
CD4+ Effective Memory T Cell Markers GBP2 and LAG3 Are Risk Factors for PTB and COVID-19 Infection: A Study Integrating Single-Cell Expression Quantitative Trait Locus and Mendelian Randomization Analyses

 , , and
, , and
Abstract
:1. Introduction
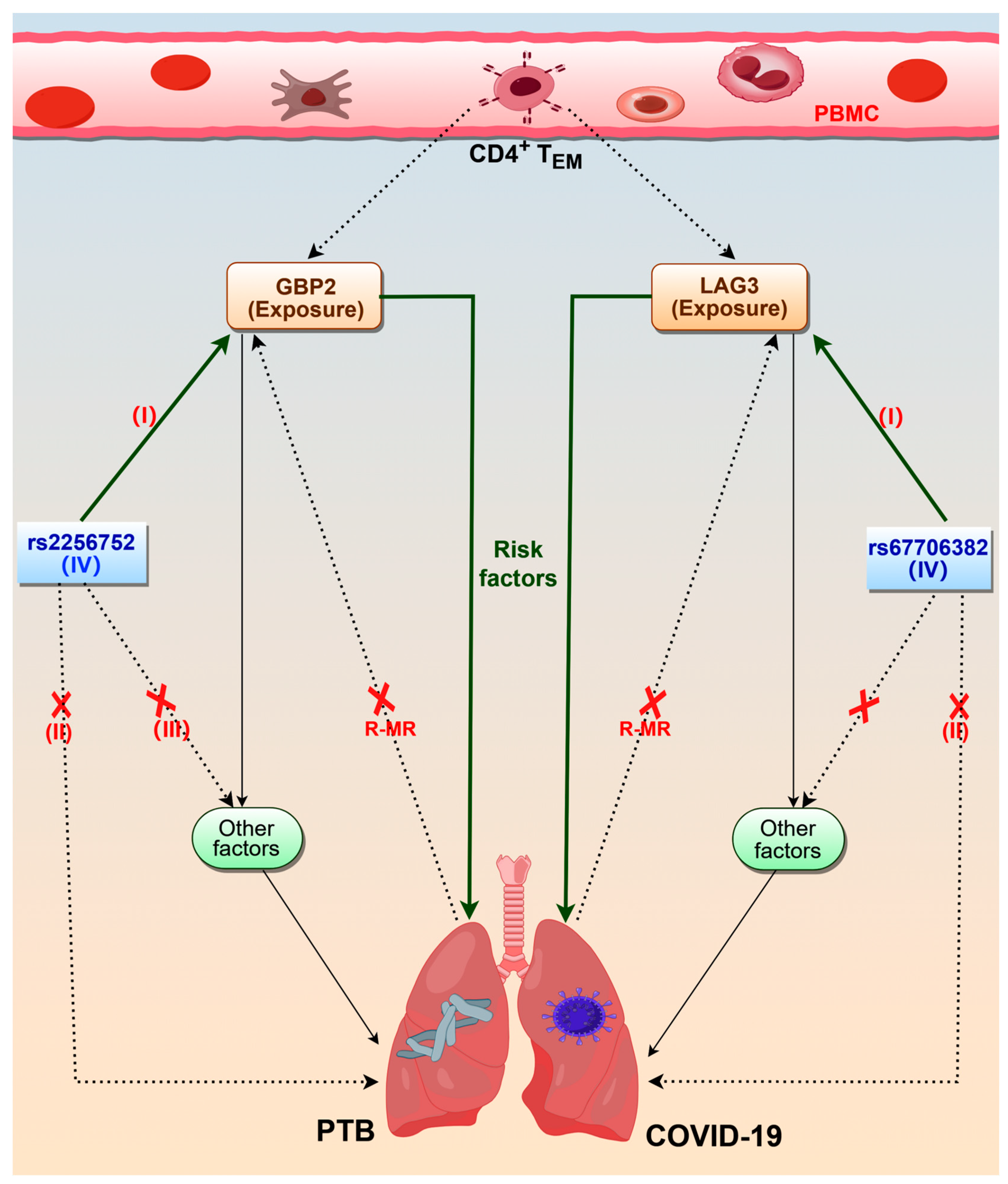
2. Results
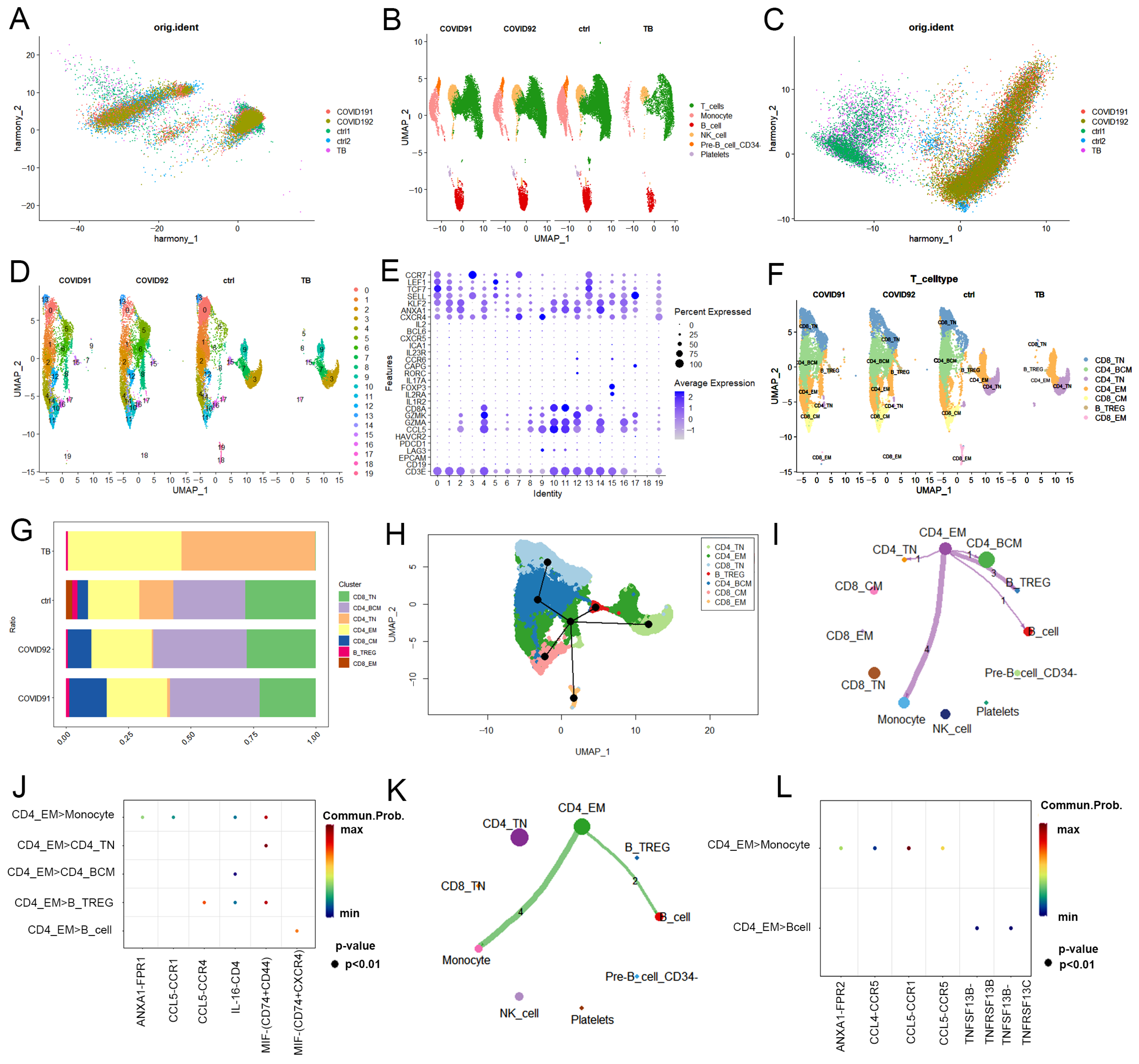
2.1. Single-Cell Transcriptional Landscape of PBMCs from PTB and COVID-19 Patients
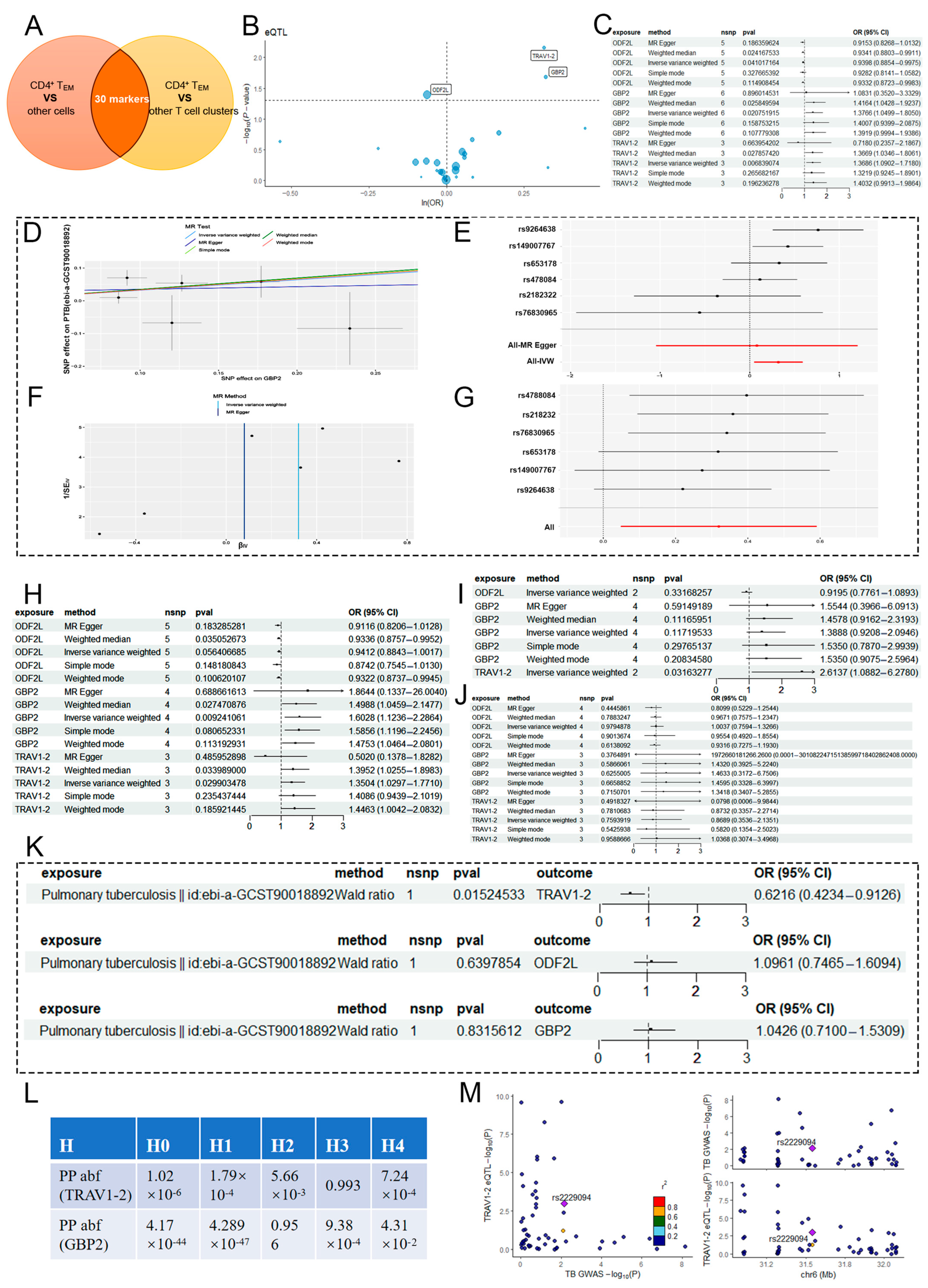
2.2. MR Analysis of CD4+ TEM Cluster Markers and PTB
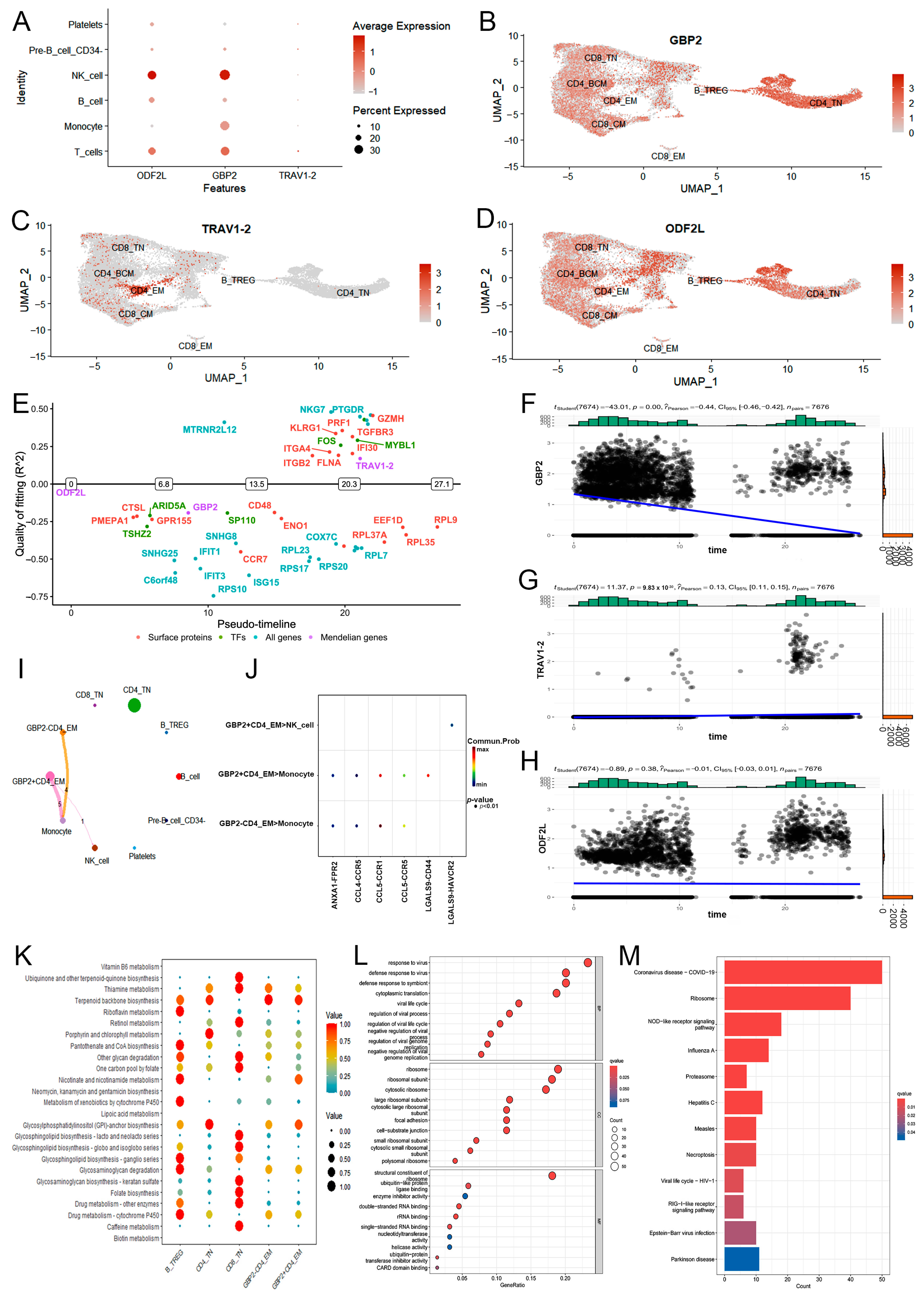
2.3. Downstream Function of CD4+ TEM Cluster Core Markers in PTB
2.4. MR Analysis of CD4+ TEM Cluster Markers and COVID-19
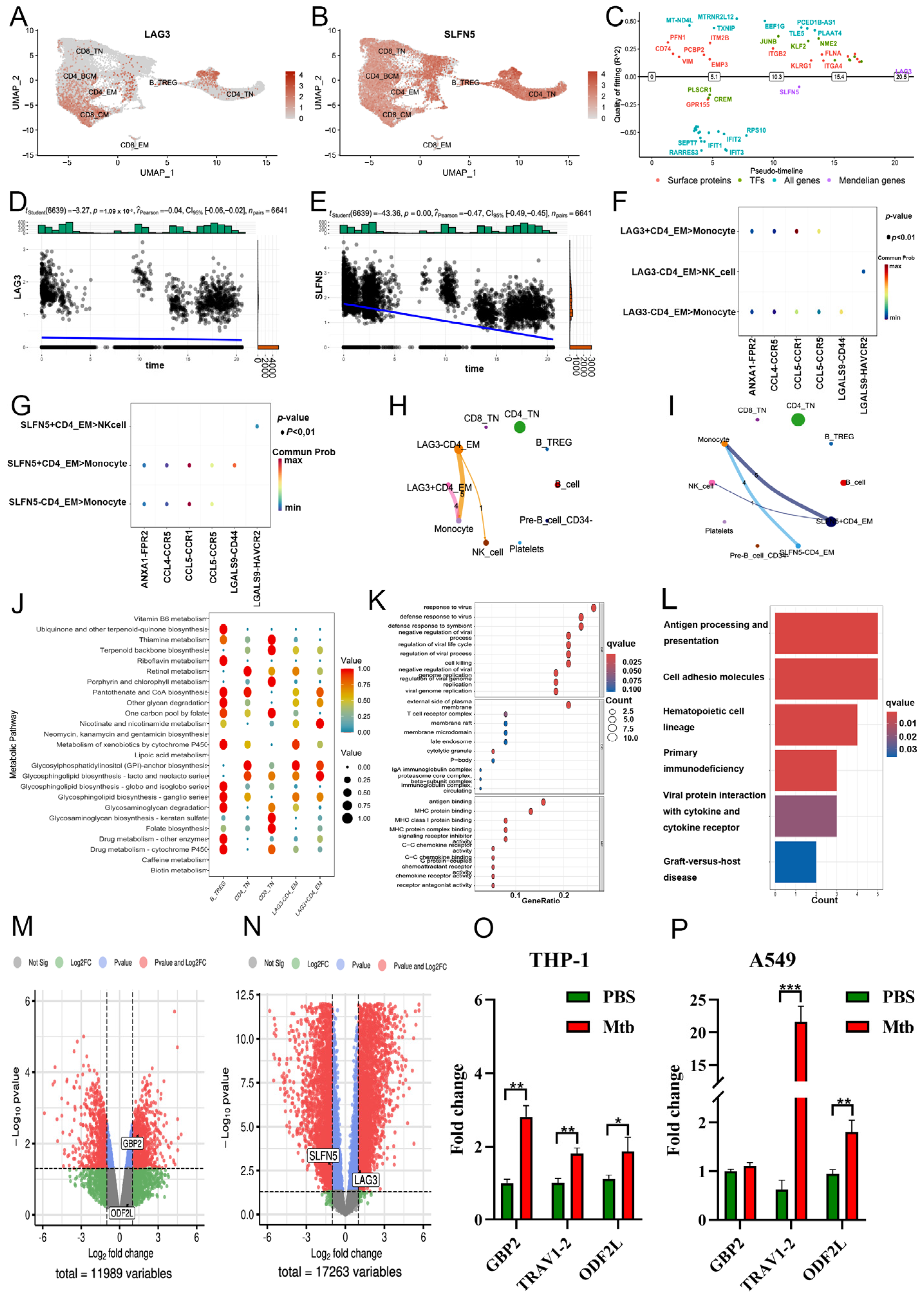
2.5. Downstream Function of CD4+ TEM Cluster’s Core Markers in COVID-19
3. Discussion
4. Materials and Methods
4.1. Study Overview
4.2. scRNA-Seq Data Processing
4.3. scRNA-Seq Data-Based Functional Analyses
4.4. Instrumental Variable (IV) Screen
4.5. MR Analyses
4.6. R-MR Analyses
4.7. Colocalization Analysis
4.8. Cell Culture and PCR Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Chai, Q.; Lu, Z.; Liu, C.H. Host defense mechanisms against Mycobacterium tuberculosis. Cell Mol. Life Sci. 2020, 77, 1859–1878. [Google Scholar] [CrossRef] [PubMed]
- Bagcchi, S. WHO’s Global Tuberculosis Report 2022. Lancet Microbe 2023, 4, e20. [Google Scholar] [CrossRef] [PubMed]
- Parker, A.; Boloko, L.; Moolla, M.S.; Ebrahim, N.; Ayele, B.T.; Broadhurst, A.G.B.; Mashigo, B.; Titus, G.; de Wet, T.; Boliter, N.; et al. Clinical features and outcomes of COVID-19 admissions in a population with a high prevalence of HIV and tuberculosis: A multicentre cohort study. BMC Infect. Dis. 2022, 22, 559. [Google Scholar] [CrossRef] [PubMed]
- Crisan-Dabija, R.; Grigorescu, C.; Pavel, C.A.; Artene, B.; Popa, I.V.; Cernomaz, A.; Burlacu, A. Tuberculosis and COVID-19: Lessons from the Past Viral Outbreaks and Possible Future Outcomes. Can. Respir. 2020, 2020, 1401053. [Google Scholar] [CrossRef]
- Freund, O.; Azolai, L.; Sror, N.; Zeeman, I.; Kozlovsky, T.; Greenberg, S.A.; Epstein Weiss, T.; Bornstein, G.; Tchebiner, J.Z.; Frydman, S. Diagnostic delays among COVID-19 patients with a second concurrent diagnosis. J. Hosp. Med. 2023, 18, 321–328. [Google Scholar] [CrossRef]
