
Network Analysis of Enhancer–Promoter Interactions Highlights Cell-Type-Specific Mechanisms of Transcriptional Regulation Variation

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
2.1. Generation of Enhancer Promoter Interactions Network
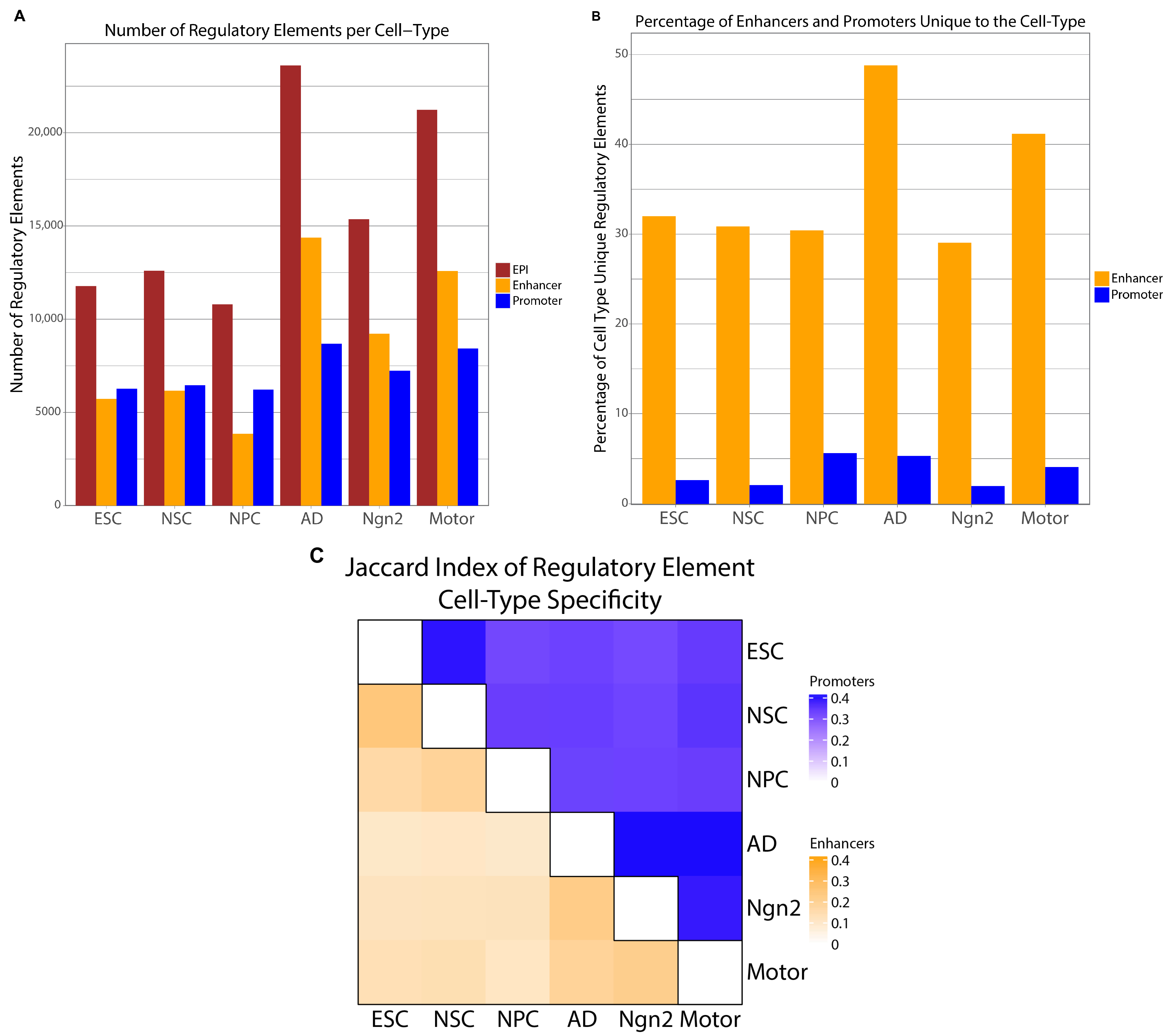
2.2. Experimental Data Validate EPIs Cell-Type Specificity
2.3. Cell-Type Specificity of EPIs Is Driven More by Enhancers than Promoters
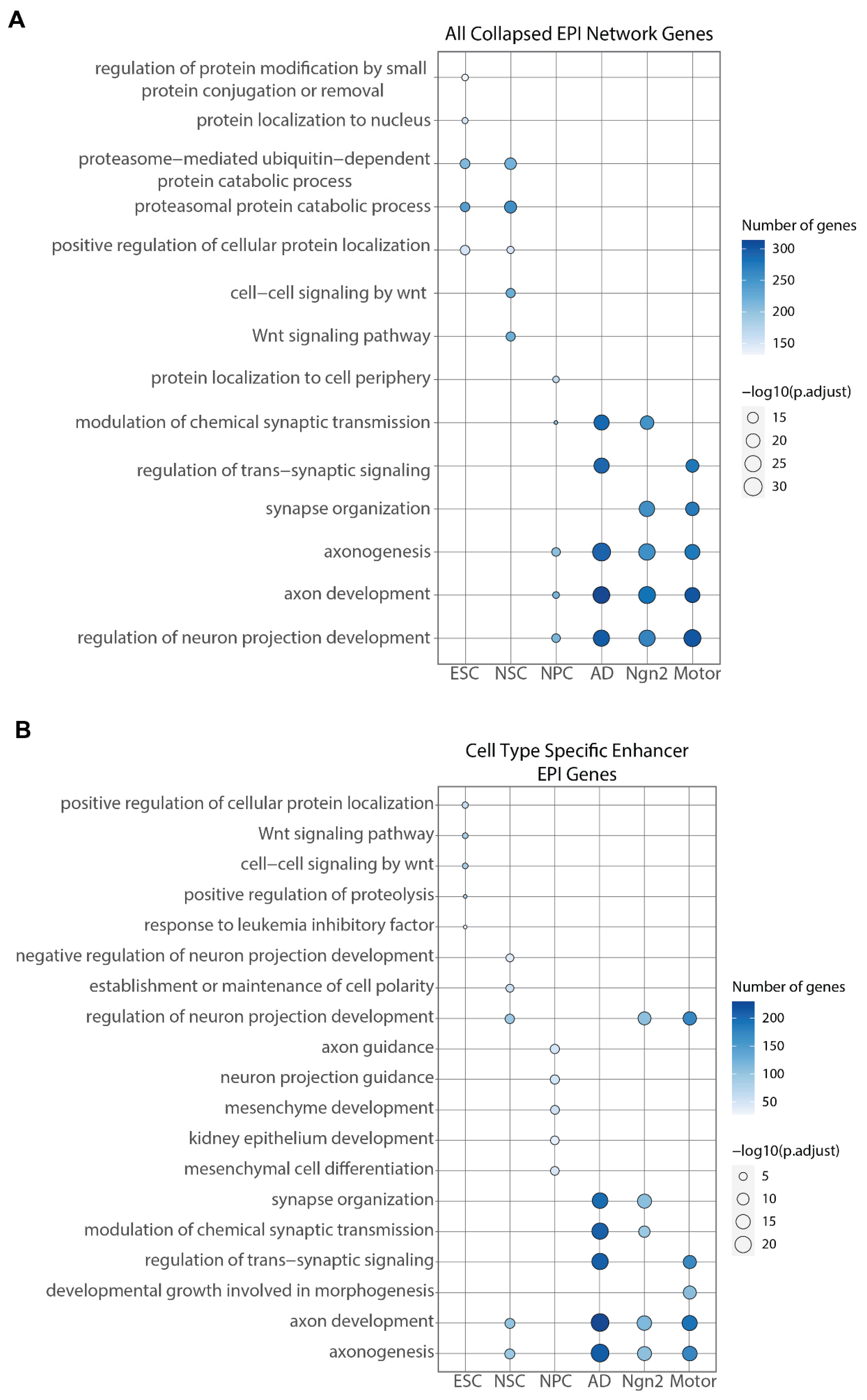
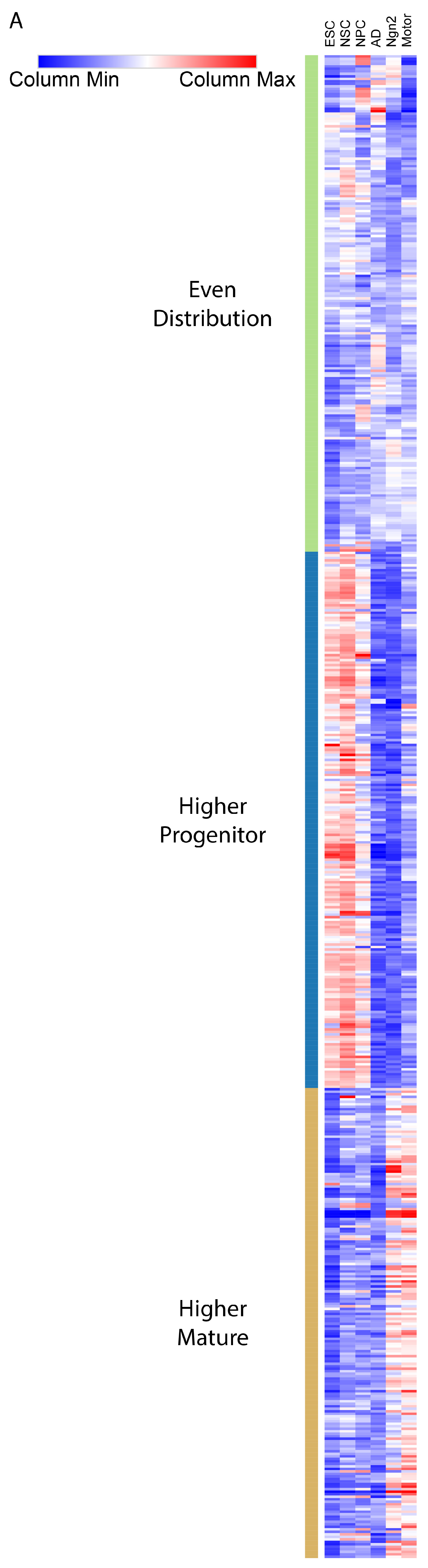
2.4. Transcription Factor Binding Presence Distinguishes between Differentiation Stages
