
Enrichment of Cis-Acting Regulatory Elements in Differentially Methylated Regions Following Lipopolysaccharide Treatment of Bovine Endometrial Epithelial Cells

 , , , , , and
, , , , , and
Abstract
:1. Introduction
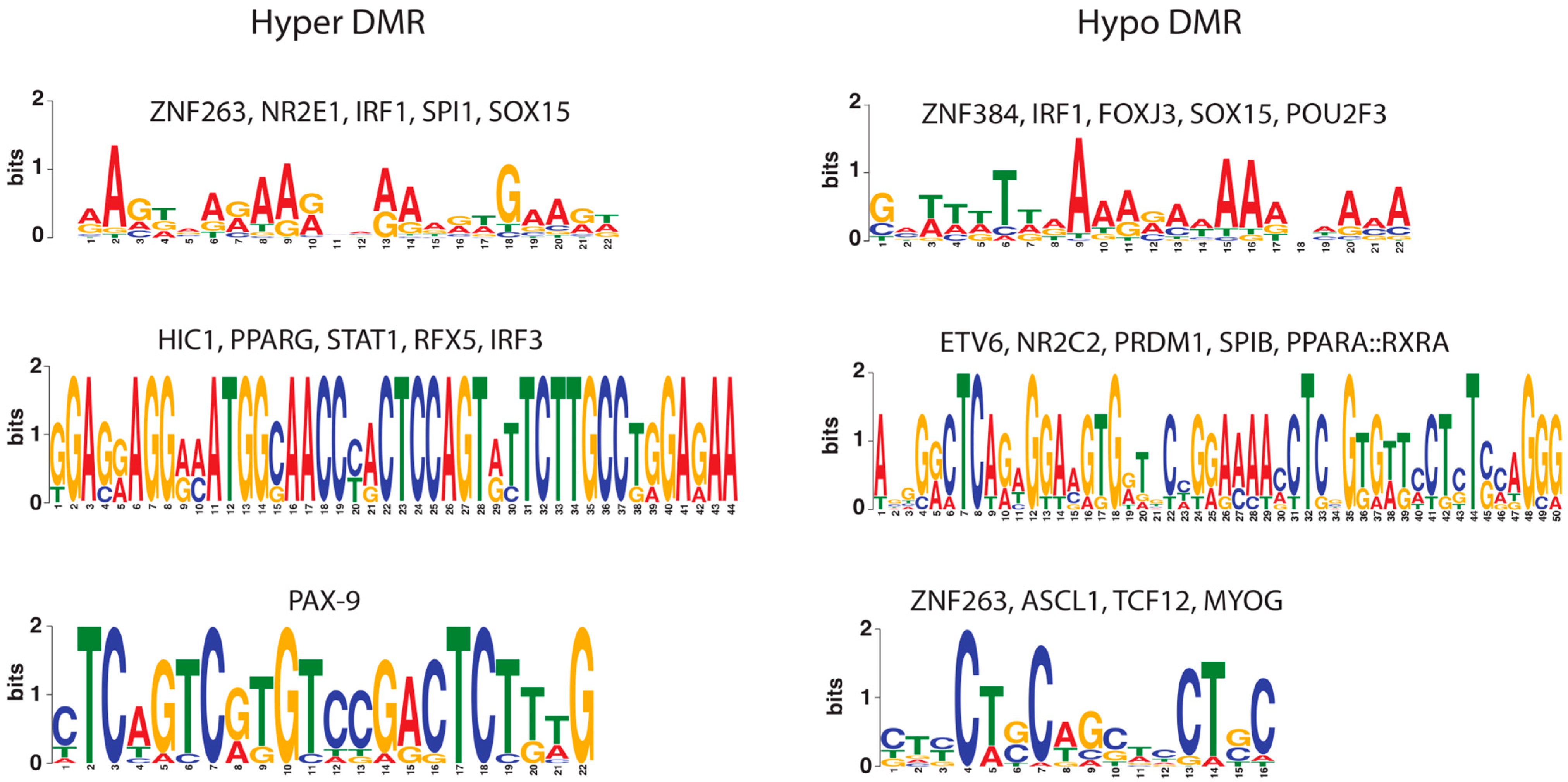
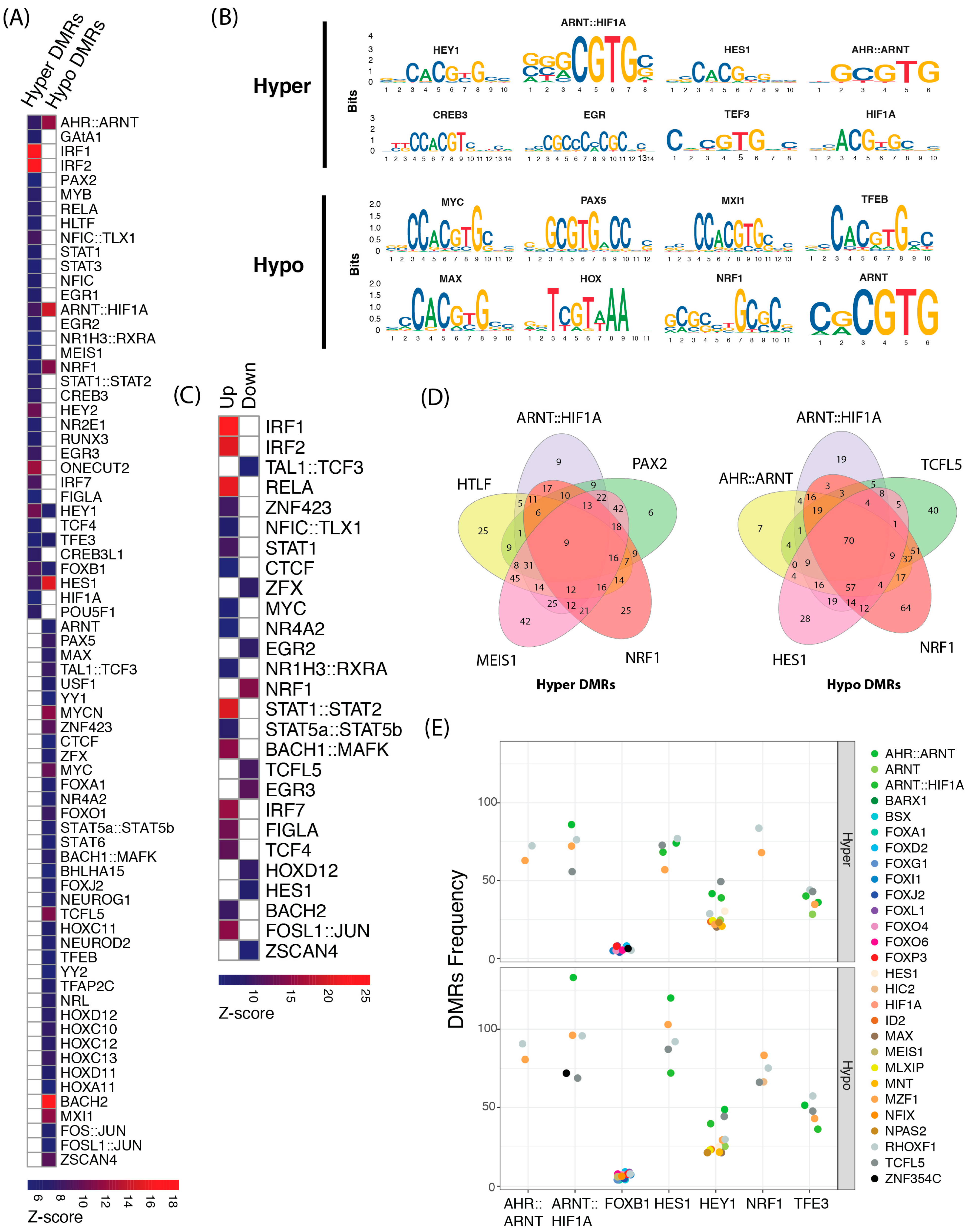
2. Results
2.1. Co-Localization of Transcription Factor Cis-Regulatory Motifs
2.2. Effects of Methylation Changes on the Activity of Transcription Factors
3. Discussion
4. Materials and Methods
4.1. Purification of Bovine Endometrial Epithelial Cells
4.2. Methylation and Gene Expression Data
4.3. Transcription Factor Binding Sites Analyses
4.4. Colocalized Motifs in DMRs
4.5. Analysis of Global Methylation Levels of TFBSs and Expression of Transcription Factors
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Platanitis, E.; Decker, T. Regulatory Networks Involving STATs, IRFs, and NFkappaB in Inflammation. Front. Immunol. 2018, 9, 2542. [Google Scholar] [CrossRef] [PubMed]
- Fischle, W.; Wang, Y.; Allis, C.D. Histone and chromatin cross-talk. Curr. Opin. Cell Biol. 2003, 15, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Gottesfeld, J.M.; Carey, M.F. Introduction to the Thematic Minireview Series: Chromatin and transcription. J. Biol. Chem. 2018, 293, 13775–13777. [Google Scholar] [CrossRef]
- Pratt, H.E.; Andrews, G.R.; Phalke, N.; Purcaro, M.J.; van der Velde, A.; Moore, J.E.; Weng, Z. Factorbook: An updated catalog of transcription factor motifs and candidate regulatory motif sites. Nucleic Acids Res. 2022, 50, D141–D149. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Braunschweig, U.; Gonatopoulos-Pournatzis, T.; Weatheritt, R.J.; Hirsch, C.L.; Ha, K.C.H.; Radovani, E.; Nabeel-Shah, S.; Sterne-Weiler, T.; Wang, J.; et al. Multilayered Control of Alternative Splicing Regulatory Networks by Transcription Factors. Mol. Cell 2017, 65, 539–553.e7. [Google Scholar] [CrossRef] [PubMed]
- Latchman, D.S. Transcription factors: An overview. Int. J. Exp. Pathol. 1993, 74, 417–422. [Google Scholar] [CrossRef]
- Roeder, R.G. The role of general initiation factors in transcription by RNA polymerase II. Trends Biochem. Sci. 1996, 21, 327–335. [Google Scholar] [CrossRef]
- Maston, G.A.; Evans, S.K.; Green, M.R. Transcriptional regulatory elements in the human genome. Annu. Rev. Genom. Hum. Genet. 2006, 7, 29–59. [Google Scholar] [CrossRef]
- Wei, W.; Yu, X.D. Comparative analysis of regulatory motif discovery tools for transcription factor binding sites. Genom. Proteom. Bioinform. 2007, 5, 131–142. [Google Scholar] [CrossRef]