- Toor, S.M.; Saleh, R.; Sasidharan Nair, V.; Taha, R.Z.; Elkord, E. T-cell responses and therapies against SARS-CoV-2 infection. Immunology 2021, 162, 30–43. [Google Scholar] [CrossRef]
- Liu, W.D.; Wang, J.T.; Hung, C.C.; Chang, S.C. Accelerated progression of pulmonary tuberculosis in a COVID-19 patient after corticosteroid treatment. J. Microbiol. Immunol. Infect. 2022, 55, 347–349. [Google Scholar] [CrossRef]
- Motta, I.; Centis, R.; D’Ambrosio, L.; García-García, J.M.; Goletti, D.; Gualano, G.; Lipani, F.; Palmieri, F.; Sánchez-Montalvá, A.; Pontali, E.; et al. Tuberculosis, COVID-19 and migrants, Preliminary analysis of deaths occurring in 69 patients from two cohorts. Pulmonology 2020, 26, 233–240. [Google Scholar] [CrossRef]
- Mousquer, G.T.; Peres, A.; Fiegenbaum, M. Pathology of TB/COVID-19 Co-Infection, The phantom menace. Tuberculosis 2021, 126, 102020. [Google Scholar] [CrossRef]
- Divangahi, M.; Desjardins, D.; Nunes-Alves, C.; Remold, H.G.; Behar, S.M. Eicosanoid pathways regulate adaptive immunity to Mycobacterium tuberculosis. Nat. Immunol. 2010, 11, 751–758. [Google Scholar] [CrossRef]
- Shafiani, S.; Tucker-Heard, G.; Kariyone, A.; Takatsu, K.; Urdahl, K.B. Pathogen-specific regulatory T cells delay the arrival of effector T cells in the lung during early tuberculosis. J. Exp. Med. 2010, 207, 1409–1420. [Google Scholar] [CrossRef] [PubMed]
- van de Vegte, Y.J.; Said, M.A.; Rienstra, M.; van der Harst, P.; Verweij, N. Genome-wide association studies and Mendelian randomization analyses for leisure sedentary behaviors. Nat. Commun. 2020, 11, 1770. [Google Scholar] [CrossRef] [PubMed]
- McHenry, M.L.; Simmons, J.; Hong, H.; Malone, L.L.; Mayanja-Kizza, H.; Bush, W.S.; Boom, W.H.; Hawn, T.R.; Williams, S.M.; Stein, C.M. Tuberculosis severity associates with variants and eQTLs related to vascular biology and infection-induced inflammation. PLoS Genet. 2023, 19, e1010387. [Google Scholar] [CrossRef]
- Hong, H.; Dill-McFarland, K.A.; Simmons, J.D.; Peterson, G.J.; Benchek, P.; Mayanja-Kizza, H.; Boom, W.H.; Stein, C.M.; Hawn, T.R. Mycobacterium tuberculosis-dependent monocyte expression quantitative trait loci, cytokine production, and TB pathogenesis. Front. Immunol. 2024, 15, 1359178. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, C.Y.; Wang, L.; Li, Y.; Xiao, X. Genetic Insights of Schizophrenia via Single Cell RNA-Sequencing Analyses. Schizophr. Bull. 2023, 49, 914–922. [Google Scholar] [CrossRef]
