

The BRD4 Inhibitor I-BET-762 Reduces HO-1 Expression in Macrophages and the Pancreas of Mice

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Activation of the NRF2-HO-1 Pathway Correlates with Poor Survival in Patients with Pancreatic Cancer
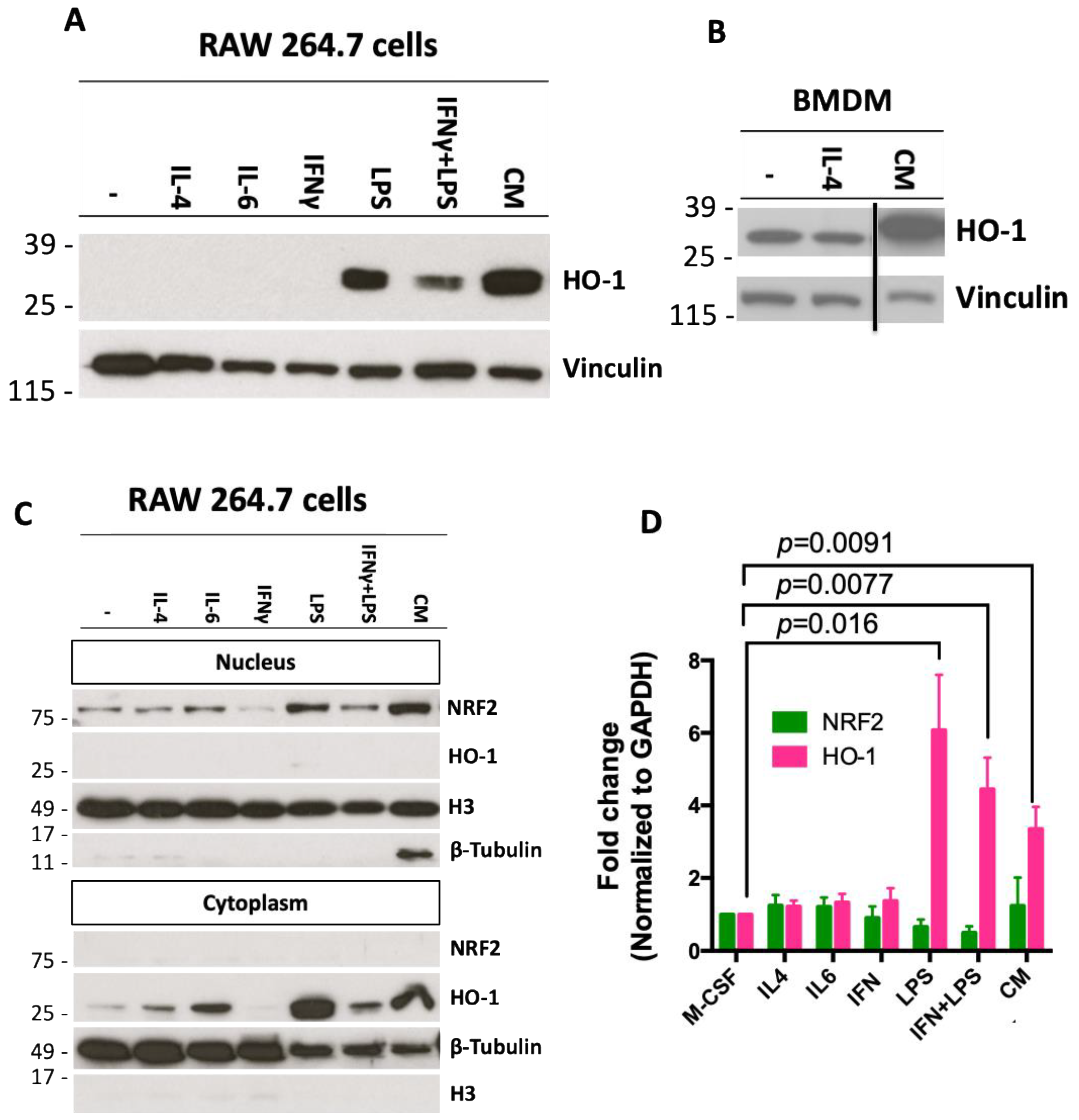
2.2. Conditioned Media from Pancreatic Cancer Cells Increases Expression of HO-1 in Macrophages
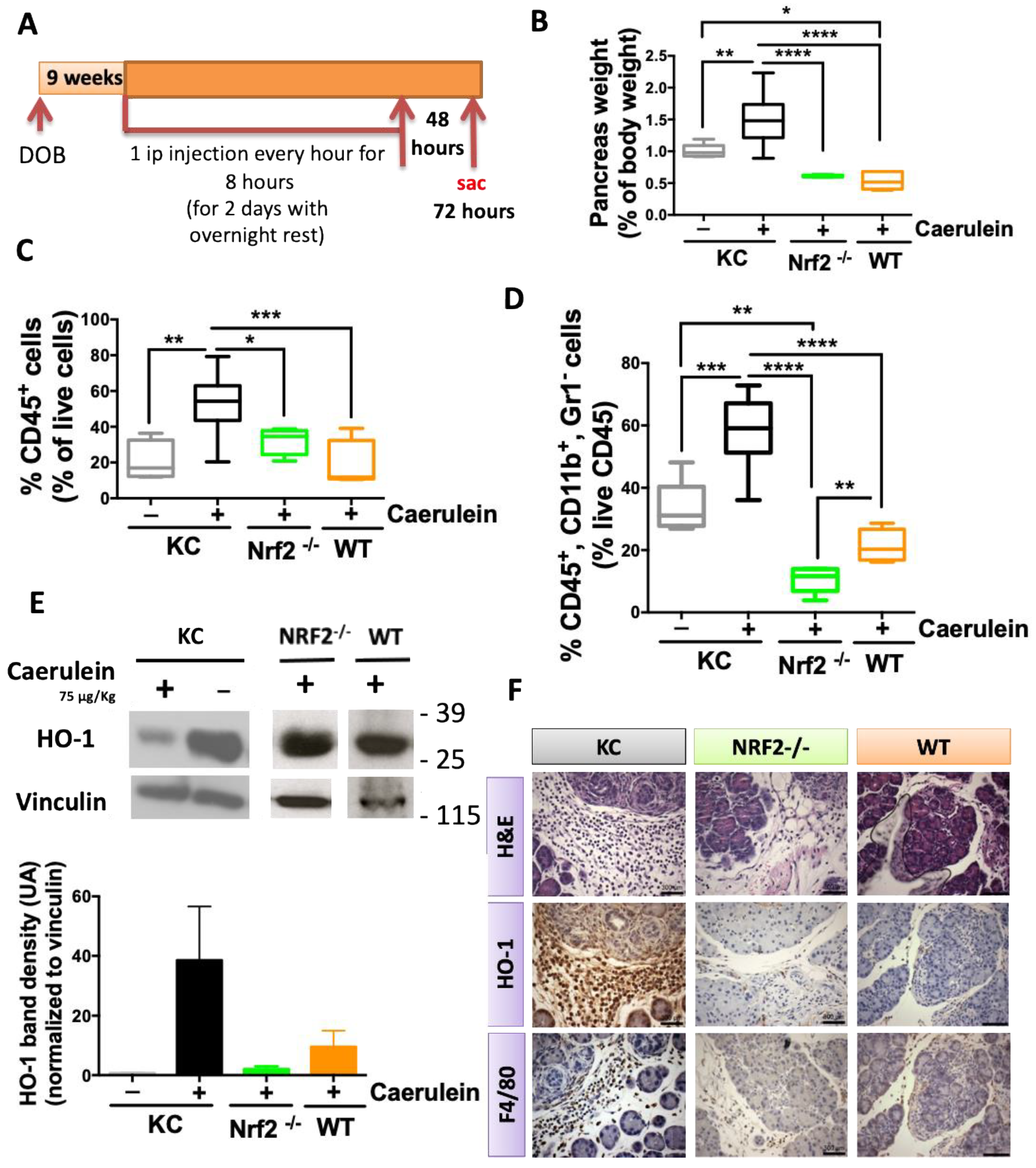
2.3. NRF2 Depletion Protects against Caerulein-Induced Pancreatic Lesions
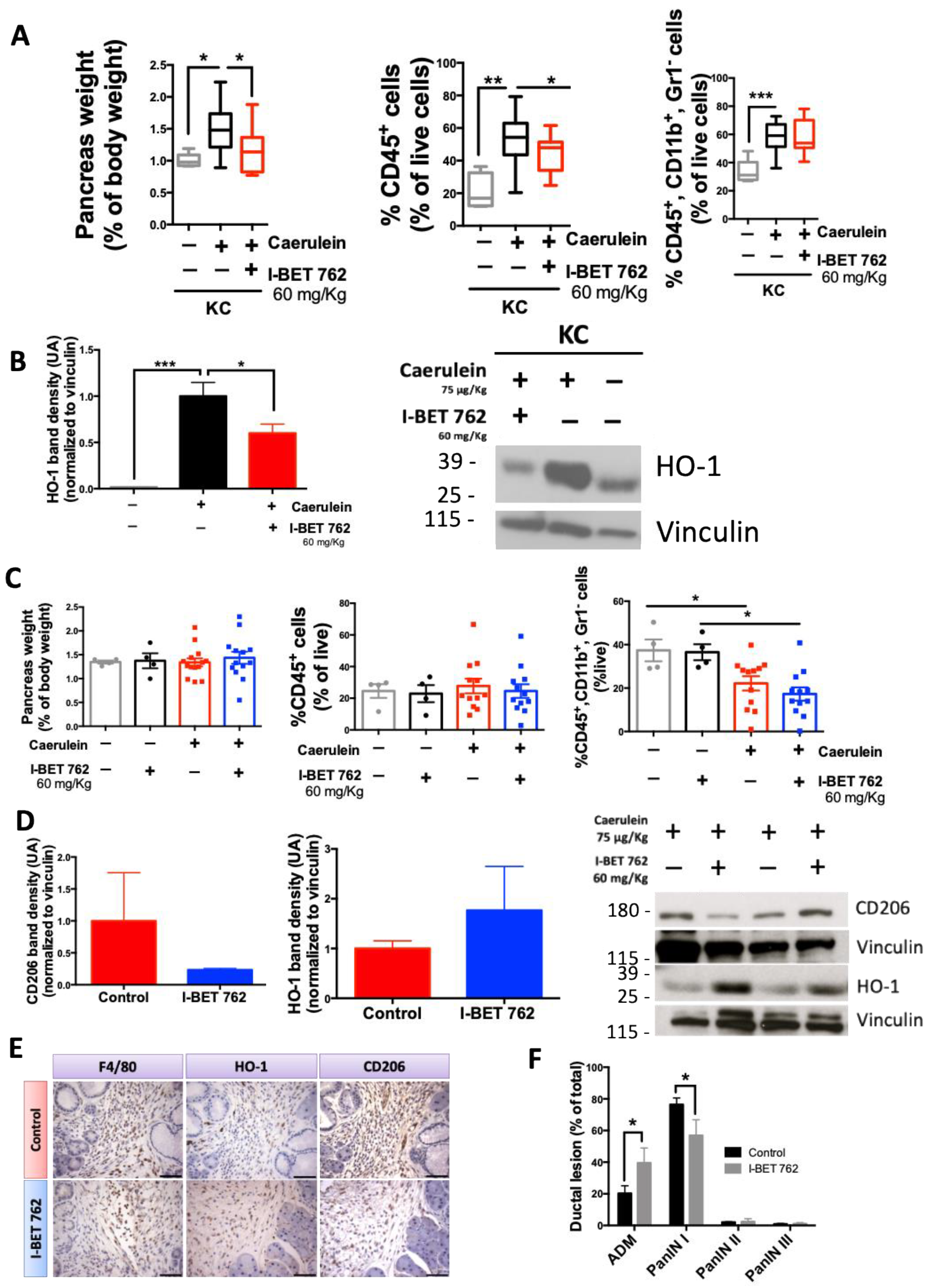
2.4. The Bromodomain Inhibitor I-BET-762 Reduces HO-1 Expression Induced by Caerulein in the Pancreas of KC Mice
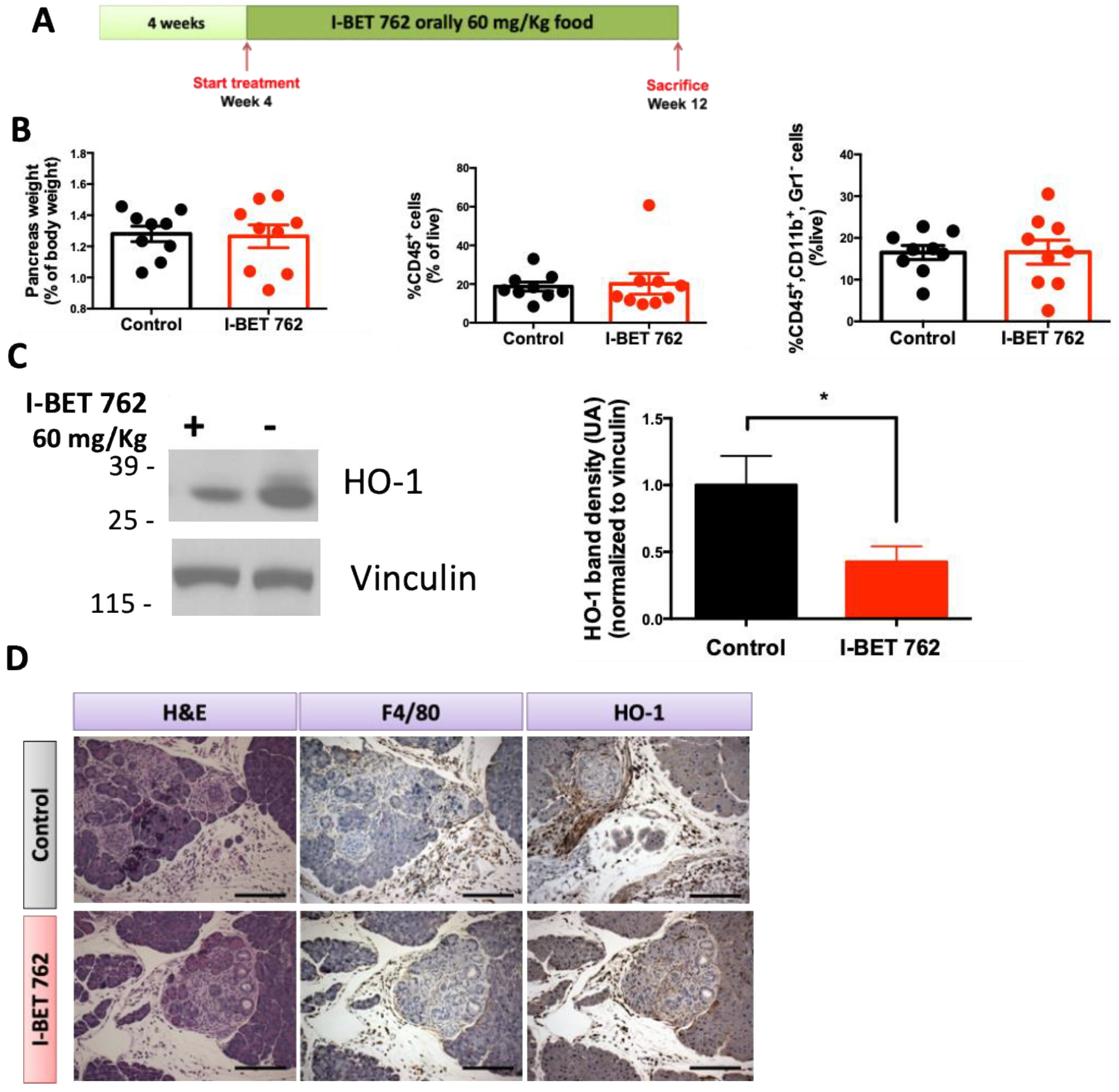
2.5. Bromodomain Inhibitor I-BET-762 Prevents HO-1 Upregulation in the Pancreas of KPC Mice
3. Discussion
4. Materials and Methods
4.1. Drugs
4.2. Cell Culture and Reagents
4.3. In Vivo Experiments
4.4. Flow Cytometry
4.5. Immunohistochemistry
4.6. Western Blotting
4.7. Multiplex Cytokine Assay
4.8. RT-PCR
4.9. Patient Survival Analysis
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer Statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Kolbeinsson, H.M.; Chandana, S.; Wright, G.P.; Chung, M. Pancreatic Cancer: A Review of Current Treatment and Novel Therapies. J. Investig. Surg. 2023, 36, 2129884. [Google Scholar] [CrossRef] [PubMed]
- McGuigan, A.; Kelly, P.; Turkington, R.C.; Jones, C.; Coleman, H.G.; McCain, R.S. Pancreatic Cancer: A Review of Clinical Diagnosis, Epidemiology, Treatment and Outcomes. World J. Gastroenterol. 2018, 24, 4846–4861. [Google Scholar] [CrossRef]
- Halbrook, C.J.; Lyssiotis, C.A.; Pasca di Magliano, M.; Maitra, A. Pancreatic Cancer: Advances and Challenges. Cell 2023, 186, 1729–1754. [Google Scholar] [CrossRef] [PubMed]
- Oberstein, P.E.; Olive, K.P. Pancreatic Cancer: Why Is It so Hard to Treat? Therap. Adv. Gastroenterol. 2013, 6, 321–337. [Google Scholar] [CrossRef]
- Manji, G.A.; Olive, K.P.; Saenger, Y.M.; Oberstein, P. Current and Emerging Therapies in Metastatic Pancreatic Cancer. Clin. Cancer Res. 2017, 23, 1670–1678. [Google Scholar] [CrossRef]
- Hu, Z.I.; O’Reilly, E.M. Therapeutic Developments in Pancreatic Cancer. Nat. Rev. Gastroenterol. Hepatol. 2024, 21, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D. The Pancreas Cancer Microenvironment. Clin. Cancer Res. 2012, 18, 4266–4276. [Google Scholar] [CrossRef]
- Ino, Y.; Yamazaki-Itoh, R.; Shimada, K.; Iwasaki, M.; Kosuge, T.; Kanai, Y.; Hiraoka, N. Immune Cell Infiltration as an Indicator of the Immune Microenvironment of Pancreatic Cancer. Br. J. Cancer 2013, 108, 914–923. [Google Scholar] [CrossRef]
