] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ung, J.; Kassai, M.; Tan, S.-F.; Loughran, T.P., Jr.; Feith, D.J.; Cabot, M.C. The Drug Transporter P-Glycoprotein and Its Impact on Ceramide Metabolism—An Unconventional Ally in Cancer Treatment. Int. J. Mol. Sci. 2024, 25, 9825. https://doi.org/10.3390/ijms25189825
Ung J, Kassai M, Tan S-F, Loughran TP Jr., Feith DJ, Cabot MC. The Drug Transporter P-Glycoprotein and Its Impact on Ceramide Metabolism—An Unconventional Ally in Cancer Treatment. International Journal of Molecular Sciences. 2024; 25(18):9825. https://doi.org/10.3390/ijms25189825
Chicago/Turabian StyleUng, Johnson, Miki Kassai, Su-Fern Tan, Thomas P. Loughran, Jr., David J. Feith, and Myles C. Cabot. 2024. "The Drug Transporter P-Glycoprotein and Its Impact on Ceramide Metabolism—An Unconventional Ally in Cancer Treatment" International Journal of Molecular Sciences 25, no. 18: 9825. https://doi.org/10.3390/ijms25189825