
Polymorphism and Pharmacological Assessment of Carbamazepine
 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Results and Discussions
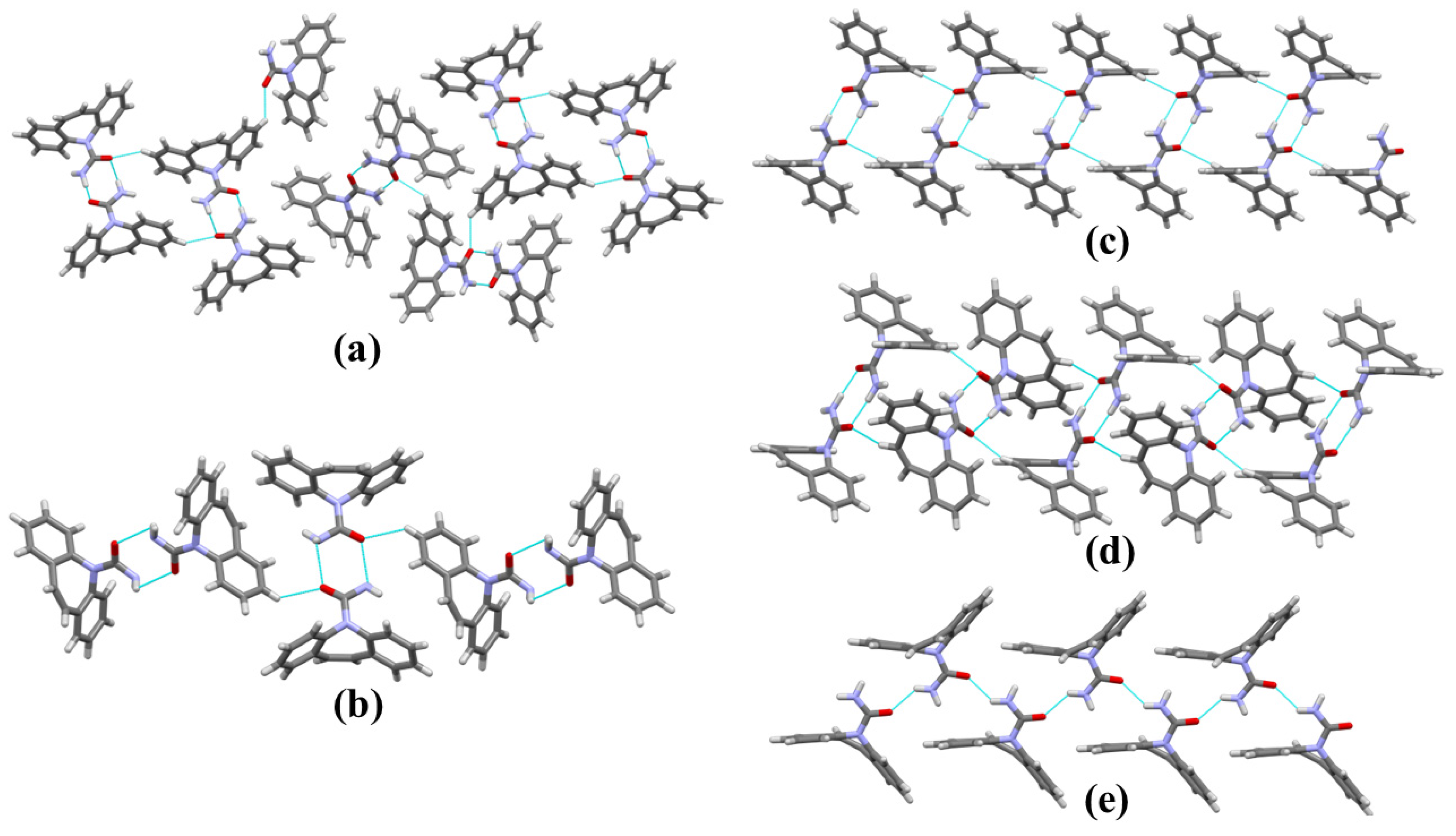
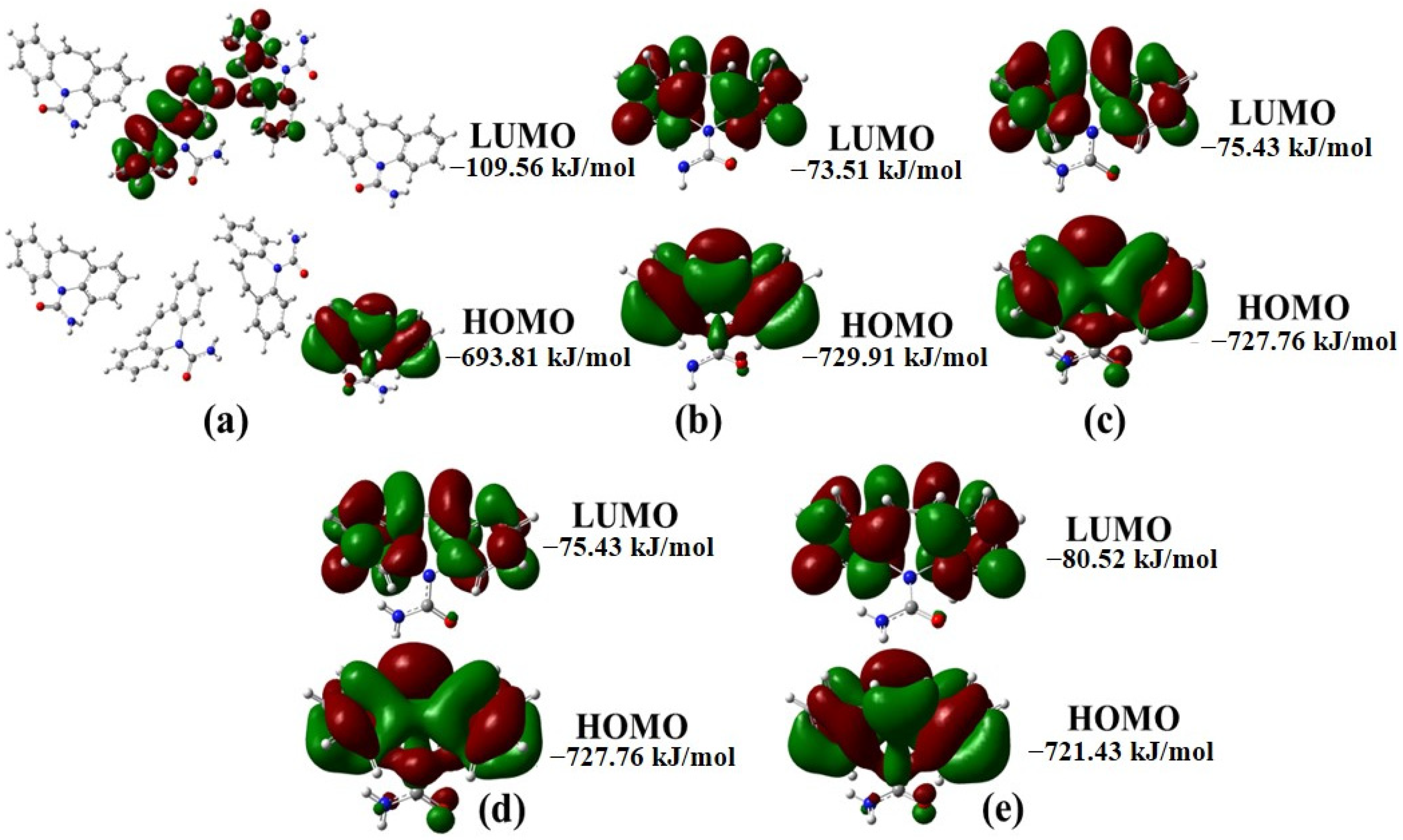
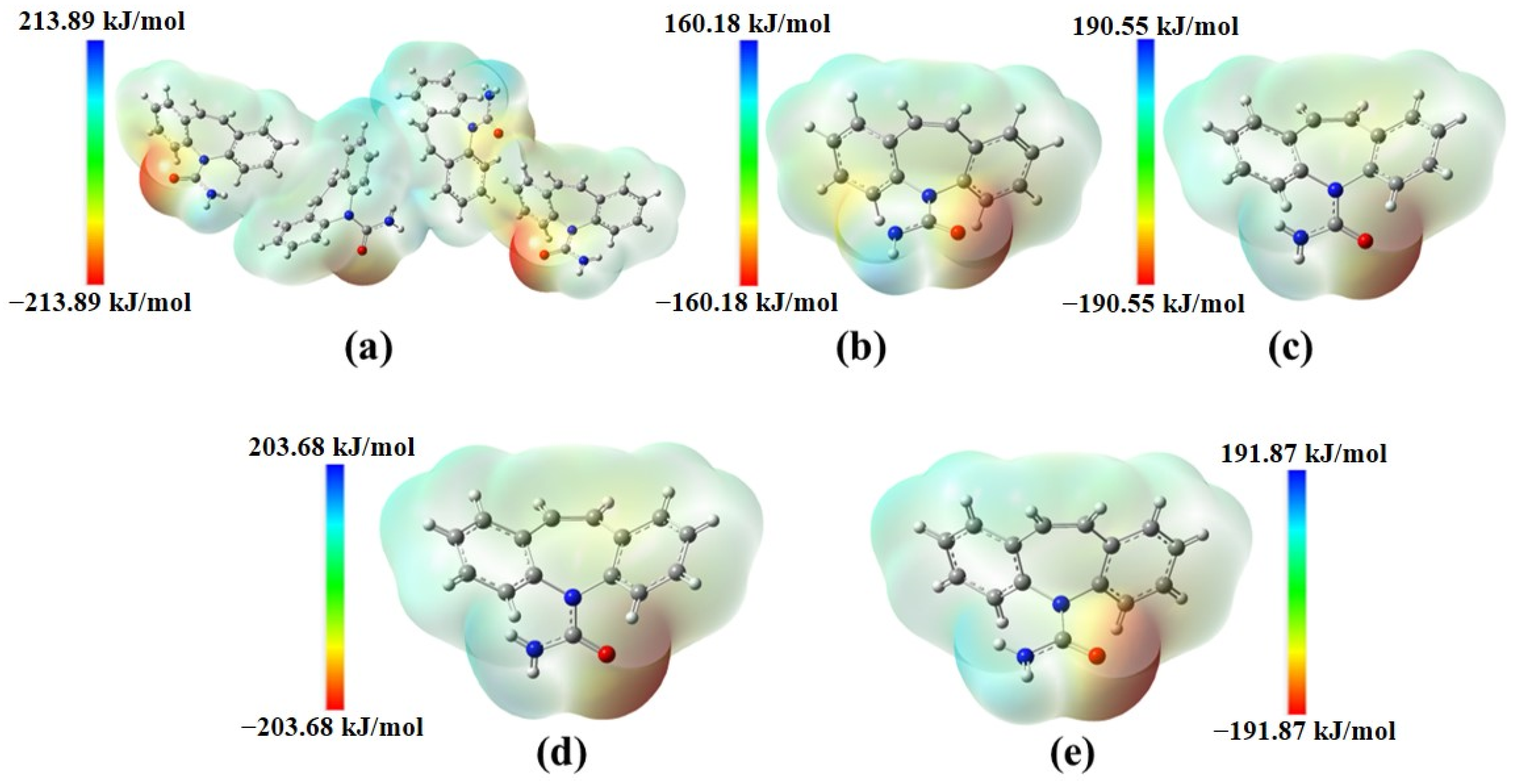
2.1. Structural Polymorphism Analysis
2.2. CBZ Formulation (CBZ-F)
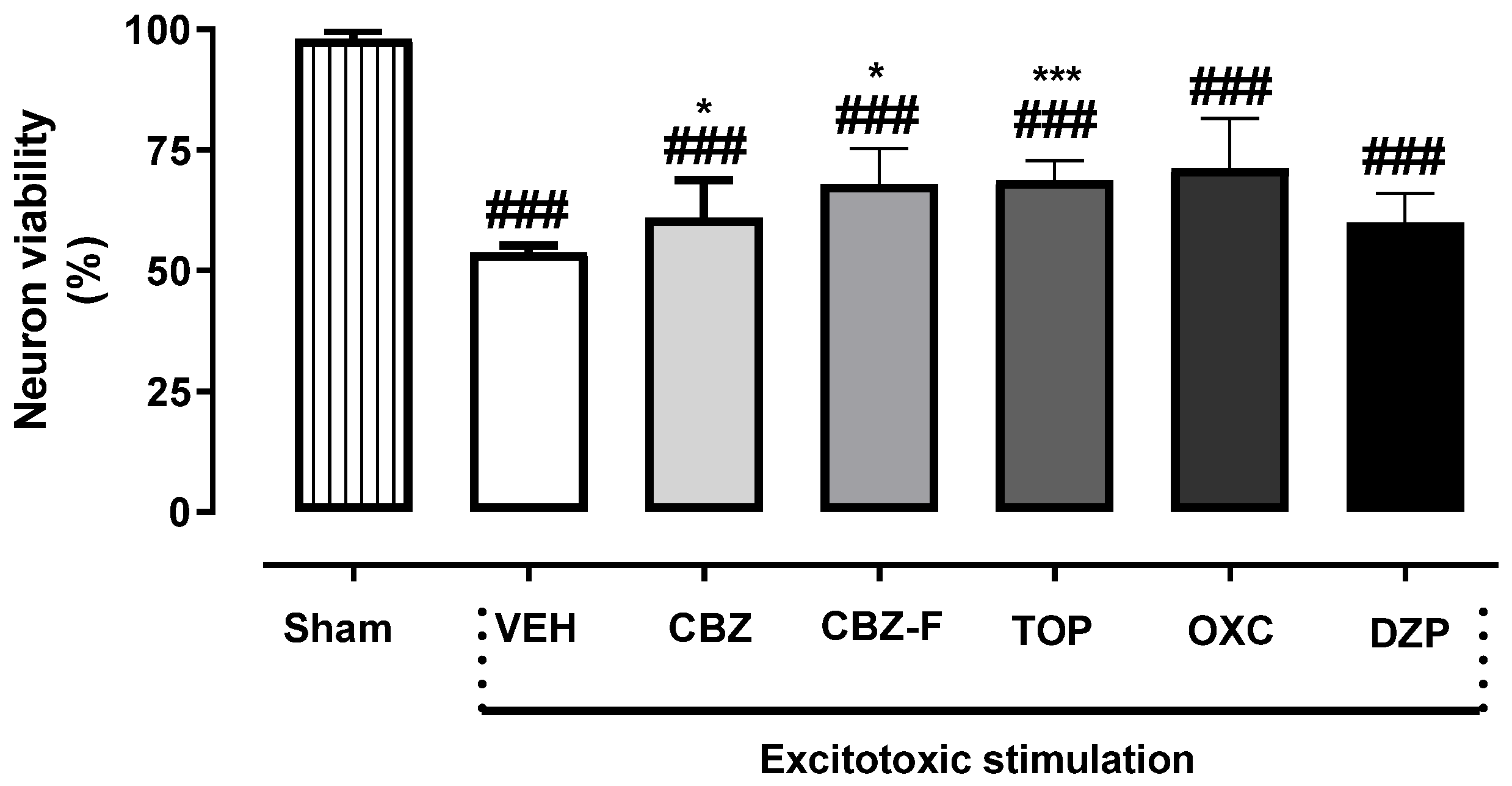
2.3. Neuron Viability Assessment In Vitro
2.4. In Vivo Assays
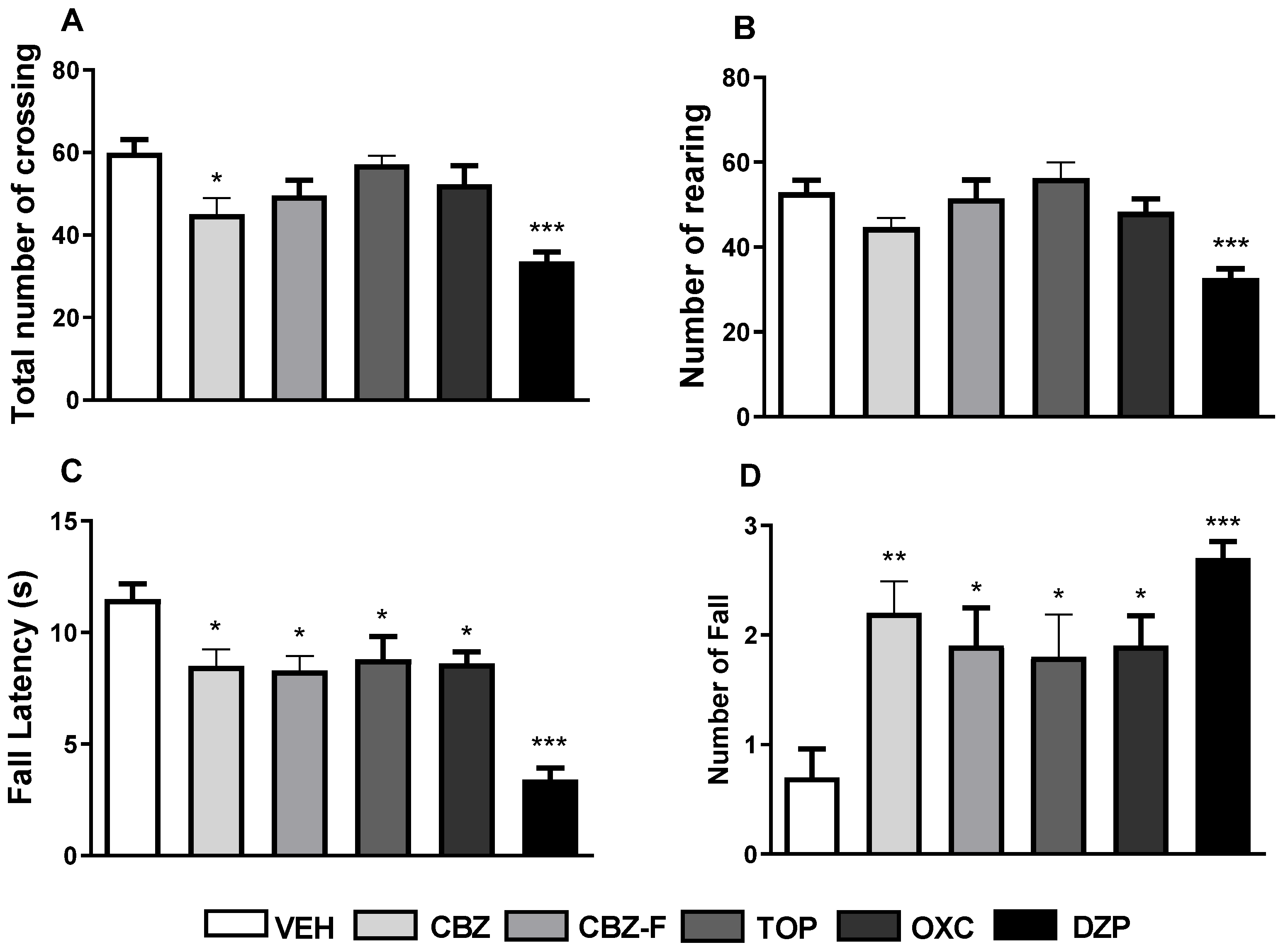
2.4.1. Screening for Exploratory and Motor Activities
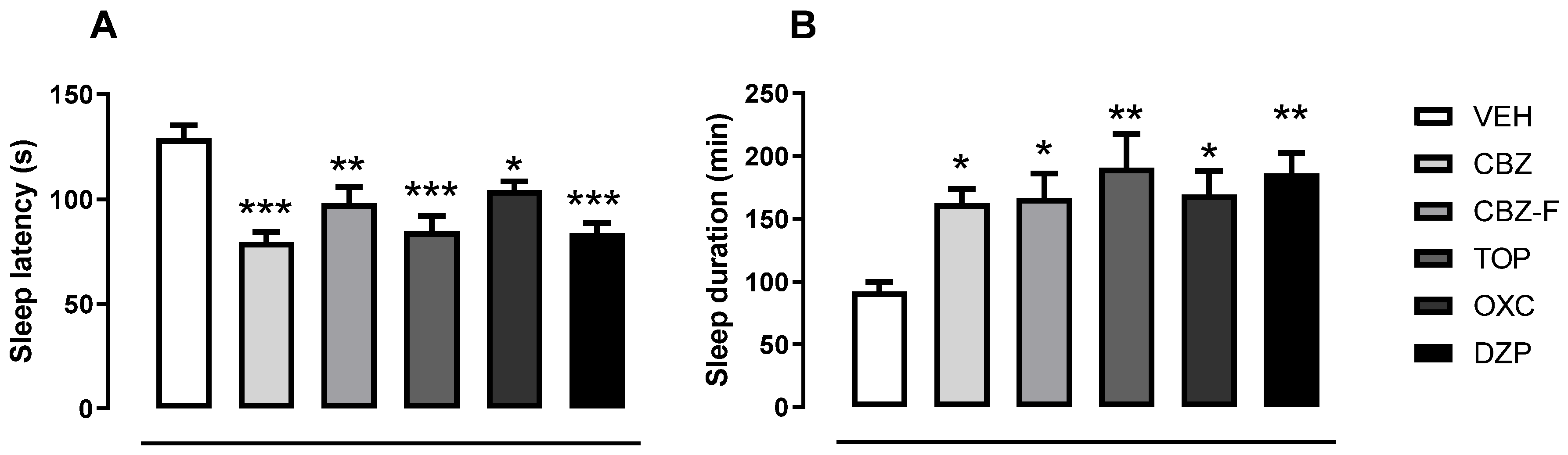
2.4.2. Screening for Barbiturate Sleep Potentiation
2.4.3. Screening for Antiseizure Activities
3. Materials and Methods
3.1. Methods
3.2. Carbamazepine (CBZ)
3.2.1. Computational Procedures
3.2.2. Preparation of CBZ-F
3.3. Pharmacological Approaches
3.3.1. Excitotoxicity Protocol and Neuron Viability Assessment In Vitro
3.3.2. Animals and Treatment
3.3.3. Screening for Exploratory and Motor Activities
3.3.4. Barbiturate Sleep Induction
3.3.5. Screening for Anticonvulsant Activity and Its Pharmacological Blockade
3.4. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Löscher, W.; Klein, P. The pharmacology and clinical efficacy of antiseizure medications: From bromide salts to cenobamate and beyond. CNS Drugs 2021, 35, 935–963. [Google Scholar] [PubMed]
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshé, S.L.; et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Marson, A.G.; Al-Kharusi, A.M.; Alwaidh, M.; Appleton, R.; Baker, G.A.; Chadwick, D.W.; Cramp, C.; Cockerell, O.C.; Cooper, P.N.; Doughty, J.; et al. The SANAD study of effectiveness of carbamazepine, gabapentin, lamotrigine, oxcarbazepine, or topiramate for treatment of partial epilepsy: An unblinded randomised controlled trial. Lancet 2007, 369, 1000–1015. [Google Scholar] [CrossRef] [PubMed]
- Bao, Z.; Bufton, J.; Hickman, R.J.; Aspuru-Guzik, A.; Bannigan, P.; Allen, C. Revolutionizing drug formulation development: The increasing impact of machine learning. Adv. Drug Deliv. Rev. 2023, 202, 115108. [Google Scholar] [CrossRef] [PubMed]
- Kovacevic, I.; Parojcic, J.; Homsek, I.; Tubic-Grozdanis, M.; Langguth, P. Justification of biowaiver for carbamazepine, a low soluble high permeable compound, in solid dosage forms based on IVIVC and gastrointestinal simulation. Mol. Pharm. 2009, 6, 40–47. [Google Scholar] [CrossRef] [PubMed]