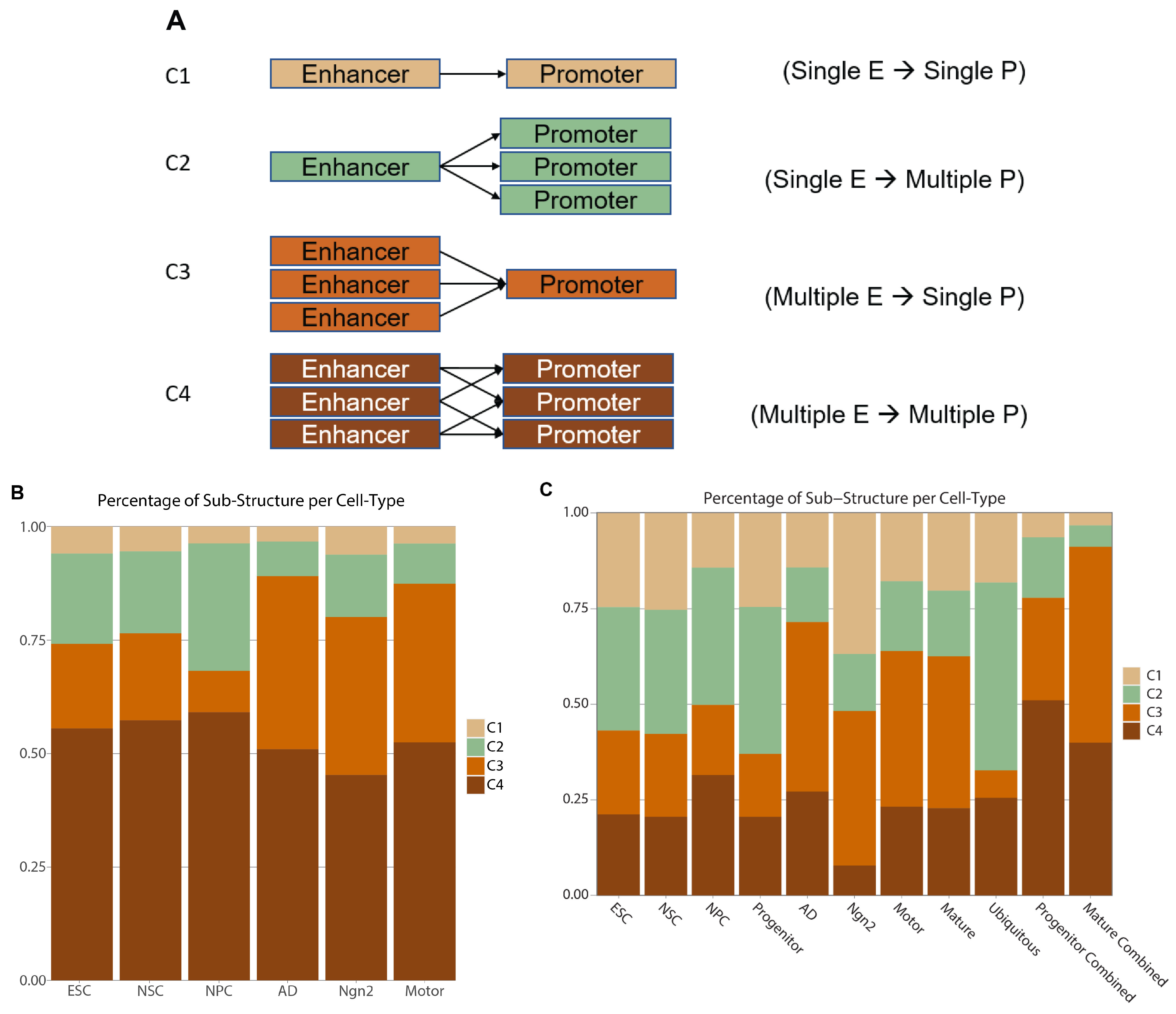
2.5. Characterization of Enhancer–Promoter Interactions Architecture Reveals Cell-Type-Specific Regulatory Mechanisms
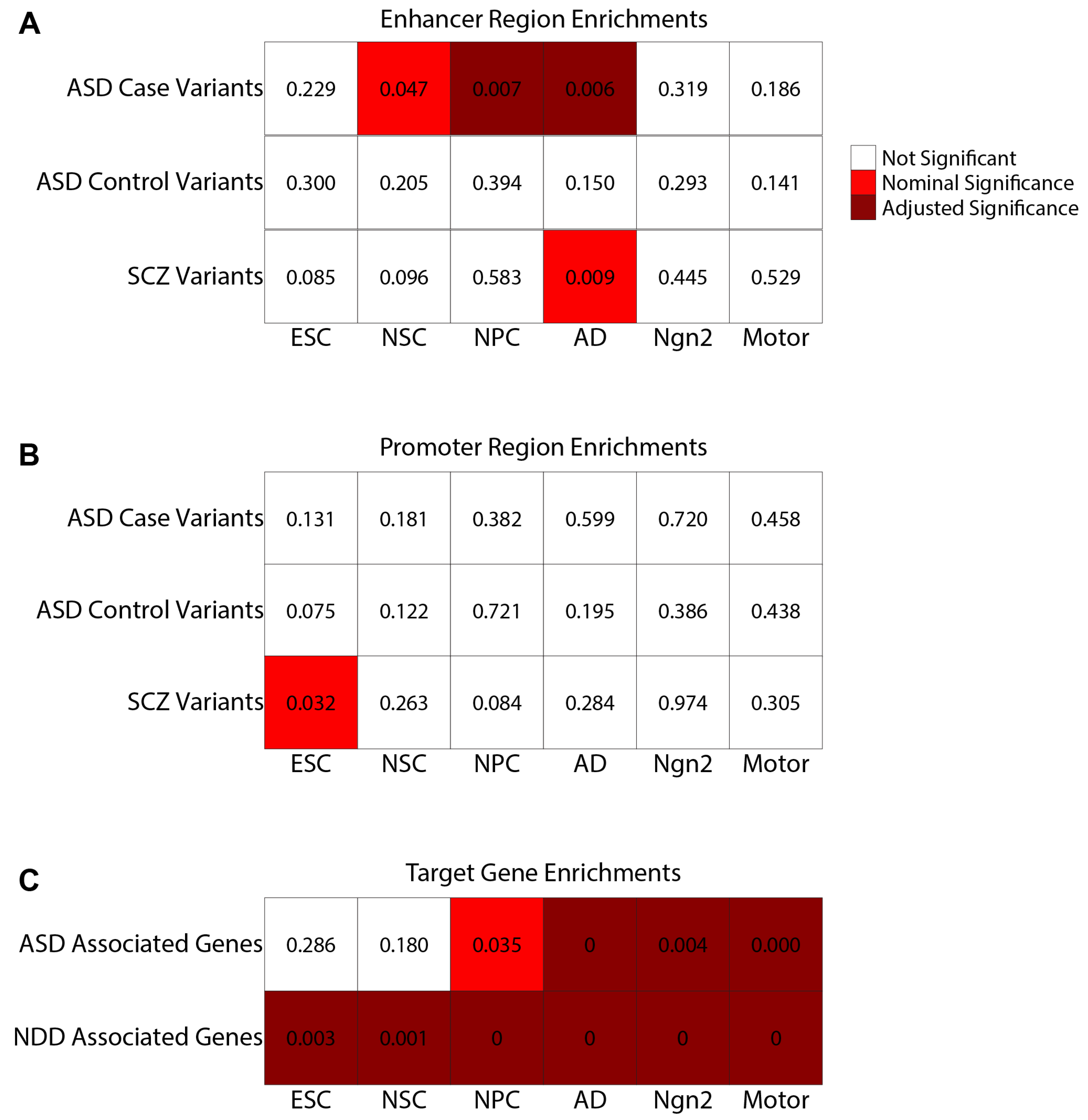
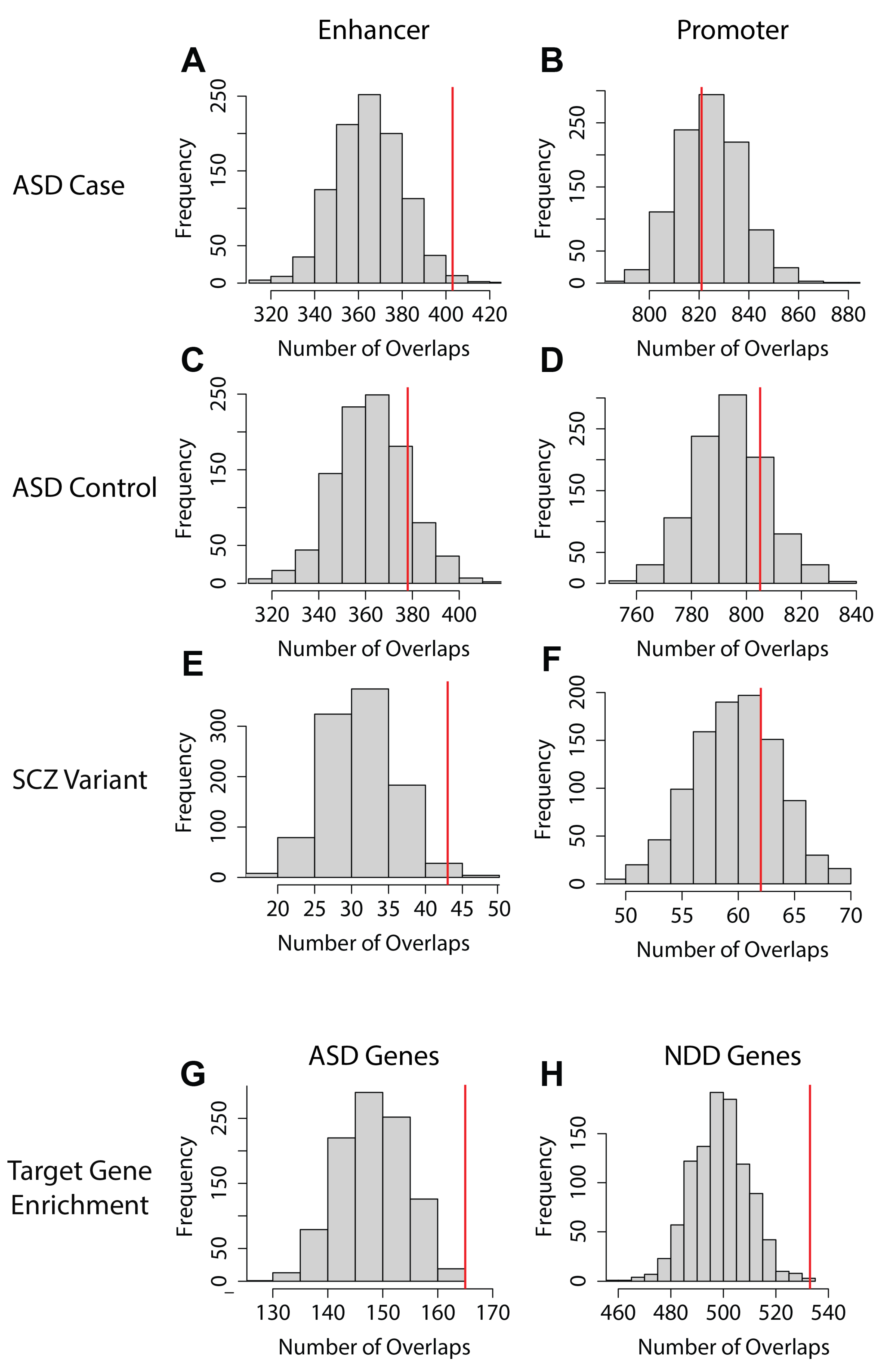
2.6. EPIs Can Be Utilized to Prioritize Potential Disease-Associated Variants
3. Discussion
4. Methods
4.1. Review of Terminologies Used in This Study
- Activity-By-Contact model (ABC model): The principle of the ABC model is that an enhancer, which is accessible, highly active, and has a high