- Guo, Y.; Gifford, D.K. Modular combinatorial binding among human trans-acting factors reveals direct and indirect factor binding. BMC Genom. 2017, 18, 45. [Google Scholar] [CrossRef]
- Toivonen, J.; Kivioja, T.; Jolma, A.; Yin, Y.; Taipale, J.; Ukkonen, E. Modular discovery of monomeric and dimeric transcription factor binding motifs for large data sets. Nucleic Acids Res. 2018, 46, e44. [Google Scholar] [CrossRef] [PubMed]
- Janson, L.; Pettersson, U. Cooperative interactions between transcription factors Sp1 and OTF-1. Proc. Natl. Acad. Sci. USA 1990, 87, 4732–4736. [Google Scholar] [CrossRef] [PubMed]
- Frietze, S.; Farnham, P.J. Transcription factor effector domains. Subcell. Biochem. 2011, 52, 261–277. [Google Scholar] [PubMed]
- Stegmaier, P.; Kel, A.E.; Wingender, E. Systematic DNA-binding domain classification of transcription factors. Genome Inform. 2004, 15, 276–286. [Google Scholar] [PubMed]
- Mitchell, P.J.; Tjian, R. Transcriptional regulation in mammalian cells by sequence-specific DNA binding proteins. Science 1989, 245, 371–378. [Google Scholar] [CrossRef]
- Jordan, I.K.; Rogozin, I.B.; Glazko, G.V.; Koonin, E.V. Origin of a substantial fraction of human regulatory sequences from transposable elements. Trends Genet. 2003, 19, 68–72. [Google Scholar] [CrossRef]
- Andrews, G.; Fan, K.; Pratt, H.E.; Phalke, N.; Zoonomia Consortium; Karlsson, E.K.; Lindblad-Toh, K.; Gazal, S.; Moore, J.E.; Weng, Z. Mammalian evolution of human cis-regulatory elements and transcription factor binding sites. Science 2023, 380, eabn7930. [Google Scholar] [CrossRef]
- Sundaram, V.; Wysocka, J. Transposable elements as a potent source of diverse cis-regulatory sequences in mammalian genomes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2020, 375, 20190347. [Google Scholar] [CrossRef]
- Lowe, C.B.; Haussler, D. 29 mammalian genomes reveal novel exaptations of mobile elements for likely regulatory functions in the human genome. PLoS ONE 2012, 7, e43128. [Google Scholar] [CrossRef]
- Grove, C.A.; De Masi, F.; Barrasa, M.I.; Newburger, D.E.; Alkema, M.J.; Bulyk, M.L.; Walhout, A.J. A multiparameter network reveals extensive divergence between C. elegans bHLH transcription factors. Cell 2009, 138, 314–327. [Google Scholar] [CrossRef]
- Reece-Hoyes, J.S.; Deplancke, B.; Shingles, J.; Grove, C.A.; Hope, I.A.; Walhout, A.J. A compendium of Caenorhabditis elegans regulatory transcription factors: A resource for mapping transcription regulatory networks. Genome Biol. 2005, 6, R110. [Google Scholar] [CrossRef] [PubMed]