- Sekine, T.; Perez-Potti, A.; Rivera-Ballesteros, O.; Stralin, K.; Gorin, J.B.; Olsson, A.; Llewellyn-Lacey, S.; Kamal, H.; Bogdanovic, G.; Muschiol, S.; et al. Robust T Cell Immunity in Convalescent Individuals with Asymptomatic or Mild COVID-19. Cell 2020, 183, 158–168.e114. [Google Scholar] [CrossRef] [PubMed]
- Nica, A.C.; Dermitzakis, E.T. Expression quantitative trait loci: Present and future. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368, 20120362. [Google Scholar] [CrossRef]
- Zhu, Y.; Yao, S.; Chen, L. Cell surface signaling molecules in the control of immune responses: A tide model. Immunity 2011, 34, 466–478. [Google Scholar] [CrossRef] [PubMed]
- Lötscher, J.; Balmer, M.L. Sensing between reactions-how the metabolic microenvironment shapes immunity. Clin. Exp. Immunol. 2019, 197, 161–169. [Google Scholar] [CrossRef]
- Habtamu, M.; Abrahamsen, G.; Aseffa, A.; Andargie, E.; Ayalew, S.; Abebe, M.; Spurkland, A. High-throughput analysis of T cell-monocyte interaction in human tuberculosis. Clin. Exp. Immunol. 2020, 201, 187–199. [Google Scholar] [CrossRef]
- Schulte-Schrepping, J.; Reusch, N.; Paclik, D.; Baßler, K.; Schlickeiser, S.; Zhang, B.; Krämer, B.; Krammer, T.; Brumhard, S.; Bonaguro, L.; et al. Severe COVID-19 Is Marked by a Dysregulated Myeloid Cell Compartment. Cell 2020, 182, 1419–1440.e1423. [Google Scholar] [CrossRef] [PubMed]
- McKinstry, K.K.; Strutt, T.M.; Swain, S.L. The potential of CD4 T-cell memory. Immunology 2010, 130, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jing, Q.; Hu, Z.; Wang, X.; Hu, Y.; Zhang, J.; Li, L. Mycobacterium tuberculosis-specific memory T cells in bronchoalveolar lavage of patients with pulmonary tuberculosis. Cytokine 2023, 171, 156374. [Google Scholar] [CrossRef]
- Al Saihati, H.A.; Hussein, H.A.M.; Thabet, A.A.; Wardany, A.A.; Mahmoud, S.Y.; Farrag, E.S.; Mohamed, T.I.A.; Fathy, S.M.; Elnosary, M.E.; Sobhy, A.; et al. Memory T Cells Discrepancies in COVID-19 Patients. Microorganisms 2023, 11, 2737. [Google Scholar] [CrossRef]