- García, M.A.; Cristofoletti, R.; Abrahamsson, B.; Groot, D.W.; Parr, A.; Polli, J.E.; Mehta, M.; Shah, V.P.; Tomakazu, T.; Dressman, J.B.; et al. Biowaiver monograph for immediate-release solid oral dosage forms: Carbamazepine. J. Pharm. Sci. 2021, 110, 1935–1947. [Google Scholar] [CrossRef] [PubMed]
- Amidon, G.L.; Lennernas, H.; Shah, V.P.; Crison, J.R. A theoretical basis for a biopharmaceutic drug classification: The correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Rustichelli, C.; Gamberini, G.; Ferioli, V.; Gamberini, M.C.; Ficarra, R.; Tommasini, S. Solid-state study of polymorphic drugs: Carbamazepine. J. Pharm. Biomed. Anal. 2000, 23, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Long, B.; Walker, G.M.; Ryan, K.M.; Padrela, L. Controlling polymorphism of carbamazepine nanoparticles in a continuous supercritical-CO2-assisted spray drying process. Cryst. Growth Des. 2019, 19, 3755–3767. [Google Scholar] [CrossRef]
- Grzesiak, A.L.; Lang, M.; Kim, K.; Matzger, A.J. Comparison of the four anhydrous polymorphs of carbamazepine and the crystal structure of form I. J. Pharm. Sci. 2003, 92, 2260–2271. [Google Scholar] [CrossRef]
- Saifee, M.; Inamda, N.; Dhamecha, D.; Rathi, A. Drug polymorphism: A review. Int. J. Health Res. 2009, 2. [Google Scholar] [CrossRef]
- Yu, L.; Reutzel, S.M.; Stephenson, G.A. Physical characterization of polymorphic drugs: An integrated characterization strategy. Pharm. Sci. Technol. Today 1998, 1, 118–127. [Google Scholar] [CrossRef]
- Brittain, H.G. Polymorphism and solvatomorphism 2010. J. Pharm. Sci. 2012, 101, 464–484. [Google Scholar] [CrossRef] [PubMed]
- Tiwary, A.K. Modification of crystal habit and its role in dosage form performance. Drug Dev. Ind. Pharm. 2001, 27, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Censi, R.; Di Martino, P. Polymorph Impact on the Bioavailability and Stability of Poorly Soluble Drugs. Molecules 2015, 20, 18759–18776. [Google Scholar] [CrossRef] [PubMed]
- Reig-López, J.; Cuquerella-Gilabert, M.; Bandín-Vilar, E.; Merino-Sanjuán, M.; Mangas-Sanjuán, V.; García-Arieta, A. Bioequivalence risk assessment of oral formulations containing racemic ibuprofen through a chiral physiologically based pharmacokinetic model of ibuprofen enantiomers. Eur. J. Pharm. Biopharm. 2024, 17, 114293. [Google Scholar] [CrossRef] [PubMed]
- Danda, L.J.A.; Batista, L.M.; Melo, V.C.S.; Soares Sobrinho, J.L.; Soares, M.F.R. Combining amorphous solid dispersions for improved kinetic solubility of posaconazole simultaneously released from soluble PVP/VA64 and an insoluble ammonio methacrylate copolymer. Eur. J. Pharm. Sci. 2019, 133, 79–85. [Google Scholar] [CrossRef]