- Simionato, E.; Ledent, V.; Richards, G.; Thomas-Chollier, M.; Kerner, P.; Coornaert, D.; Degnan, B.M.; Vervoort, M. Origin and diversification of the basic helix-loop-helix gene family in metazoans: Insights from comparative genomics. BMC Evol. Biol. 2007, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Loots, G.G.; Ovcharenko, I. rVISTA 2.0: Evolutionary analysis of transcription factor binding sites. Nucleic Acids Res. 2004, 32, W217–W221. [Google Scholar] [CrossRef] [PubMed]
- Jager, R.; Werling, U.; Rimpf, S.; Jacob, A.; Schorle, H. Transcription factor AP-2gamma stimulates proliferation and apoptosis and impairs differentiation in a transgenic model. Mol. Cancer Res. 2003, 1, 921–929. [Google Scholar] [PubMed]
- Kalaitzidis, D.; Gilmore, T.D. Transcription factor cross-talk: The estrogen receptor and NF-kappaB. Trends Endocrinol. Metab. 2005, 16, 46–52. [Google Scholar] [CrossRef]
- Pritsker, M.; Liu, Y.C.; Beer, M.A.; Tavazoie, S. Whole-genome discovery of transcription factor binding sites by network-level conservation. Genome Res. 2004, 14, 99–108. [Google Scholar] [CrossRef]
- Boerno, S.T.; Grimm, C.; Lehrach, H.; Schweiger, M.R. Next-generation sequencing technologies for DNA methylation analyses in cancer genomics. Epigenomics 2010, 2, 199–207. [Google Scholar] [CrossRef]
- Grada, A.; Weinbrecht, K. Next-generation sequencing: Methodology and application. J. Investig. Dermatol. 2013, 133, e11. [Google Scholar] [CrossRef]
- Lindblad-Toh, K.; Garber, M.; Zuk, O.; Lin, M.F.; Parker, B.J.; Washietl, S.; Kheradpour, P.; Ernst, J.; Jordan, G.; Mauceli, E.; et al. A high-resolution map of human evolutionary constraint using 29 mammals. Nature 2011, 478, 476–482. [Google Scholar] [CrossRef]
- McGuire, A.M.; Hughes, J.D.; Church, G.M. Conservation of DNA regulatory motifs and discovery of new motifs in microbial genomes. Genome Res. 2000, 10, 744–757. [Google Scholar] [CrossRef]
- Wingender, E.; Dietze, P.; Karas, H.; Knuppel, R. TRANSFAC: A database on transcription factors and their DNA binding sites. Nucleic Acids Res. 1996, 24, 238–241. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Fornes, O.; Stigliani, A.; Gheorghe, M.; Castro-Mondragon, J.A.; van der Lee, R.; Bessy, A.; Cheneby, J.; Kulkarni, S.R.; Tan, G.; et al. JASPAR 2018: Update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 2018, 46, D260–D266. [Google Scholar] [CrossRef] [PubMed]