- Apte, M.V.; Wilson, J.S.; Lugea, A.; Pandol, S.J. A Starring Role for Stellate Cells in the Pancreatic Cancer Microenvironment. Gastroenterology 2013, 144, 1210–1219. [Google Scholar] [CrossRef] [PubMed]
- Joyce, J.A.; Fearon, D.T. T Cell Exclusion, Immune Privilege, and the Tumor Microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Xue, J.; Jaffee, E.M.; Habtezion, A. Role of Immune Cells and Immune-Based Therapies in Pancreatitis and Pancreatic Ductal Adenocarcinoma. Gastroenterology 2013, 144, 1230–1240. [Google Scholar] [CrossRef]
- de Visser, K.E.; Joyce, J.A. The Evolving Tumor Microenvironment: From Cancer Initiation to Metastatic Outgrowth. Cancer Cell 2023, 41, 374–403. [Google Scholar] [CrossRef]
- Chakkera, M.; Foote, J.B.; Farran, B.; Nagaraju, G.P. Breaking the Stromal Barrier in Pancreatic Cancer: Advances and Challenges. Biochim. Biophys. Acta Rev. Cancer 2024, 1879, 189065. [Google Scholar] [CrossRef]
- Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. The Yin-Yang of Tumor-Associated Macrophages in Neoplastic Progression and Immune Surveillance. Immunol. Rev. 2008, 222, 155–161. [Google Scholar] [CrossRef]
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Pancreatic Stellate Cells Support Tumour Metabolism through Autophagic Alanine Secretion. Nature 2016, 536, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K. Metabolic Reprogramming of Immune Cells in Cancer Progression. Immunity 2015, 43, 435–449. [Google Scholar] [CrossRef]
- McCleary-Wheeler, A.L.; Lomberk, G.; Weiss, F.U.; Schneider, G.; Fabbri, M.; Poshusta, T.L.; Dusetti, N.J.; Baumgart, S.; Iovanna, J.L.; Ellenrieder, V.; et al. Insights into the Epigenetic Mechanisms Controlling Pancreatic Carcinogenesis. Cancer Lett. 2013, 328, 212–221. [Google Scholar] [CrossRef]
- Lomberk, G.; Blum, Y.; Nicolle, R.; Nair, A.; Gaonkar, K.S.; Marisa, L.; Mathison, A.; Sun, Z.; Yan, H.; Elarouci, N.; et al. Distinct Epigenetic Landscapes Underlie the Pathobiology of Pancreatic Cancer Subtypes. Nat. Commun. 2018, 9, 1978. [Google Scholar] [CrossRef]
- Lomberk, G.; Dusetti, N.; Iovanna, J.; Urrutia, R. Emerging Epigenomic Landscapes of Pancreatic Cancer in the Era of Precision Medicine. Nat. Commun. 2019, 10, 3875. [Google Scholar] [CrossRef]
- Kawakubo, K.; Castillo, C.F.D.; Liss, A.S. Epigenetic Regulation of Pancreatic Adenocarcinoma in the Era of Cancer Immunotherapy. J. Gastroenterol. 2022, 57, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Orlacchio, A.; Muzyka, S.; Gonda, T.A. Epigenetic Therapeutic Strategies in Pancreatic Cancer. Int. Rev. Cell Mol. Biol. 2024, 383, 1–40. [Google Scholar] [PubMed]
- Kim, H.S.; Gweon, T.G.; Park, S.H.; Kim, T.H.; Kim, C.W.; Chang, J.H. Incidence and Risk of Pancreatic Cancer in Patients with Chronic Pancreatitis: Defining the Optimal Subgroup for Surveillance. Sci. Rep. 2023, 13, 106. [Google Scholar] [CrossRef] [PubMed]