- Celebioglu, A.; Uyar, T. Metronidazole/Hydroxypropyl-beta-Cyclodextrin inclusion complex nanofibrous webs as fast-dissolving oral drug delivery system. Int. J. Pharm. 2019, 572, 118828. [Google Scholar] [CrossRef] [PubMed]
- Jeevanandam, J.; Chan, Y.S.; Danquah, M.K. Nano-formulations of drugs: Recent developments, impact and challenges. Biochimie 2016, 128, 99–112. [Google Scholar] [CrossRef]
- Strandjord, R.E.; Johannessen, S.I. Single-drug therapy with carbamazepine in patients with epilepsy: Serum levels and clinical effect. Epilepsia 1980, 21, 655–662. [Google Scholar] [CrossRef]
- Abdelsayed, M.; Sokolov, S. Voltage-gated sodium channels: Pharmaceutical targets via anticonvulsants to treat epileptic syndromes. Channels 2013, 7, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Žiburkus, J.; Cressman, J.R.; Schiff, S.J. Seizures as imbalanced up states: Excitatory and inhibitory conductances during seizure-like events. J. Neurophysiol. 2013, 109, 1296–1306. [Google Scholar] [CrossRef] [PubMed]
- Sills, G.J.; Rogawski, M.A. Mechanisms of action of currently used antiseizure drugs. Neuropharmacology 2020, 168, 107966. [Google Scholar] [CrossRef] [PubMed]
- Akduran, N. Crystal structure of 2-hy-droxy-2-phenyl-aceto-phenone oxime. Acta Crystallogr. E Crystallogr. Commun. 2021, 77 Pt 1, 66–69. [Google Scholar] [CrossRef] [PubMed]
- van der Merwe, J.; Steenekamp, J.; Steyn, D.; Hamman, J. The Role of Functional Excipients in Solid Oral Dosage Forms to Overcome Poor Drug Dissolution and Bioavailability. Pharmaceutics 2020, 12, 393. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Park, S.-A.; Sah, H. Surfactant effects upon dissolution patterns of carbamazepine immediate release tablet. Arch. Pharm. Res. 2005, 28, 120–126. [Google Scholar] [CrossRef] [PubMed]
- El-Massik, M.A.; Abdallah, O.Y.; Galal, S.; Daabis, N.A. Towards a universal dissolution medium for carbamazepine. Drug Dev. Ind. Pharm. 2006, 32, 893–905. [Google Scholar] [CrossRef] [PubMed]
- Chert, L.R.; Wesley, J.A.; Bhattachar, S.; Ruiz, B.; Bahash, K.; Babu, S.R. Dissolution behavior of a poorly water soluble compound in the presence of Tween 80. Pharm. Res. 2003, 20, 797–801. [Google Scholar]
- Crison, J.R.; Weiner, N.D.; Amidon, G.L. Dissolution media for in vitro testing of water-insoluble drugs: Effect of surfactant purity and electrolyte on in vitro dissolution of carbamazepine in aqueous solutions of sodium lauryl sulfate. J. Pharm. Sci. 1997, 86, 384–388. [Google Scholar] [CrossRef]