contact frequency with a promoter, is also likely to have a regulatory impact on that promoter’s gene.
- Activity-By-Contact score (ABC score) is utilized to describe the activity of the EPI. To calculate the scores, the necessary inputs are to replicate separated ATAC-Seq peak files, replicate merged H3K27ac peak files, and the RNA-Seq-processed TPM table file (described in the following Section). The formula of ABC score is the activity of the enhancer, defined by the read counts of the ATAC-seq and H3K27ac peaks, multiplied by the contact frequency between the enhancer and target-gene promoter, defined by the Hi-C contact frequency or estimated distance frequency, divided by the sum of all enhancer scores between the target gene promoter and enhancers within 5 mega-bases of the target gene promoter.
- The Enhancer–Promoter Interaction, abbreviated as EPI, is defined as “interactions between cis-acting enhancer and promoter that facilitate enhancer-mediated upregulation of gene transcription”. In our study, an active EPI is confirmed when its Activity-By-Contact score (described below) is greater than the threshold 0.02 and the enhancer is within 5 mega-bases of the target-gene promoter.
- Collapsed EPI network: To investigate EPIs identified in different cell types, we concatenated the 6 cell-type EPI networks into a single collapsed EPI network without additional filtering to investigate differences between cell-type EPIs and as a pooled resource to select from for the empirical analysis of overlapping potential disease-associated variants and genes. Further details regarding the generation of the EPI network and the empirical analysis are described below.