- Raphael, I.; Joern, R.R.; Forsthuber, T.G. Memory CD4(+) T Cells in Immunity and Autoimmune Diseases. Cells 2020, 9, 531. [Google Scholar] [CrossRef]
- Jain, A.; Song, R.; Wakeland, E.K.; Pasare, C. T cell-intrinsic IL-1R signaling licenses effector cytokine production by memory CD4 T cells. Nat. Commun. 2018, 9, 3185. [Google Scholar] [CrossRef]
- MacLeod, M.K.; David, A.; McKee, A.S.; Crawford, F.; Kappler, J.W.; Marrack, P. Memory CD4 T cells that express CXCR5 provide accelerated help to B cells. J. Immunol. 2011, 186, 2889–2896. [Google Scholar] [CrossRef]
- MacLennan, I.C.; Gulbranson-Judge, A.; Toellner, K.M.; Casamayor-Palleja, M.; Chan, E.; Sze, D.M.; Luther, S.A.; Orbea, H.A. The changing preference of T and B cells for partners as T-dependent antibody responses develop. Immunol. Rev. 1997, 156, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Westhorpe, C.L.V.; Norman, M.U.; Hall, P.; Snelgrove, S.L.; Finsterbusch, M.; Li, A.; Lo, C.; Tan, Z.H.; Li, S.; Nilsson, S.K.; et al. Effector CD4(+) T cells recognize intravascular antigen presented by patrolling monocytes. Nat. Commun. 2018, 9, 747. [Google Scholar] [CrossRef]
- Corrêa, R.D.S.; Leal-Calvo, T.; Mafort, T.T.; Santos, A.P.; Leung, J.; Pinheiro, R.O.; Rufino, R.; Moraes, M.O.; Rodrigues, L.S. Reanalysis and validation of the transcriptional pleural fluid signature in pleural tuberculosis. Front. Immunol. 2023, 14, 1256558. [Google Scholar] [CrossRef]
- Long, N.P.; Phat, N.K.; Yen, N.T.H.; Park, S.; Park, Y.; Cho, Y.S.; Shin, J.G. A 10-gene biosignature of tuberculosis treatment monitoring and treatment outcome prediction. Tuberculosis 2021, 131, 102138. [Google Scholar] [CrossRef]
- Martin-Sancho, L.; Lewinski, M.K.; Pache, L.; Stoneham, C.A.; Yin, X.; Becker, M.E.; Pratt, D.; Churas, C.; Rosenthal, S.B.; Liu, S.; et al. Functional landscape of SARS-CoV-2 cellular restriction. Mol. Cell 2021, 81, 2656–2668.e2658. [Google Scholar] [CrossRef] [PubMed]
- Mesner, D.; Reuschl, A.K.; Whelan, M.V.X.; Bronzovich, T.; Haider, T.; Thorne, L.G.; Ragazzini, R.; Bonfanti, P.; Towers, G.J.; Jolly, C. SARS-CoV-2 evolution influences GBP and IFITM sensitivity. Proc. Natl. Acad. Sci. USA 2023, 120, e2212577120. [Google Scholar] [CrossRef] [PubMed]