- Coyle, J.T.; Puttfarcken, P. Oxidative stress, glutamate, and neurodegenerative disorders. Science 1993, 262, 689–695. [Google Scholar] [CrossRef]
- Benjumea, D.M.; Gómez-Betancur, I.C.; Vásquez, J.; Alzate, F.; García-Silva, A.; Fontenla, J.A. Neuropharmacological effects of the ethanolic extract of Sida acuta. Rev. Bras. Farmacogn. 2016, 26, 209–215. [Google Scholar] [CrossRef]
- Simon, P.; Panissaud, C.; Costentin, J. The stimulant effect of modafinil on wakefulness is not associated with an increase in anxiety in mice. A comparison with dexamphetamine. Psychopharmacology 1994, 114, 597–600. [Google Scholar] [CrossRef] [PubMed]
- Rocha, F.F.; Almeida, C.S.; Santos RT, D.; Santana, S.A.; Costa, E.A.; Paula JR, D.; Vanderlinde, F.A. Anxiolytic-like and sedative effects of Hydrocotyle umbellata L., Araliaceae, extract in mice. Rev. Bras. Farmacogn. 2011, 21, 115–120. [Google Scholar] [CrossRef]
- Fajemiroye, J.O.; Galdino, P.M.; Alves, S.F.; de Paula JA, M.; de Paula, J.R.; Ghedini, P.C.; Costa, E.A. Involvement of 5-HT1A in the anxiolytic-like effect of dichloromethane fraction of Pimenta pseudocaryophyllus. J. Ethnopharmacol. 2012, 141, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Kwan, P.; Sills, G.J.; Brodie, M.J. The mechanisms of action of commonly used antiepileptic drugs. Pharmacol. Ther. 2001, 90, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Thapliyal, S.; Babu, K. Pentylenetetrazole (PTZ)-induced Convulsion Assay to Determine GABAergic Defects in Caenorhabditis elegans. Bio Protoc. 2018, 8, e2989. [Google Scholar] [CrossRef] [PubMed]
- White, H.S.; Brown, S.D.; Woodhead, J.H.; Skeen, G.A.; Wolf, H.H. Topiramate enhances GABA-mediated chloride flux and GABA-evoked chloride currents in murine brain neurons and increases seizure threshold. Epilepsy Res. 1997, 28, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Lowes, M.M.; Caira, M.R.; Lotter, A.P.; Van der Watt, J.G. Physicochemical properties and X-ray structural studies of the trigonal polymorph of carbamazepine. J. Pharm. Sci. 1987, 76, 744–752. [Google Scholar] [CrossRef]
- Eccles, K.S.; Stokes, S.P.; Daly, C.A.; Barry, N.M.; McSweeney, S.P.; O’Neill, D.J.; Kelly, D.M.; Jennings, W.B.; Dhubhghaill, O.M.N.; Moynihan, H.A.; et al. Evaluation of the Bruker SMART X2S: Crystallography for the nonspecialist? J. Appl. Crystallogr. 2011, 44, 213–215. [Google Scholar] [CrossRef]