- Yadav, D.; Lowenfels, A.B. The Epidemiology of Pancreatitis and Pancreatic Cancer. Gastroenterology 2013, 144, 1252–1261. [Google Scholar] [CrossRef]
- Lowenfels, A.B.; Maisonneuve, P.; Cavallini, G.; Ammann, R.W.; Lankisch, P.G.; Andersen, J.R.; Dimagno, E.P.; Andrén-Sandberg, A.; Domellof, L. Pancreatitis and the Risk of Pancreatic Cancer. International Study Group. N. Engl. J. Med. 1993, 328, 1433–1437. [Google Scholar] [CrossRef]
- Malka, D.; Hammel, P.; Maire, F.; Rufat, P.; Madeira, I.; Pessione, F.; Lévy, P.; Ruszniewski, P. Risk of Pancreatic Adenocarcinoma in Chronic Pancreatitis. Gut 2002, 51, 849–852. [Google Scholar] [CrossRef] [PubMed]
- Munigala, S.; Kanwal, F.; Xian, H.; Scherrer, J.F.; Agarwal, B. Increased Risk of Pancreatic Adenocarcinoma after Acute Pancreatitis. Clin. Gastroenterol. Hepatol. 2014, 12, 1143–1150. [Google Scholar] [CrossRef]
- Nicodeme, E.; Jeffrey, K.L.; Schaefer, U.; Beinke, S.; Dewell, S.; Chung, C.-W.; Chandwani, R.; Marazzi, I.; Wilson, P.; Coste, H.; et al. Suppression of Inflammation by a Synthetic Histone Mimic. Nature 2010, 468, 1119–1123. [Google Scholar] [CrossRef]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET Bromodomain Inhibition as a Therapeutic Strategy to Target C-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef]
- Belkina, A.C.; Denis, G. V BET Domain Co-Regulators in Obesity, Inflammation and Cancer. Nat. Rev. Cancer 2012, 12, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Nishiyama, A.; Karpova, T.; Mcnally, J.; Ozato, K. Brd4 Marks Select Genes on Mitotic Chromatin and Directs Postmitotic Transcription. Mol. Cell Biol. Cell 2009, 20, 4899–4909. [Google Scholar] [CrossRef] [PubMed]
- Mertz, J.; Conery, A.R.; Bryant, B.M.; Sandy, P.; Balasubramanian, S.; Mele, D.; Bergeron, L.; Sims, R.J. Targeting MYC Dependence in Cancer by Inhibiting BET Bromodomains. Proc. Natl. Acad. Sci. USA 2011, 108, 16669–16674. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.-W.; Coste, H.; White, J.H.; Mirguet, O.; Wilde, J.; Gosmini, R.L.; Delves, C.; Magny, S.M.; Woodward, R.; Hughes, S.A.; et al. Discovery and Characterization of Small Molecule Inhibitors of the BET Family Bromodomains. J. Med. Chem. 2011, 54, 3827–3838. [Google Scholar] [CrossRef]
- Shimamura, T.; Chen, Z.; Soucheray, M.; Carretero, J.; Kikuchi, E.; Tchaicha, J.H.; Gao, Y.; Cheng, K.A.; Cohoon, T.J.; Qi, J.; et al. Efficacy of BET Bromodomain Inhibition in Kras-Mutant Non-Small Cell Lung Cancer. Clin. Cancer Res. 2013, 19, 6183–6192. [Google Scholar] [CrossRef]