- Paine, S.K.; Choudhury, P.; Alam, M.; Bhattacharyya, C.; Pramanik, S.; Tripathi, D.; Das, C.; Patel, V.; Ghosh, S.; Chatterjee, S.; et al. Multi-faceted dysregulated immune response for COVID-19 infection explaining clinical heterogeneity. Cytokine 2024, 174, 156434. [Google Scholar] [CrossRef] [PubMed]
- Lafuse, W.P.; Wu, Q.; Kumar, N.; Saljoughian, N.; Sunkum, S.; Ahumada, O.S.; Turner, J.; Rajaram, M.V.S. Psychological stress creates an immune suppressive environment in the lung that increases susceptibility of aged mice to Mycobacterium tuberculosis infection. Front. Cell. Infect. Microbiol. 2022, 12, 990402. [Google Scholar] [CrossRef]
- Naranbhai, V. The Role of Host Genetics (and Genomics) in Tuberculosis. Microbiol. Spectr. 2016, 4, 10–1128. [Google Scholar] [CrossRef]
- Deng, W.; Zeng, J.; Xiang, X.; Xie, J. Insights into the distribution and functions of the eukaryotic GPI-like anchored genes among Mycobacterium from a comparative genomic perspective. Crit. Rev. Eukaryot. Gene Expr. 2012, 22, 299–307. [Google Scholar] [CrossRef]
- Zhang, L.; Yu, X.; Zheng, L.; Zhang, Y.; Li, Y.; Fang, Q.; Gao, R.; Kang, B.; Zhang, Q.; Huang, J.Y.; et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature 2018, 564, 268–272. [Google Scholar] [CrossRef]
- Zhang, X.; Lan, Y.; Xu, J.; Quan, F.; Zhao, E.; Deng, C.; Luo, T.; Xu, L.; Liao, G.; Yan, M.; et al. CellMarker: A manually curated resource of cell markers in human and mouse. Nucleic Acids Res. 2019, 47, D721–D728. [Google Scholar] [CrossRef] [PubMed]
- Street, K.; Risso, D.; Fletcher, R.B.; Das, D.; Ngai, J.; Yosef, N.; Purdom, E.; Dudoit, S. Slingshot: Cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genom. 2018, 19, 477. [Google Scholar] [CrossRef]
- Skrivankova, V.W.; Richmond, R.C.; Woolf, B.A.R.; Yarmolinsky, J.; Davies, N.M.; Swanson, S.A.; VanderWeele, T.J.; Higgins, J.P.T.; Timpson, N.J.; Dimou, N.; et al. Strengthening the Reporting of Observational Studies in Epidemiology Using Mendelian Randomization, The STROBE-MR Statement. JAMA 2021, 326, 1614–1621. [Google Scholar] [CrossRef] [PubMed]
- Hemani, G.; Tilling, K.; Davey Smith, G. Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLoS Genet. 2017, 13, e1007081. [Google Scholar]