- Lang, M.; Kampf, J.W.; Matzger, A.J. Form IV of carbamazepine. J. Pharm. Sci. 2002, 91, 1186–1190. [Google Scholar] [CrossRef]
- Arlin, J.B.; Price, L.S.; Price, S.L.; Florence, A.J. A strategy for producing predicted polymorphs: Catemeric carbamazepine form V. Chem. Commun. 2011, 47, 7074–7076. [Google Scholar] [CrossRef] [PubMed]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Spackman, M.A.; Mitchell, A.S. Novel tools for visualizing and exploring intermolecular interactions in molecular crystals. Acta Crystallogr. B 2004, 60, 627–668. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, J.J.; Jayatilaka, D.; Spackman, M.A. Towards quantitative analysis of intermolecular interactions with Hirshfeld surfaces. Chem. Commun. 2007, 3814–3816. [Google Scholar] [CrossRef] [PubMed]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Spackman, M.A.; McKinnon, J. Fingerprinting intermolecular interactions in molecular crystals. CrystEngComm 2002, 4, 378–392. [Google Scholar] [CrossRef]
- Burke, K. Perspective on density functional theory. J. Chem. Phys. 2012, 136, 150901. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- McLean, A.; Chandler, G. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z= 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Hohenstein, E.G.; Chill, S.T.; Sherrill, C.D. Assessment of the Performance of the M05-2X and M06-2X Exchange-Correlation Functionals for Noncovalent Interactions in Biomolecules. J. Chem. Theory Comput. 2008, 4, 1996–2000. [Google Scholar] [CrossRef] [PubMed]
- Wiberg, K.B. Basis set effects on calculated geometries: 6-311++G** vs. aug-cc-pVDZ. J. Comput. Chem. 2004, 25, 1342–1346. [Google Scholar] [CrossRef]
- Sousa, S.F.; Fernandes, P.A.; Ramos, M.J. General performance of density functionals. J. Phys. Chem. A 2007, 111, 10439–10452. [Google Scholar] [CrossRef] [PubMed]
- Pereira, D.H.; La Porta, F.; Santiago, R.; Garcia, D.; Ramalho, T. New perspectives on the role of frontier molecular orbitals in the study of chemical reactivity: A review. Rev. Virtual Quimica 2016, 425–453. [Google Scholar] [CrossRef]
- Murray, J.S.; Politzer, P. The electrostatic potential: An overview. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 153–163. [Google Scholar] [CrossRef]
- Weiner, P.K.; Langridge, R.; Blaney, J.M.; Schaefer, R.; Kollman, P.A. Electrostatic potential molecular surfaces. Proc. Natl. Acad. Sci. USA 1982, 79, 3754–3758. [Google Scholar] [CrossRef] [PubMed]