- Mouse, E.C.; Stamatoyannopoulos, J.A.; Snyder, M.; Hardison, R.; Ren, B.; Gingeras, T.; Gilbert, D.M.; Groudine, M.; Bender, M.; Kaul, R.; et al. An encyclopedia of mouse DNA elements (Mouse ENCODE). Genome Biol. 2012, 13, 418. [Google Scholar]
- Guo, Y.; van Schaik, T.; Jhamat, N.; Niazi, A.; Chanrot, M.; Charpigny, G.; Valarcher, J.F.; Bongcam-Rudloff, E.; Andersson, G.; Humblot, P. Differential gene expression in bovine endometrial epithelial cells after challenge with LPS; specific implications for genes involved in embryo maternal interactions. PLoS ONE 2019, 14, e0222081. [Google Scholar] [CrossRef]
- Guo, Y.; Chankeaw, W.; Chanrot, M.; Valarcher, J.F.; Chantarapratep, P.; Bage, R.; Bongcam-Rudloff, E.; Andersson, G.; Charpigny, G.; Humblot, P. Changes Induced by Pathogens and Metabolic Stress on Endometrial Function in cattle: Possible Impacts of Increased Inflammation on Fertility; Anais do XXIII Congresso Brasileiro de Reprodução Animal (CBRA-2019); Revista Brasileira de Reprodução Animal: Gramado, Brazil, 2019; Volume 43, pp. 295–307. [Google Scholar]
- Chanrot, M.; Guo, Y.; Dalin, A.M.; Persson, E.; Bage, R.; Svensson, A.; Gustafsson, H.; Humblot, P. Dose related effects of LPS on endometrial epithelial cell populations from dioestrus cows. Anim. Reprod. Sci. 2017, 177, 12–24. [Google Scholar] [CrossRef]
- Pereira, G.; Guo, Y.; Silva, E.; Silva, M.F.; Bevilacqua, C.; Charpigny, G.; Lopes-da-Costa, L.; Humblot, P. Subclinical endometritis differentially affects the transcriptomic profiles of endometrial glandular, luminal, and stromal cells of postpartum dairy cows. J. Dairy Sci. 2022, 105, 6125–6143. [Google Scholar] [CrossRef]
- Jhamat, N.; Niazi, A.; Guo, Y.; Chanrot, M.; Ivanova, E.; Kelsey, G.; Bongcam-Rudloff, E.; Andersson, G.; Humblot, P. LPS-treatment of bovine endometrial epithelial cells causes differential DNA methylation of genes associated with inflammation and endometrial function. BMC Genom. 2020, 21, 385. [Google Scholar] [CrossRef]
- Alsamman, K.; El-Masry, O.S. Interferon regulatory factor 1 inactivation in human cancer. Biosci. Rep. 2018, 38, BSR20171672. [Google Scholar] [CrossRef]
- Levy, D.E.; Darnell, J.E., Jr. Stats: Transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 2002, 3, 651–662. [Google Scholar] [CrossRef]
- Ng, C.K.; Li, N.X.; Chee, S.; Prabhakar, S.; Kolatkar, P.R.; Jauch, R. Deciphering the Sox-Oct partner code by quantitative cooperativity measurements. Nucleic Acids Res. 2012, 40, 4933–4941. [Google Scholar] [CrossRef]