- Shu, S.; Lin, C.Y.; He, H.H.; Witwicki, R.M.; Tabassum, D.P.; Roberts, J.M.; Janiszewska, M.; Jin Huh, S.; Liang, Y.; Ryan, J.; et al. Response and Resistance to BET Bromodomain Inhibitors in Triple-Negative Breast Cancer. Nature 2016, 529, 413–417. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective Inhibition of BET Bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef]
- Wu, X.; Liu, D.; Tao, D.; Xiang, W.; Xiao, X.; Wang, M.; Wang, L.; Luo, G.; Li, Y.; Zeng, F.; et al. Cancer Biology and Signal Transduction BRD4 Regulates EZH2 Transcription through Upregulation of C-MYC and Represents a Novel Therapeutic Target in Bladder Cancer. Mol. Cancer Ther. 2016, 15, 1029–1042. [Google Scholar] [CrossRef]
- Leal, A.S.A.S.; Williams, C.R.C.R.; Royce, D.B.D.B.; Pioli, P.A.P.A.; Sporn, M.B.M.B.; Liby, K.T.K.T. Bromodomain Inhibitors, JQ1 and I-BET 762, as Potential Therapies for Pancreatic Cancer. Cancer Lett. 2017, 394, 76–87. [Google Scholar] [CrossRef]
- Mantovani, A.; Schioppa, T.; Porta, C.; Allavena, P.; Sica, A. Role of Tumor-Associated Macrophages in Tumor Progression and Invasion. Cancer Metastasis Rev. 2006, 25, 315–322. [Google Scholar] [CrossRef]
- Chen, S.; Saeed, A.F.U.H.; Liu, Q.; Jiang, Q.; Xu, H.; Xiao, G.G.; Rao, L.; Duo, Y. Macrophages in Immunoregulation and Therapeutics. Signal Transduct. Target. Ther. 2023, 8, 207. [Google Scholar] [CrossRef] [PubMed]
- Pérez, S.; Rius-Pérez, S. Macrophage Polarization and Reprogramming in Acute Inflammation: A Redox Perspective. Antioxidants 2022, 11, 1394. [Google Scholar] [CrossRef]
- Cairo, G.; Recalcati, S.; Mantovani, A.; Locati, M. Iron Trafficking and Metabolism in Macrophages: Contribution to the Polarized Phenotype. Trends Immunol. 2011, 32, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, P.; Vijayan, V.; Gueler, F.; Immenschuh, S. Interplay of Heme with Macrophages in Homeostasis and Inflammation. Int. J. Mol. Sci. 2020, 270, 740. [Google Scholar] [CrossRef]
- Naito, Y.; Takagi, T.; Higashimura, Y. Heme Oxygenase-1 and Anti-Inflammatory M2 Macrophages. Arch. Biochem. Biophys. 2014, 564, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 Suppresses Macrophage Inflammatory Response by Blocking Proinflammatory Cytokine Transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic Targeting of the NRF2 and KEAP1 Partnership in Chronic Diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef]
- de la Vega, M.R.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 2, 1–23. [Google Scholar]
- Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules 2020, 25, 5474. [Google Scholar] [CrossRef]