- Guo, H.; Fortune, M.D.; Burren, O.S.; Schofield, E.; Todd, J.A.; Wallace, C. Integration of disease association and eQTL data using a Bayesian colocalisation approach highlights six candidate causal genes in immune-mediated diseases. Hum. Mol. Genet. 2015, 24, 3305–3313. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Source | Characteristic |
|---|---|---|
| PTB ScRNA-seq | GSE218065 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE218065 (accessed on 13 December 2023) | Male, PBMC |
| PTB ScRNA-seq | GSE192483 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE192483 (accessed on 13 December 2023) | Lung tissue |
| COVID-19 scRNA-seq | GSE171555 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE171555 (accessed on 13 December 2023) | Male, PBMC |
| COVID-19 scRNA-seq | GSE192391 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE192391 (accessed on 13 December 2023) | PBMC |
| PTB bulk | GSE65517 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE65517 (accessed on 5 February 2024) | Male, PBMC |
| COVID-19 bulk | GSE215262 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE215262 (accessed on 5 February 2024) | PBMC |
| PTB GWAS | ebi-a-GCST90018892 https://gwas.mrcieu.ac.uk/datasets/ebi-a-GCST90018892/ (accessed on 2 January 2024) | 477,386 sample, European |
| PTB validation-1 GWAS | ebi-a-GCST90018672 https://gwas.mrcieu.ac.uk/datasets/ebi-a-GCST90018672/ (accessed on 2 January 2024) | 178,671 sample, East Asian |
| PTB validation-2 GWAS | bbj-a-149 https://gwas.mrcieu.ac.uk/datasets/bbj-a-149/ (accessed on 2 January 2024) | 212,453 sample, East Asian, Males, and Females |
| PTB validation-3 GWAS | finn-b-TBC_RESP https://gwas.mrcieu.ac.uk/datasets/finn-b-TBC_RESP/ (accessed on 2 January 2024) | European, Males, and Females |
| COVID-19 GWAS | ebi-a-GCST011081 https://gwas.mrcieu.ac.uk/datasets/ebi-a-GCST011081/ (accessed on 10 January 2024) | 1,887,658 sample, European |
| COVID-19 validation-1 GWAS | ebi-a-GCST011082 https://gwas.mrcieu.ac.uk/datasets/ebi-a-GCST011082/ (accessed on 10 January 2024) | 1,557,411 sample, European |
| COVID-19 validation-2 GWAS | ebi-a-GCST011075 https://gwas.mrcieu.ac.uk/datasets/ebi-a-GCST011075/ (accessed on 10 January 2024) | 1,388,342 sample, European, severe COVID-19 |
| Marker | Ensemble ID | Outcome | SNP | EA | OR (95% CI) | p Value | PVE | F Statistic |