- Cabral, I.B.; de Lima Moreira, C.V.; Rodrigues, A.C.C.; da Silva Moreira, L.K.; Pereira, J.K.A.; Gomides, C.D.; Lião, L.M.; Machado, L.S.; Vaz, B.G.; da Cunha, L.C.; et al. Preclinical data on morpholine (3,5-di-tertbutyl-4-hydroxyphenyl) methanone induced anxiolysis. Naunyn Schmiedebergs Arch. Pharmacol. 2023, 396, 2957–2975. [Google Scholar] [CrossRef]
- Fajemiroye, J.O.; Galdino, P.M.; Florentino, I.F.; Da Rocha, F.F.; Ghedini, P.C.; Polepally, P.R.; Zjawiony, J.K.; Costa, E.A. Plurality of anxiety and depression alteration mechanism by oleanolic acid. J. Psychopharmacol. 2014, 28, 923–934. [Google Scholar] [CrossRef]
- Brito, A.F.; Fajemiroye, J.O.; Neri, H.F.S.; Silva, D.M.; Silva, D.P.B.; Sanz, G.; Vaz, B.G.; de Carvalho, F.S.; Ghedini, P.C.; Lião, L.M.; et al. Anxiolytic-like effect of 2-(4-((1-phenyl-1H-pyrazol-4-yl)methyl)piperazin-1-yl)ethan-1-ol is mediated through the benzodiazepine and nicotinic pathways. Chem. Biol. Drug Des. 2017, 90, 432–442. [Google Scholar] [CrossRef]
- Fajemiroye, J.O.; Galdino, P.M.; De Paula, J.A.M.; Rocha, F.F.; Akanmu, M.A.; Vanderlinde, F.A.; Zjawionye, J.K.; Costa, E.A. Anxiolytic and antidepressant like effects of natural food flavour (E)-methyl isoeugenol. Food Funct. 2014, 5, 1819–1828. [Google Scholar] [CrossRef]
- Shiotsuki, H.; Yoshimi, K.; Shimo, Y.; Funayama, M.; Takamatsu, Y.; Ikeda, K.; Takahashi, R.; Kitazawa, S.; Hattori, N. A rotarod test for evaluation of motor skill learning. J. Neurosci. Methods 2010, 189, 180–185. [Google Scholar] [CrossRef]
- Shi, X.; Bai, H.; Wang, J.; Wang, J.; Huang, L.; He, M.; Zheng, X.; Duan, Z.; Chen, D.; Zhang, J.; et al. Behavioral Assessment of Sensory, Motor, Emotion, and Cognition in Rodent Models of Intracerebral Hemorrhage. Front. Neurol. 2021, 12, 667511. [Google Scholar] [CrossRef]
- de Brito, A.F.; Martins JL, R.; Fajemiroye, J.O.; Galdino, P.M.; De Lima TC, M.; Menegatti, R.; Costa, E.A. Central pharmacological activity of a new piperazine derivative: 4-(1-Phenyl-1h-pyrazol-4-ylmethyl)-piperazine-1-carboxylic acid ethyl ester. J. Life Sci. 2012, 90, 910–916. [Google Scholar] [CrossRef]
- Jones, B.J.; Roberts, D.J. A rotarod suitable for quantitative measurements of motor incoordination in naive mice. Naunyn Schmiedebergs Arch. Exp. Pathol. Pharmakol. 1968, 259, 211. [Google Scholar] [CrossRef]
- Carlini, E.; Burgos, V. Screening farmacológico de ansiolíticos: Metodologia laboratorial e comparação entre o diazepam e o clorobenzapam. Rev. Bras. Psiquiatr. 1979, 1, 25–31. [Google Scholar]