- Subset EPI: To study more cell-type-specific EPIs, we subset the EPIs that were initially identified in this study. Briefly, the criteria for sub-setting are based on ABC scores in specific combinations of cell types such as only one cell type, at least two progenitors and no mature neurons or two mature neurons and no progenitor cell types, or ubiquitously across most cell types. Details regarding sub-setting are described in the following Sections.
4.2. Dataset Overview
4.3. Processing of ATAC-Seq
4.4. Processing of ChIP-Seq and CUT&RUN Data
4.5. Description of Cell-Type-Specific Hi-C Data
4.6. Description of Cell-Type-Specific RNA-Seq Data
4.7. Predicting Enhancer–Promoter Interactions Using the Activity-by-Contact Model
4.8. Generation of the Collapsed EPI Network
4.9. Generation of Subset EPI Network
4.10. Fisher’s Exact Test of Enriched Overlap of Cell-Type-Predicted Active EPI with Log-Fold Change
4.11. Controlling for Batch Effects across Studies
4.12. Identification of Enhancers and Genes Predicted Active across Different Cell Types
4.13. Gene Ontology (GO) Enrichment
4.14. Identifying Sub-Structures of EPIs
4.15. Prediction of TFBSs Using FIMO
4.16. Clustering TFs Based on Gene Expression
4.17. Identifying the Presence of TFs in Different Enhancer Regions
4.18. Selection of Disease-Associated Variants for Empirical Analysis
4.19. Generation of Regulatory Element Regions for Empirical Analysis
4.20. Empirical Analysis of Overlap
4.21. Code Availability Statement
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, F.; Lupski, J.R. Non-coding genetic variants in human disease. Hum. Mol. Genet. 2015, 24, R102–R110. [Google Scholar] [CrossRef] [PubMed]
- Akbarian, S.; Liu, C.; Knowles, J.A.; Vaccarino, F.M.; Farnham, P.J.; Crawford, G.E.; Jaffe, A.E.; Pinto, D.; Dracheva, S.; Geschwind, D.H.; et al. The PsychENCODE project. Nat. Neurosci. 2015, 18, 1707–1712. [Google Scholar] [CrossRef] [PubMed]
- Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; Ziller, M.J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Sloutskin, A.; Shir-Shapira, H.; Freiman, R.N.; Juven-Gershon, T. The Core Promoter Is a Regulatory Hub for Developmental Gene Expression. Front. Cell Dev. Biol. 2021, 9, 666508. [Google Scholar] [CrossRef] [PubMed]
- Furlong, E.E.M.; Levine, M. Developmental enhancers and chromosome topology. Science 2018, 361, 1341–1345. [Google Scholar] [CrossRef]
- Zuin, J.; Roth, G.; Zhan, Y.; Cramard, J.; Redolfi, J.; Piskadlo, E.; Mach, P.; Kryzhanovska, M.; Tihanyi, G.; Kohler, H.; et al. Nonlinear control of transcription through enhancer-promoter interactions. Nature 2022, 604, 571–577. [Google Scholar] [CrossRef]
- Fulco, C.P.; Nasser, J.; Jones, T.R.; Munson, G.; Bergman, D.T.; Subramanian, V.; Grossman, S.R.; Anyoha, R.; Doughty, B.R.; Patwardhan, T.A.; et al. Activity-by-contact model of enhancer–promoter regulation from thousands of CRISPR perturbations. Nat. Genet. 2019, 51, 1664–1669. [Google Scholar] [CrossRef]
- Heintzman, N.D.; Hon, G.C.; Hawkins, R.D.; Kheradpour, P.; Stark, A.; Harp, L.F.; Ye, Z.; Lee, L.K.; Stuart, R.K.; Ching, C.W.; et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009, 459, 108–112. [Google Scholar] [CrossRef]
- Heinz, S.; Romanoski, C.E.; Benner, C.; Glass, C.K. The selection and function of cell type-specific enhancers. Nat. Rev. Mol. Cell Biol. 2015, 16, 144–154. [Google Scholar] [CrossRef]
- Song, M.; Yang, X.; Ren, X.; Maliskova, L.; Li, B.; Jones, I.R.; Wang, C.; Jacob, F.; Wu, K.; Traglia, M.; et al. Mapping cis-regulatory chromatin contacts in neural cells links neuropsychiatric disorder risk variants to target genes. Nat. Genet. 2019, 51, 1252–1262. [Google Scholar] [CrossRef]
- Zaugg, J.B.; Sahlén, P.; Andersson, R.; Alberich-Jorda, M.; de Laat, W.; Deplancke, B.; Ferrer, J.; Mandrup, S.; Natoli, G.; Plewczynski, D.; et al. Current challenges in understanding the role of enhancers in disease. Nat. Struct. Mol. Biol. 2022, 29, 1148–1158. [Google Scholar] [CrossRef] [PubMed]