- Kwon, A.T.; Arenillas, D.J.; Worsley Hunt, R.; Wasserman, W.W. oPOSSUM-3: Advanced analysis of regulatory motif over-representation across genes or ChIP-Seq datasets. G3 2012, 2, 987–1002. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Morgunova, E.; Jolma, A.; Kaasinen, E.; Sahu, B.; Khund-Sayeed, S.; Das, P.K.; Kivioja, T.; Dave, K.; Zhong, F.; et al. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science 2017, 356, eaaj2239. [Google Scholar] [CrossRef] [PubMed]
- Wapinski, O.L.; Vierbuchen, T.; Qu, K.; Lee, Q.Y.; Chanda, S.; Fuentes, D.R.; Giresi, P.G.; Ng, Y.H.; Marro, S.; Neff, N.F.; et al. Hierarchical mechanisms for direct reprogramming of fibroblasts to neurons. Cell 2013, 155, 621–635. [Google Scholar] [CrossRef] [PubMed]
- Zaret, K.S.; Carroll, J.S. Pioneer transcription factors: Establishing competence for gene expression. Genes Dev. 2011, 25, 2227–2241. [Google Scholar] [CrossRef] [PubMed]
- Babon, J.J.; Lucet, I.S.; Murphy, J.M.; Nicola, N.A.; Varghese, L.N. The molecular regulation of Janus kinase (JAK) activation. Biochem. J. 2014, 462, 1–13. [Google Scholar] [CrossRef]
- Aaronson, D.S.; Horvath, C.M. A road map for those who don’t know JAK-STAT. Science 2002, 296, 1653–1655. [Google Scholar] [CrossRef]
- Villarino, A.V.; Kanno, Y.; O’Shea, J.J. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat. Immunol. 2017, 18, 374–384. [Google Scholar] [CrossRef]
- Maj, T.; Chelmonska-Soyta, A. Pleiotropy and redundancy of STAT proteins in early pregnancy. Reprod. Domest. Anim. 2007, 42, 343–353. [Google Scholar] [CrossRef]
- Catalano, R.D.; Johnson, M.H.; Campbell, E.A.; Charnock-Jones, D.S.; Smith, S.K.; Sharkey, A.M. Inhibition of Stat3 activation in the endometrium prevents implantation: A nonsteroidal approach to contraception. Proc. Natl. Acad. Sci. USA 2005, 102, 8585–8590. [Google Scholar] [CrossRef]
- Teng, C.B.; Diao, H.L.; Ma, X.H.; Xu, L.B.; Yang, Z.M. Differential expression and activation of Stat3 during mouse embryo implantation and decidualization. Mol. Reprod. Dev. 2004, 69, 1–10. [Google Scholar] [CrossRef]
- Nakamura, H.; Kimura, T.; Koyama, S.; Ogita, K.; Tsutsui, T.; Shimoya, K.; Taniguchi, T.; Koyama, M.; Kaneda, Y.; Murata, Y. Mouse model of human infertility: Transient and local inhibition of endometrial STAT-3 activation results in implantation failure. FEBS Lett. 2006, 580, 2717–2722. [Google Scholar] [CrossRef] [PubMed]