- Chio, I.I.C.; Jafarnejad, S.M.; Ponz-Sarvise, M.; Park, Y.; Rivera, K.; Palm, W.; Wilson, J.; Sangar, V.; Hao, Y.; Öhlund, D.; et al. NRF2 Promotes Tumor Maintenance by Modulating MRNA Translation in Pancreatic Cancer. Cell 2016, 166, 963–976. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-Induced Nrf2 Transcription Promotes ROS Detoxification and Tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Occhiuto, C.J.; Moerland, J.A.; Leal, A.S.; Gallo, K.A.; Liby, K.T. The Multi-Faceted Consequences of NRF2 Activation throughout Carcinogenesis. Mol. Cells 2023, 46, 176–186. [Google Scholar] [CrossRef]
- Hussong, M.; Börno, S.T.; Kerick, M.; Wunderlich, A.; Franz, A.; Sültmann, H.; Timmermann, B.; Lehrach, H.; Hirsch-Kauffmann, M.; Schweiger, M.R. The Bromodomain Protein BRD4 Regulates the KEAP1/NRF2-Dependent Oxidative Stress Response. Cell Death Dis. 2014, 5, e1195. [Google Scholar] [CrossRef] [PubMed]
- Michaeloudes, C.; Mercado, N.; Clarke, C.; Bhavsar, P.K.; Adcock, I.M.; Barnes, P.J.; Chung, K.F. Bromodomain and Extraterminal Proteins Suppress NF-E2-Related Factor 2-Mediated Antioxidant Gene Expression. J. Immunol. 2014, 192, 4913–4920. [Google Scholar] [CrossRef]
- Arnold, J.N.; Magiera, L.; Kraman, M.; Fearon, D.T. Tumoral Immune Suppression by Macrophages Expressing Fibroblast Activation Protein- and Heme Oxygenase-1. Cancer Immunol. Res. 2014, 2, 121–126. [Google Scholar] [CrossRef]
- Bartha, Á.; Győrffy, B. Tnmplot.Com: A Web Tool for the Comparison of Gene Expression in Normal, Tumor and Metastatic Tissues. Int. J. Mol. Sci. 2021, 22, 2622. [Google Scholar] [CrossRef]
- Lánczky, A.; Győrffy, B. Web-Based Survival Analysis Tool Tailored for Medical Research (KMplot): Development and Implementation. J. Med. Internet Res. 2021, 23, e27633. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D. Trp53R172H and KrasG12D Cooperate to Promote Chromosomal Instability and Widely Metastatic Pancreatic Ductal Adenocarcinoma in Mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage Polarization: Tumor-Associated Macrophages as a Paradigm for Polarized M2 Mononuclear Phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Guerra, C.; Collado, M.; Navas, C.; Schuhmacher, A.J.; Hernández-Porras, I.; Cañamero, M.; Rodriguez-Justo, M.; Serrano, M.; Barbacid, M. Pancreatitis-Induced Inflammation Contributes to Pancreatic Cancer by Inhibiting Oncogene-Induced Senescence. Cancer Cell 2011, 19, 728–739. [Google Scholar] [CrossRef]