- Grandi, F.C.; Modi, H.; Kampman, L.; Corces, M.R. Chromatin accessibility profiling by ATAC-seq. Nat. Protoc. 2022, 17, 1518–1552. [Google Scholar] [CrossRef] [PubMed]
- Thurman, R.E.; Rynes, E.; Humbert, R.; Vierstra, J.; Maurano, M.T.; Haugen, E.; Sheffield, N.C.; Stergachis, A.B.; Wang, H.; Vernot, B.; et al. The accessible chromatin landscape of the human genome. Nature 2012, 489, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Kreimer, A.; Ashuach, T.; Inoue, F.; Khodaverdian, A.; Deng, C.; Yosef, N.; Ahituv, N. Massively parallel reporter perturbation assays uncover temporal regulatory architecture during neural differentiation. Nat. Commun. 2022, 13, 1504. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Priego, C.; Hu, R.; Boshans, L.L.; Lalli, M.; Janas, J.A.; Williams, S.E.; Dong, Z.; Yang, N. Mapping cis-regulatory elements in human neurons links psychiatric disease heritability and activity-regulated transcriptional programs. Cell Rep. 2022, 39, 110877. [Google Scholar] [CrossRef] [PubMed]
- Whalen, S.; Inoue, F.; Ryu, H.; Fair, T.; Markenscoff-Papadimitriou, E.; Keough, K.; Kircher, M.; Martin, B.; Alvarado, B.; Elor, O.; et al. Machine learning dissection of human accelerated regions in primate neurodevelopment. Neuron 2023, 111, 857–873.e858. [Google Scholar] [CrossRef] [PubMed]
- Nott, A.; Holtman, I.R.; Coufal, N.G.; Schlachetzki, J.C.M.; Yu, M.; Hu, R.; Han, C.Z.; Pena, M.; Xiao, J.; Wu, Y.; et al. Brain cell type-specific enhancer-promoter interactome maps and disease-risk association. Science 2019, 366, 1134–1139. [Google Scholar] [CrossRef]
- Hait, T.A.; Amar, D.; Shamir, R.; Elkon, R. FOCS: A novel method for analyzing enhancer and gene activity patterns infers an extensive enhancer–promoter map. Genome Biol. 2018, 19, 56. [Google Scholar] [CrossRef]
- Mulvey, B.; Lagunas, T., Jr.; Dougherty, J.D. Massively Parallel Reporter Assays: Defining Functional Psychiatric Genetic Variants Across Biological Contexts. Biol. Psychiatry 2021, 89, 76–89. [Google Scholar] [CrossRef]
- Rao, S.S.P.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based Analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed]
- Inoue, F.; Kreimer, A.; Ashuach, T.; Ahituv, N.; Yosef, N. Identification and Massively Parallel Characterization of Regulatory Elements Driving Neural Induction. Cell Stem Cell 2019, 25, 713–727.e710. [Google Scholar] [CrossRef] [PubMed]
- Chung, N.C.; Miasojedow, B.; Startek, M.; Gambin, A. Jaccard/Tanimoto similarity test and estimation methods for biological presence-absence data. BMC Bioinform. 2019, 20, 644. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Liao, Y.; Qiu, M.; Shen, W. Wnt/β-Catenin Signaling in Neural Stem Cell Homeostasis and Neurological Diseases. Neuroscientist 2021, 27, 58–72. [Google Scholar] [CrossRef]
- Maston, G.A.; Landt, S.G.; Snyder, M.; Green, M.R. Characterization of Enhancer Function from Genome-Wide Analyses. Annu. Rev. Genom. Hum. Genet. 2012, 13, 29–57. [Google Scholar] [CrossRef]
- Levo, M.; Segal, E. In pursuit of design principles of regulatory sequences. Nat. Rev. Genet. 2014, 15, 453–468. [Google Scholar] [CrossRef]
- Grant, C.E.; Bailey, T.L.; Noble, W.S. FIMO: Scanning for occurrences of a given motif. Bioinformatics 2011, 27, 1017–1018. [Google Scholar] [CrossRef]
- Houbaviy, H.B.; Murray, M.F.; Sharp, P.A. Embryonic Stem Cell-Specific MicroRNAs. Dev. Cell 2003, 5, 351–358. [Google Scholar] [CrossRef]
- Huang, J.; Liu, X.; Li, D.; Shao, Z.; Cao, H.; Zhang, Y.; Trompouki, E.; Bowman, T.V.; Zon, L.I.; Yuan, G.C.; et al. Dynamic Control of Enhancer Repertoires Drives Lineage and Stage-Specific Transcription during Hematopoiesis. Dev. Cell 2016, 36, 9–23. [Google Scholar] [CrossRef]