- Bode, J.G.; Ehlting, C.; Haussinger, D. The macrophage response towards LPS and its control through the p38(MAPK)-STAT3 axis. Cell Signal. 2012, 24, 1185–1194. [Google Scholar] [CrossRef] [PubMed]
- Greenhill, C.J.; Rose-John, S.; Lissilaa, R.; Ferlin, W.; Ernst, M.; Hertzog, P.J.; Mansell, A.; Jenkins, B.J. IL-6 trans-signaling modulates TLR4-dependent inflammatory responses via STAT3. J. Immunol. 2011, 186, 1199–1208. [Google Scholar] [CrossRef]
- Duncan, S.A.; Zhong, Z.; Wen, Z.; Darnell, J.E., Jr. STAT signaling is active during early mammalian development. Dev. Dyn. 1997, 208, 190–198. [Google Scholar] [CrossRef]
- Vitorino Carvalho, A.; Eozenou, C.; Healey, G.D.; Forde, N.; Reinaud, P.; Chebrout, M.; Gall, L.; Rodde, N.; Padilla, A.L.; Delville, C.G.; et al. Analysis of STAT1 expression and biological activity reveals interferon-tau-dependent STAT1-regulated SOCS genes in the bovine endometrium. Reprod. Fertil. Dev. 2016, 28, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Li, B.; Wu, Y.; Wang, X.; Deng, G. Interferon-tau increases BoLA-I for implantation during early pregnancy in dairy cows. Oncotarget 2017, 8, 95095–95107. [Google Scholar] [CrossRef] [PubMed]
- Suman, P.; Malhotra, S.S.; Gupta, S.K. LIF-STAT signaling and trophoblast biology. JAKSTAT 2013, 2, e25155. [Google Scholar] [CrossRef]
- Fu, Y.; Liu, B.; Feng, X.; Liu, Z.; Liang, D.; Li, F.; Li, D.; Cao, Y.; Feng, S.; Zhang, X.; et al. Lipopolysaccharide increases Toll-like receptor 4 and downstream Toll-like receptor signaling molecules expression in bovine endometrial epithelial cells. Vet. Immunol. Immunopathol. 2013, 151, 20–27. [Google Scholar] [CrossRef]
- Oguejiofor, C.F.; Cheng, Z.; Abudureyimu, A.; Fouladi-Nashta, A.A.; Wathes, D.C. Global transcriptomic profiling of bovine endometrial immune response in vitro. I. Effect of lipopolysaccharide on innate immunity. Biol. Reprod. 2015, 93, 100. [Google Scholar]
- Canaff, L.; Zhou, X.; Hendy, G.N. The proinflammatory cytokine, interleukin-6, up-regulates calcium-sensing receptor gene transcription via Stat1/3 and Sp1/3. J. Biol. Chem. 2008, 283, 13586–13600. [Google Scholar] [CrossRef]
- Taylor, C.T.; Colgan, S.P. Regulation of immunity and inflammation by hypoxia in immunological niches. Nat. Rev. Immunol. 2017, 17, 774–785. [Google Scholar] [CrossRef] [PubMed]
- Tan, G.; Lenhard, B. TFBSTools: An R/bioconductor package for transcription factor binding site analysis. Bioinformatics 2016, 32, 1555–1556. [Google Scholar] [CrossRef] [PubMed]