|---|---|---|---|---|---|---|---|---|
| ODF2L | ENSG00000122417 | PTB | rs7523135 | G | 0.94 (0.89–1.00) | 0.041 | 10.36% | 2597.99 |
| ODF2L | ENSG00000122417 | PTB | rs6576834 | C | 0.94 (0.89–1.00) | 0.041 | 10.36% | 670.87 |
| ODF2L | ENSG00000122417 | PTB | rs5744305 | G | 0.94 (0.89–1.00) | 0.041 | 10.36% | 118.33 |
| ODF2L | ENSG00000122417 | PTB | rs61161313 | T | 0.94 (0.89–1.00) | 0.041 | 10.36% | 40.94 |
| ODF2L | ENSG00000122417 | PTB | rs4512701 | A | 0.94 (0.89–1.00) | 0.041 | 10.36% | 40.92 |
| GBP2 | ENSG00000162645 | PTB | rs2182322 | G | 1.38 (1.05–1.81) | 0.0208 | 1.60% | 47.99 |
| GBP2 | ENSG00000162645 | PTB | rs76830965 | A | 1.38 (1.05–1.81) | 0.0208 | 1.60% | 40.69 |
| GBP2 | ENSG00000162645 | PTB | rs9264638 | A | 1.38 (1.05–1.81) | 0.0208 | 1.60% | 52.07 |
| GBP2 | ENSG00000162645 | PTB | rs149007767 | T | 1.38 (1.05–1.81) | 0.0208 | 1.60% | 57.79 |
| GBP2 | ENSG00000162645 | PTB | rs653178 | T | 1.38 (1.05–1.81) | 0.0208 | 1.60% | 223.35 |
| GBP2 | ENSG00000162645 | PTB | rs4788084 | T | 1.38 (1.05–1.81) | 0.0208 | 1.60% | 51.35 |
| TRAV1-2 | ENSG00000256553 | PTB | rs13325613 | T | 1.37 (1.09–1.72) | 0.00684 | 0.75% | 60.48 |
| TRAV1-2 | ENSG00000256553 | PTB | rs3130559 | T | 1.37 (1.09–1.72) | 0.00684 | 0.75% | 40.1 |
| TRAV1-2 | ENSG00000256553 | PTB | rs2256752 | C | 1.37 (1.09–1.72) | 0.00684 | 0.75% | 61.35 |
| LAG3 | ENSG00000089692 | COVID-19 | rs9420589 | T | 1.46 (1.11–1.92) | 0.00692 | 0.48% | 42.93 |
| LAG3 | ENSG00000089692 | COVID-19 | rs3809272 | T | 1.46 (1.11–1.92) | 0.00692 | 0.48% | 38.49 |
| LAG3 | ENSG00000089692 | COVID-19 | rs67706382 | A | 1.46 (1.11–1.92) | 0.00692 | 0.48% | 65.38 |
| SLFN5 | ENSG00000166750 | COVID-19 | rs7215469 | A | 0.91 (0.86–0.96) | 0.00126 | 9.10% | 2796.81 |
| SLFN5 | ENSG00000166750 | COVID-19 | rs76240782 | T | 0.91 (0.86–0.96) | 0.00126 | 9.10% | 256.05 |
| SLFN5 | ENSG00000166750 | COVID-19 | rs8076768 | T | 0.91 (0.86–0.96) | 0.00126 | 9.10% | 48.33 |
| Exposure | Outcome | Method | Q | P (Heterogeneity) | P (Pleiotropy) |
|---|---|---|---|---|---|
| GBP2 (eqtl-a-ENSG00000162645) | PTB (ebi-a-GCST90018892) | MR Egger | 7.45 | 0.11 | 0.69 |
| GBP2 (eqtl-a-ENSG00000162645) | PTB (ebi-a-GCST90018892) | IVW | 7.80 | 0.17 | - |