- Guerra, C.; Schuhmacher, A.J.; Cañamero, M.; Grippo, P.J.; Verdaguer, L.; Pérez-Gallego, L.; Dubus, P.; Sandgren, E.P.; Barbacid, M. Chronic Pancreatitis Is Essential for Induction of Pancreatic Ductal Adenocarcinoma by K-Ras Oncogenes in Adult Mice. Cancer Cell 2007, 11, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Clark, C.E.; Hingorani, S.R.; Mick, R.; Combs, C.; Tuveson, D.A.; Vonderheide, R.H. Dynamics of the Immune Reaction to Pancreatic Cancer from Inception to Invasion. Cancer Res. 2007, 67, 9518–9527. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Herndon, J.M.; Sojka, D.K.; Kim, K.W.; Knolhoff, B.L.; Zuo, C.; Cullinan, D.R.; Luo, J.; Bearden, A.R.; Lavine, K.J.; et al. Correction: Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity 2017, 47, 597. [Google Scholar] [CrossRef]
- Leal, A.S.; Liu, P.; Krieger-Burke, T.; Ruggeri, B.; Liby, K.T. The Bromodomain Inhibitor, INCB057643, Targets Both Cancer Cells and the Tumor Microenvironment in Two Preclinical Models of Pancreatic Cancer. Cancers 2021, 13, 96. [Google Scholar] [CrossRef] [PubMed]
- Garcia, P.L.; Miller, A.L.; Kreitzburg, K.M.; Council, L.N.; Gamblin, T.L.; Christein, J.D.; Heslin, M.J.; Arnoletti, J.P.; Richardson, J.H.; Chen, D.; et al. The BET Bromodomain Inhibitor JQ1 Suppresses Growth of Pancreatic Ductal Adenocarcinoma in Patient-Derived Xenograft Models. Oncogene 2015, 35, 833–845. [Google Scholar] [CrossRef]
- Mazur, P.K.; Herner, A.; Mello, S.S.; Wirth, M.; Hausmann, S.; Sánchez-Rivera, F.J.; Lofgren, S.M.; Kuschma, T.; Hahn, S.A.; Vangala, D.; et al. Combined Inhibition of BET Family Proteins and Histone Deacetylases as a Potential Epigenetics-Based Therapy for Pancreatic Ductal Adenocarcinoma. Nat. Med. 2015, 21, 1163–1171. [Google Scholar] [CrossRef]
- Wang, S.; Wang, J.; Chen, Z.; Luo, J.; Guo, W.; Sun, L.; Lin, L. Targeting M2-like Tumor-Associated Macrophages Is a Potential Therapeutic Approach to Overcome Antitumor Drug Resistance. NPJ Precis. Oncol. 2024, 8, 31. [Google Scholar] [CrossRef]
- Gocheva, V.; Wang, H.W.; Gadea, B.B.; Shree, T.; Hunter, K.E.; Garfall, A.L.; Berman, T.; Joyce, J.A. IL-4 Induces Cathepsin Protease Activity in Tumor-Associated Macrophages to Promote Cancer Growth and Invasion. Genes Dev. 2010, 24, 241–255. [Google Scholar] [CrossRef]
- Candido, J.B.; Morton, J.P.; Bailey, P.; Campbell, A.D.; Karim, S.A.; Jamieson, T.; Lapienyte, L.; Gopinathan, A.; Clark, W.; McGhee, E.J.; et al. CSF1R+ Macrophages Sustain Pancreatic Tumor Growth through T Cell Suppression and Maintenance of Key Gene Programs That Define the Squamous Subtype. Cell Rep. 2018, 23, 1448–1460. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Petricoin, E.F.; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and Invasive Ductal Pancreatic Cancer and Its Early Detection in the Mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef]