- An, J.-Y.; Lin, K.; Zhu, L.; Werling, D.M.; Dong, S.; Brand, H.; Wang, H.Z.; Zhao, X.; Schwartz, G.B.; Collins, R.L.; et al. Genome-wide de novo risk score implicates promoter variation in autism spectrum disorder. Science 2018, 362, eaat6576. [Google Scholar] [CrossRef] [PubMed]
- Koesterich, J.; An, J.Y.; Inoue, F.; Sohota, A.; Ahituv, N.; Sanders, S.J.; Kreimer, A. Characterization of De Novo Promoter Variants in Autism Spectrum Disorder with Massively Parallel Reporter Assays. Int. J. Mol. Sci. 2023, 24, 3509. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.M.; Satterstrom, F.K.; Peng, M.; Brand, H.; Collins, R.L.; Dong, S.; Wamsley, B.; Klei, L.; Wang, L.; Hao, S.P.; et al. Rare coding variation provides insight into the genetic architecture and phenotypic context of autism. Nat. Genet. 2022, 54, 1320–1331. [Google Scholar] [CrossRef] [PubMed]
- Pantelis, C.; Papadimitriou, G.N.; Papiol, S.; Parkhomenko, E.; Pato, M.T.; Paunio, T.; Pejovic-Milovancevic, M.; Perkins, D.O.; Pietiläinen, O.; Pimm, J. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421–427. [Google Scholar]
- Notaras, M.; Lodhi, A.; Dündar, F.; Collier, P.; Sayles, N.M.; Tilgner, H.; Greening, D.; Colak, D. Schizophrenia is defined by cell-specific neuropathology and multiple neurodevelopmental mechanisms in patient-derived cerebral organoids. Mol. Psychiatry 2022, 27, 1416–1434. [Google Scholar] [CrossRef]
- Volk, D.W.; Pierri, J.N.; Fritschy, J.-M.; Auh, S.; Sampson, A.R.; Lewis, D.A. Reciprocal alterations in pre- and postsynaptic inhibitory markers at chandelier cell inputs to pyramidal neurons in schizophrenia. Cereb. Cortex 2002, 12, 1063–1070. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Amemiya, H.M.; Kundaje, A.; Boyle, A.P. The ENCODE Blacklist: Identification of Problematic Regions of the Genome. Sci. Rep. 2019, 9, 9354. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.11–11.10.33. [Google Scholar] [CrossRef]
- Zhu, Q.; Liu, N.; Orkin, S.H.; Yuan, G.-C. CUT&RUNTools: A flexible pipeline for CUT&RUN processing and footprint analysis. Genome Biol. 2019, 20, 192. [Google Scholar]
- Quinlan, A.R. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr. Protoc. Bioinform. 2014, 47, 11.12.1–11.12.34. [Google Scholar] [CrossRef] [PubMed]
- Morpheus. Available online: https://software.broadinstitute.org/morpheus (accessed on 6 June 2023).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | Labeling in Original Publication | Data Source |
|---|---|---|
| Embryonic stem cells (ESC) | 0 h | GSE115046 |
| Neural stem cells (NSC) | 72 h | |
| Neural Progenitor Cell (NPC) | N2 | GSE110758 |
| GABAergic Neurons | AD | GSE196856 |
| Glutamatergic Neurons | Ngn2 | |
| Motor Neurons | Motor | GSE113483 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koesterich, J.; Liu, J.; Williams, S.E.; Yang, N.; Kreimer, A. Network Analysis of Enhancer–Promoter Interactions Highlights Cell-Type-Specific Mechanisms of Transcriptional Regulation Variation. Int. J. Mol. Sci. 2024, 25, 9840. https://doi.org/10.3390/ijms25189840
Koesterich J, Liu J, Williams SE, Yang N, Kreimer A. Network Analysis of Enhancer–Promoter Interactions Highlights Cell-Type-Specific Mechanisms of Transcriptional Regulation Variation. International Journal of Molecular Sciences. 2024; 25(18):9840. https://doi.org/10.3390/ijms25189840
Chicago/Turabian StyleKoesterich, Justin, Jiayi Liu, Sarah E. Williams, Nan Yang, and Anat Kreimer. 2024. "Network Analysis of Enhancer–Promoter Interactions Highlights Cell-Type-Specific Mechanisms of Transcriptional Regulation Variation" International Journal of Molecular Sciences 25, no. 18: 9840. https://doi.org/10.3390/ijms25189840
APA StyleKoesterich, J., Liu, J., Williams, S. E., Yang, N., & Kreimer, A. (2024). Network Analysis of Enhancer–Promoter Interactions Highlights Cell-Type-Specific Mechanisms of Transcriptional Regulation Variation. International Journal of Molecular Sciences, 25(18), 9840. https://doi.org/10.3390/ijms25189840