| TRAV1-2 (eqtl-a-ENSG00000256553) | PTB (ebi-a-GCST90018892) | MR Egger | 0.24 | 0.63 | 0.45 |
| TRAV1-2 (eqtl-a-ENSG00000256553) | PTB (ebi-a-GCST90018892) | IVW | 1.58 | 0.45 | - |
| ODF2L (eqtl-a-ENSG00000122417) | PTB (ebi-a-GCST90018892) | MR Egger | 2.06 | 0.56 | 0.57 |
| ODF2L (eqtl-a-ENSG00000122417) | PTB (ebi-a-GCST90018892) | IVW | 2.46 | 0.65 | - |
| LAG3 (eqtl-a-ENSG00000089692) | COVID-19 (ebi-a-GCST011071) | MR Egger | 1.96 | 0.16 | 0.76 |
| LAG3 (eqtl-a-ENSG00000089692) | COVID-19 (ebi-a-GCST011071) | IVW | 2.28 | 0.32 | - |
| SLFN5 (eqtl-a-ENSG00000166750) | COVID-19 (ebi-a-GCST011071) | MR Egger | 0.52 | 0.47 | 0.97 |
| SLFN5 (eqtl-a-ENSG00000166750) | COVID-19 (ebi-a-GCST011071) | IVW | 0.52 | 0.77 | - |
| Gene | Forward (5′-3′) | Reverse (5′-3′) |
|---|---|---|
| GBP2 | AATTAGGGGCCCAGTTGGAAG | AAGAGACGGTAACCTCCTGGT |
| TRAV1-2 | GCTACGGAAGGTGCCATTGT | AATGTAGGTGCTTCGCCAGC |
| ODF2L | AAAGCAAACCGTTTTTCCCAATC | CGTTCTCGGCTTCCCTTTTATG |
| β-actin | CACTCTTCCAGCCTTCCTTC | GTACAGGTCTTTGCGGATGT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, L.; Wu, H.; Peng, L.; Huang, X.; Yang, R.; Ma, W.; Zhong, L.; Li, B.; Song, J.; Luo, S.; et al. CD4+ Effective Memory T Cell Markers GBP2 and LAG3 Are Risk Factors for PTB and COVID-19 Infection: A Study Integrating Single-Cell Expression Quantitative Trait Locus and Mendelian Randomization Analyses. Int. J. Mol. Sci. 2024, 25, 9971. https://doi.org/10.3390/ijms25189971
Zhu L, Wu H, Peng L, Huang X, Yang R, Ma W, Zhong L, Li B, Song J, Luo S, et al. CD4+ Effective Memory T Cell Markers GBP2 and LAG3 Are Risk Factors for PTB and COVID-19 Infection: A Study Integrating Single-Cell Expression Quantitative Trait Locus and Mendelian Randomization Analyses. International Journal of Molecular Sciences. 2024; 25(18):9971. https://doi.org/10.3390/ijms25189971
Chicago/Turabian StyleZhu, Liangyu, Hanxin Wu, Li Peng, Xun Huang, Rui Yang, Weijie Ma, Lei Zhong, Bingxue Li, Jieqin Song, Suyi Luo, and et al. 2024. "CD4+ Effective Memory T Cell Markers GBP2 and LAG3 Are Risk Factors for PTB and COVID-19 Infection: A Study Integrating Single-Cell Expression Quantitative Trait Locus and Mendelian Randomization Analyses" International Journal of Molecular Sciences 25, no. 18: 9971. https://doi.org/10.3390/ijms25189971