- Sanford, D.E.; Belt, B.; Panni, R.Z.; Mayer, A.; Deshpande, A.D.; Carpenter, D.; Mitchem, J.B.; Plambeck-Suess, S.M.; Worley, L.; Goetz, B.D.; et al. Inflammatory Monocyte Mobilization Decreases Patient Survival in Pancreatic Cancer: A Role for Targeting the CCL2/CCR2 Axis. Clin. Cancer Res. 2013, 19, 3404–3415. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Knolhoff, B.L.; Meyer, M.A.; Nywening, T.M.; West, B.L.; Luo, J.; Wang-Gillam, A.; Goedegebuure, S.P.; Linehan, D.C.; DeNardo, D.G. CSF1/CSF1R Blockade Reprograms Tumor-Infiltrating Macrophages and Improves Response to T-Cell Checkpoint Immunotherapy in Pancreatic Cancer Models. Cancer Res. 2014, 74, 5057–5069. [Google Scholar] [CrossRef]
- Hu, F.; Lou, N.; Jiao, J.; Guo, F.; Xiang, H.; Shang, D. Macrophages in Pancreatitis: Mechanisms and Therapeutic Potential. Biomed. Pharmacother. 2020, 131, 110693. [Google Scholar] [CrossRef] [PubMed]
- Nakamichi, I.; Butcher, E.C.; Omary, M.B.; Nakamichi, I.; Habtezion, A.; Zhong, B.; Contag, C.H. Hemin-Activated Macrophages Home to the Pancreas and Protect from Acute Pancreatitis via Heme Oxygenase-1 Induction Find the Latest Version: Hemin-Activated Macrophages Home to the Pancreas and Protect from Acute Pancreatitis via Heme Oxygenase-1 Inducti. J. Clin. Invest. 2005, 115, 3007–3014. [Google Scholar] [CrossRef]
- Sato, H.; Siow, R.C.M.; Bartlett, S.; Taketani, S.; Ishii, T.; Bannai, S.; Mann, G.E. Expression of Stress Proteins Heme Oxygenase-1 and -2 in Acute Pancreatitis and Pancreatic Islet ΒTC3 and Acinar AR42J Cells. FEBS Lett. 1997, 405, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Noh, S.J.; Bae, J.S.; Jamiyandorj, U.; Park, H.S.; Kwon, K.S.; Jung, S.H.; Youn, H.J.; Lee, H.; Park, B.-H.; Chung, M.J.; et al. Expression of Nerve Growth Factor and Heme Oxygenase-1 Predict Poor Survival of Breast Carcinoma Patients. BMC Cancer 2013, 13, 516. [Google Scholar] [CrossRef]
- Nitti, M.; Ivaldo, C.; Traverso, N.; Furfaro, A.L. Clinical Significance of Heme Oxygenase 1 in Tumor Progression. Antioxidants 2021, 10, 789. [Google Scholar] [CrossRef]
- Consonni, F.M.; Bleve, A.; Totaro, M.G.; Storto, M.; Kunderfranco, P.; Termanini, A.; Pasqualini, F.; Alì, C.; Pandolfo, C.; Sgambelluri, F.; et al. Heme Catabolism by Tumor-Associated Macrophages Controls Metastasis Formation. Nat. Immunol. 2021, 22, 595–606. [Google Scholar] [CrossRef]
- Sporn, M.B.; Liby, K.T. NRF2 and Cancer: The Good, the Bad and the Importance of Context. Nat. Rev. Cancer 2012, 12, 564–571. [Google Scholar] [CrossRef]
- Kitajima, S.; Thummalapalli, R.; Barbie, D.A. Inflammation as a Driver and Vulnerability of KRAS Mediated Oncogenesis. Semin. Cell Dev. Biol. 2016, 58, 127–135. [Google Scholar] [CrossRef]
- de Visser, K.E.; Eichten, A.; Coussens, L.M. Paradoxical Roles of the Immune System during Cancer Development. Nat. Rev. Cancer 2006, 6, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Surace, A.E.A.; Hedrich, C.M. The Role of Epigenetics in Autoimmune/Inflammatory Disease. Front. Immunol. 2019, 10, 1525. [Google Scholar] [CrossRef]