- Rekawiecki, R.; Rutkowska, J.; Kotwica, J. Identification of optimal housekeeping genes for examination of gene expression in bovine corpus luteum. Reprod. Biol. 2012, 12, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.G.; Meier, S.; Mitchell, M.D.; Roche, J.R.; Littlejohn, M. Evaluation of real-time PCR endogenous control genes for analysis of gene expression in bovine endometrium. BMC Mol. Biol. 2009, 10, 100. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Class | Transcription Factors |
|---|---|
| Basic helix-loop-helix factors (bHLH) | ARNT, Bhlha15, HES1, AHR, HEY1, HEY2, MYC, MXI1, MYCN, TFE3, MAX, NEUROD2, HIF-1α, USF1, TCFL5, TAL1, TCF3, NEUROG1, TFEB, TCF4, FIGLA |
| Homeo-domain factors | POU5F1, MEIS1, HOXC10, HOXC11, HOXC12, HOXC13, ONECUT2, HOXA11, HOXD11, HOXD12, TLX1 |
| Basic leucine zipper factors (bZIP) | CREB3, CREB3L1, MAFK, BACH1, BACH2, FOS, NRL, FOSL1, JUN, NRF1 |
| C2H2 zinc finger | ZSCAN4, YY1, YY2, ZNF423, CTCF, ZFX, EGR1, EGR2, EGR3 |
| STAT domain factors | STAT1, STAT2, STAT3, STAT5A, STAT5B, STAT6 |
| Tryptophan clusters | IRF1, IRF2, IRF7, MYB, HLTF |
| Nuclear receptors with C4 zinc fingers | NR1H3, RXRA, NR4A2, NR2E1 |
| Fork head/Winged helix | FOXB1, FOXA1, FOXJ2, FOXO1 |
| Paired box | PAX2, PAX5 |
| Basic helix-span-helix factors (bHSH) | TFAP2C |
| Runt domain | RUNX3 |
| C4 zinc finger-type factors | GATA1 |
| Rel homology region (RHR) factors | RELA |
| SMAD/NF-1 DNA-binding domain factors | NFIC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jhamat, N.; Guo, Y.; Han, J.; Humblot, P.; Bongcam-Rudloff, E.; Andersson, G.; Niazi, A. Enrichment of Cis-Acting Regulatory Elements in Differentially Methylated Regions Following Lipopolysaccharide Treatment of Bovine Endometrial Epithelial Cells. Int. J. Mol. Sci. 2024, 25, 9832. https://doi.org/10.3390/ijms25189832
Jhamat N, Guo Y, Han J, Humblot P, Bongcam-Rudloff E, Andersson G, Niazi A. Enrichment of Cis-Acting Regulatory Elements in Differentially Methylated Regions Following Lipopolysaccharide Treatment of Bovine Endometrial Epithelial Cells. International Journal of Molecular Sciences. 2024; 25(18):9832. https://doi.org/10.3390/ijms25189832
Chicago/Turabian StyleJhamat, Naveed, Yongzhi Guo, Jilong Han, Patrice Humblot, Erik Bongcam-Rudloff, Göran Andersson, and Adnan Niazi. 2024. "Enrichment of Cis-Acting Regulatory Elements in Differentially Methylated Regions Following Lipopolysaccharide Treatment of Bovine Endometrial Epithelial Cells" International Journal of Molecular Sciences 25, no. 18: 9832. https://doi.org/10.3390/ijms25189832