- Mesdaghinia, A.; Alinejad, M.; Abed, A.; Heydari, A.; Banafshe, H.R. Anticonvulsant effects of thiamine on pentylenetetrazole-induced seizure in mice. Nutr. Neurosci. 2019, 22, 165–173. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Form I | Form II | Form III | Form IV | Form V | |
|---|---|---|---|---|---|---|
| Chemical formula | C15H12N2O | C15H12N2O | C15H12N2O | C15H12N2O | C15H12N2O | |
| Crystal system | Triclinic | Trigonal | Monoclinic | Monoclinic | Orthorhombic | |
| Space group | P | R | P21/n | C2/c | Pbca | |
| Z, Z′ | 8, 4 | 18, 1 | 4, 1 | 8, 1 | 8, 1 | |
| Unit cell parameters | a (Å) | 5.1705 (6) | 35.454 (3) | 7.5500 (16) | 26.609 (4) | 9.1245 (5) |
| b (Å) | 20.574 (2) | 35.454 (3) | 11.186 (3) | 6.9269 (10) | 10.4518 (5) | |
| c (Å) | 22.245 (2) | 5.253 (1) | 13.954 (3) | 13.957 (2) | 24.8224 (11) | |
| α (°) | 84.124 (4) | 90 | 90.00 | 90.00 | 90.00 | |
| β (°) | 88.008 (4) | 90 | 92.938 (8) | 109.702 (2) | 90.00 | |
| γ (°) | 85.187 (4) | 120 | 90.00 | 90.00 | 90.00 | |
| Volume cell (Å3) | 2344.8 (5) | 5718.32 | 1176.9 (5) | 2421.9 (6) | 2367.2 (2) | |
| Crystalline Form | ) | d (N-H…O) (°) | ||
|---|---|---|---|---|
| Form I | 0.908 | 1.978 | 2.864 | 176.93 |
| Form II | - | - | 2.890 | - |
| Form III | 0.862 | 2.075 | 2.937 | 176.77 |
| Form IV | 0.928 | 1.943 | 2.847 | 177.55 |
| Form V | - | - | - | - |
| Crystalline Form | H…H | C…H | O…H | C…C | Others |
|---|---|---|---|---|---|
| Form I | 47.5% | 34.3% | 13.7% | 1.8% | 2.7% |
| Form II | - | - | - | - | - |
| Form III | 53.7% | 22.6% | 13.3% | 8.2% | 2.2% |
| Form IV | 50.6% | 29.5% | 13.2% | 4.6% | 2.7% |
| Form V | 48% | 35.7% | 13.% | 0.8% | 2.5% |
| Table A | VEH + VEH | VEH + FLU | VEH + OXC | FLU + OXC | VEH + CBZ | FLU + CBZ | VEH + CBZ-F | FLU + CBZ-F | VEH + TOP | FLU + TOP |
|---|---|---|---|---|---|---|---|---|---|---|
| VEH + VEH | - | p = 0.999 | p = 0.300 | p = 0.001 | p = 0.0001 | p = 0.0001 | p = 0.0001 | p = 0.0001 | p = 0.0001 | p = 0.011 |
| VEH + FLU | - | p = 0.999 | p = 0.021 | p = 0.0001 | p = 0.0001 | p = 0.0001 | p = 0.0001 | p = 0.0001 | p = 0.107 | |
| VEH + OXC | - | p = 0.999 | p = 0.0008 | p = 0.173 | p = 0.0001 | p = 0.0001 | p = 0.0041 | p = 0.999 | ||
| FLU + OXC | - | p = 0.1532 | p = 0.999 | p = 0.0001 | p = 0.0098 | p = 0.5386 | p = 0.999 | |||
| VEH + CBZ | - | p = 0.999 | p = 0.1737 | p = 0.999 | p = 0.999 | p = 0.0311 | ||||
| FLU + CBZ | - | p = 0.0008 | p = 0.7944 | p = 0.999 | p = 0.999 | |||||