- Shen, S.; Li, B.; Dai, J.; Wu, Z.; He, Y.; Wen, L.; Wang, X.; Hu, G. BRD4 Inhibition Protects Against Acute Pancreatitis Through Restoring Impaired Autophagic Flux. Front. Pharmacol. 2020, 11, 618. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Vakoc, C.R. The Mechanisms behind the Therapeutic Activity of BET Bromodomain Inhibition. Mol. Cell 2014, 54, 728–736. [Google Scholar] [CrossRef]
- Deeney, J.T.; Belkina, A.C.; Shirihai, O.S.; Corkey, B.E.; Denis, G.V. BET Bromodomain Proteins Brd2, Brd3 and Brd4 Selectively Regulate Metabolic Pathways in the Pancreatic β-cell. PLoS ONE 2016, 11, 1–16. [Google Scholar] [CrossRef]
- Cassetta, L.; Pollard, J.W. Targeting Macrophages: Therapeutic Approaches in Cancer. Nat. Rev. Drug Discov. 2018, 17, 887–904. [Google Scholar] [CrossRef]
- Steele, C.W.; Jamieson, N.B.; Evans, T.R.J.; McKay, C.J.; Sansom, O.J.; Morton, J.P.; Carter, C.R. Exploiting Inflammation for Therapeutic Gain in Pancreatic Cancer. Br. J. Cancer 2013, 108, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Hsu, F.F.; Chiang, M.T.; Li, F.A.; Yeh, C.T.; Lee, W.H.; Chau, L.Y. Acetylation Is Essential for Nuclear Heme Oxygenase-1-Enhanced Tumor Growth and Invasiveness. Oncogene 2017, 36, 6805–6814. [Google Scholar] [CrossRef]
- Liby, K.T.; Royce, D.B.; Risingsong, R.; Williams, C.R.; Maitra, A.; Hruban, R.H.; Sporn, M.B. Synthetic Triterpenoids Prolong Survival in a Transgenic Mouse Model of Pancreatic Cancer. Cancer Prev. Res. 2010, 3, 1427–1434. [Google Scholar] [CrossRef]
- Liby, K.; Risingsong, R.; Royce, D.B.; Williams, C.R.; Yore, M.M.; Honda, T.; Gribble, G.W.; Lamph, W.W.; Vannini, N.; Sogno, I.; et al. Prevention and Treatment of Experimental Estrogen Receptor-Negative Mammary Carcinogenesis by the Synthetic Triterpenoid CDDO-Methyl Ester and the Rexinoid LG100268. Clin. Cancer Res. 2008, 14, 4556–4563. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leal, A.S.; Liby, K.T. The BRD4 Inhibitor I-BET-762 Reduces HO-1 Expression in Macrophages and the Pancreas of Mice. Int. J. Mol. Sci. 2024, 25, 9985. https://doi.org/10.3390/ijms25189985
Leal AS, Liby KT. The BRD4 Inhibitor I-BET-762 Reduces HO-1 Expression in Macrophages and the Pancreas of Mice. International Journal of Molecular Sciences. 2024; 25(18):9985. https://doi.org/10.3390/ijms25189985
Chicago/Turabian StyleLeal, Ana S., and Karen T. Liby. 2024. "The BRD4 Inhibitor I-BET-762 Reduces HO-1 Expression in Macrophages and the Pancreas of Mice" International Journal of Molecular Sciences 25, no. 18: 9985. https://doi.org/10.3390/ijms25189985