| VEH + CBZ-F | - | p = 0.999 | p = 0.0439 | p = 0.0001 | ||||||
| FLU + CBZ-F | - | p = 0.999 | p = 0.0016 | |||||||
| VEH + TOP | - | p = 0.1267 | ||||||||
| FLU + TOP | - | |||||||||
| Table B | ||||||||||
| VEH + VEH | - | p = 0.999 | p = 0.0118 | p = 0.191 | p = 0.051 | p = 0.2994 | p = 0.0001 | p = 0.0003 | p = 0.0001 | p = 0.999 |
| VEH + FLU | - | p = 0.0240 | p = 0.353 | p = 0.0984 | p = 0.540 | p = 0.0001 | p = 0.0008 | p = 0.0001 | p = 0.999 | |
| VEH + OXC | - | p = 0.999 | p = 0.999 | p = 0.999 | p = 0.999 | p = 0.999 | p = 0.0073 | p = 0.999 | ||
| FLU + OXC | - | p = 0.999 | p = 0.999 | p = 0.424 | p = 0.999 | p = 0.0003 | p = 0.999 | |||
| VEH + CBZ | - | p = 0.999 | p = 0.999 | p = 0.999 | p = 0.0015 | p = 0.999 | ||||
| FLU + CBZ | - | p = 0.275 | p = 0.999 | p = 0.0001 | p = 0.999 | |||||
| VEH + CBZ-F | - | p = 0.999 | p = 0.999 | p = 0.0314 | ||||||
| FLU + CBZ-F | - | p = 0.162 | p = 0.399 | |||||||
| VEH + TOP | - | p = 0.0001 | ||||||||
| FLU + TOP | - |
| Group | Pharmacological Doses and Routes |
|---|---|
| 1 | VEH (10 mL/kg, i.p) + VEH (10 mL/kg, p.o) |
| 2 | VEH (10 mL/kg, i.p) + CBZ (5 mg/kg, p.o) |
| 3 | FLU (2 mg/kg, i.p) + CBZ (5 mg/kg, p.o) |
| 4 | VEH (10 mL/kg, i.p) + FLU (2 mg/kg, p.o) |
| 5 | VEH (10 mL/kg, i.p) + OXC (5 mg/kg, p.o) |
| 6 | FLU (2 mg/kg, i.p) + OXC (5 mg/kg, p.o) |
| 7 | VEH (10 mL/kg, i.p) + CBZ-F (5 mg/kg, p.o) |
| 8 | FLU (2 mg/kg, i.p) + CBZ-F (5 mg/kg, p.o) |
| 9 | VEH (10 mL/kg, i.p) + TOP (5 mg/kg, p.o) |
| 10 | FLU (2 mg/kg, i.p) + TOP (5 mg/kg, p.o) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sá Filho, A.; Martins, J.L.R.; Costa, R.F.; Pedrino, G.R.; Duarte, V.S.; Silva, O.N.; Napolitano, H.B.; Fajemiroye, J.O. Polymorphism and Pharmacological Assessment of Carbamazepine. Int. J. Mol. Sci. 2024, 25, 9835. https://doi.org/10.3390/ijms25189835
Sá Filho A, Martins JLR, Costa RF, Pedrino GR, Duarte VS, Silva ON, Napolitano HB, Fajemiroye JO. Polymorphism and Pharmacological Assessment of Carbamazepine. International Journal of Molecular Sciences. 2024; 25(18):9835. https://doi.org/10.3390/ijms25189835
Chicago/Turabian StyleSá Filho, Alberto, Jose Luis Rodrigues Martins, Rafael Fernandes Costa, Gustavo Rodrigues Pedrino, Vitor Santos Duarte, Osmar Nascimento Silva, Hamilton Barbosa Napolitano, and James Oluwagbamigbe Fajemiroye. 2024. "Polymorphism and Pharmacological Assessment of Carbamazepine" International Journal of Molecular Sciences 25, no. 18: 9835. https://doi.org/10.3390/ijms25189835