
Genome Instability Induced by Topoisomerase Misfunction


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
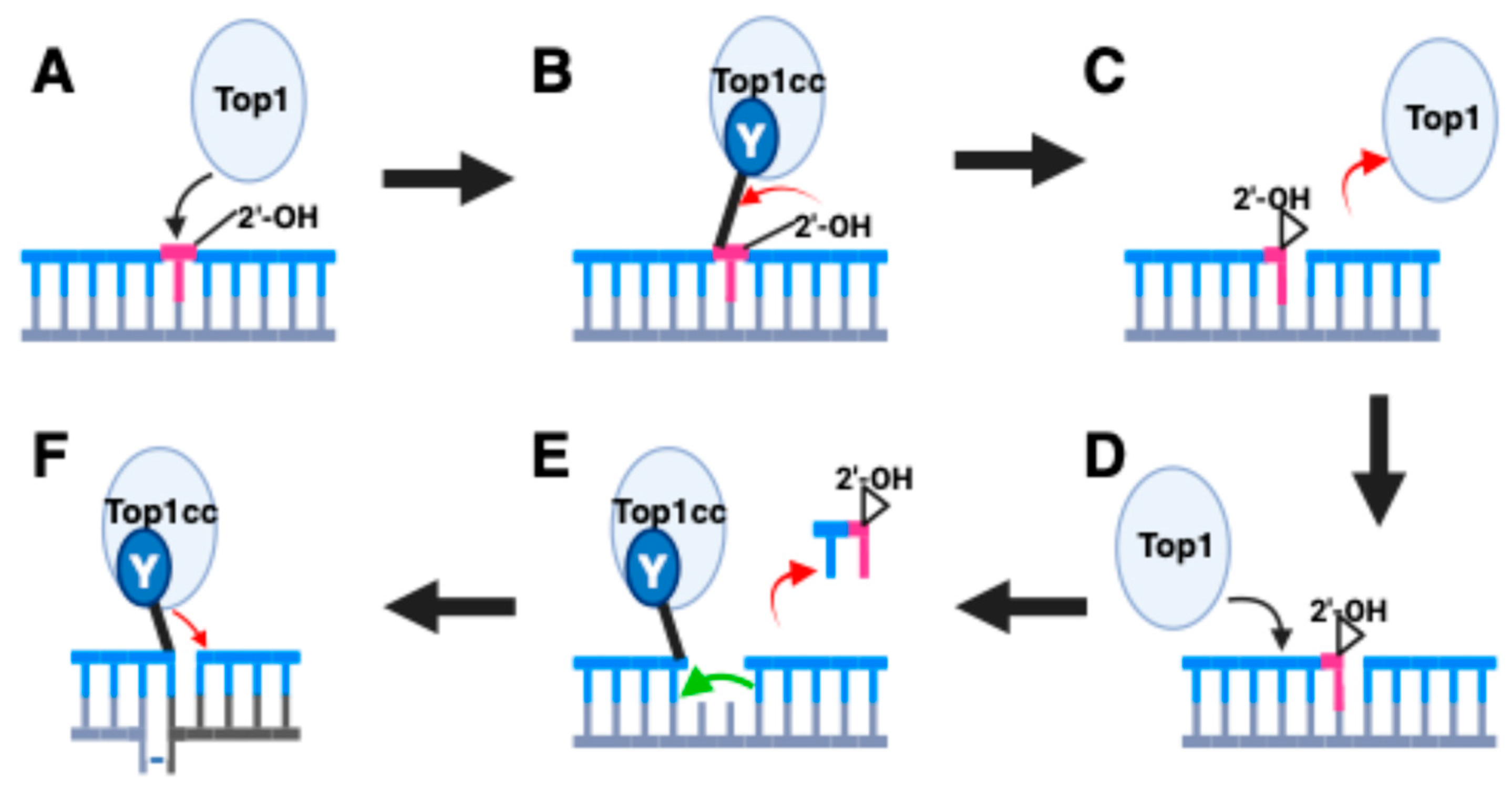
2. Top1 and Genome Instability
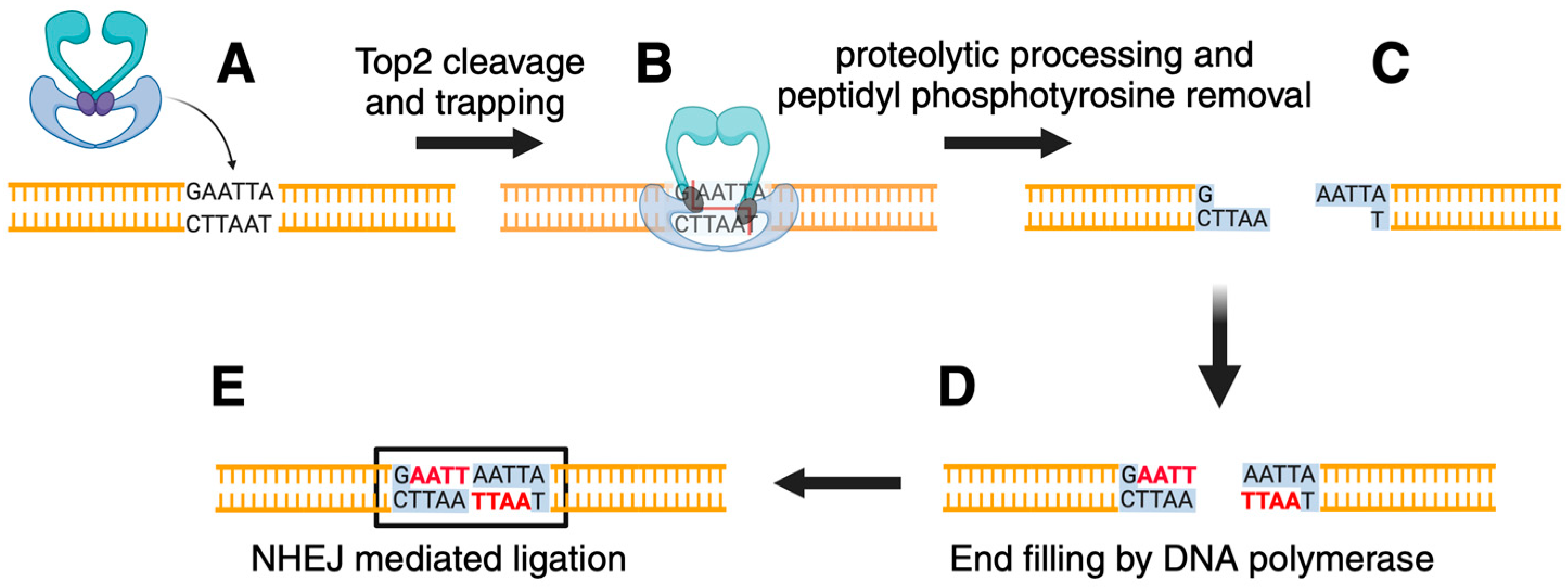
3. Top2 and Genome Stability
4. Top2 and Genome Instability: The Damage Induced by Defective Enzymes
5. Hyper Cleavage Topoisomerases: Biochemical and Structural Views
6. Pathways for Repairing Top2 Damage: The View from Mutation Induction
7. Consequences of Defective Topoisomerase Action: Genome Instability Leading to Cancer and Other Diseases
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, L.F.; Wang, J.C. Supercoiling of the DNA template during transcription. Proc. Natl. Acad. Sci. USA 1987, 84, 7024–7027. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.Y.; Shyy, S.H.; Wang, J.C.; Liu, L.F. Transcription generates positively and negatively supercoiled domains in the template. Cell 1988, 53, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Nitiss, J.L. DNA topoisomerase II and its growing repertoire of biological functions. Nat. Rev. Cancer 2009, 9, 327–337. [Google Scholar] [CrossRef]
- Vos, S.M.; Tretter, E.M.; Schmidt, B.H.; Berger, J.M. All tangled up: How cells direct, manage and exploit topoisomerase function. Nat. Rev. Mol. Cell Biol. 2011, 12, 827–841. [Google Scholar] [CrossRef]
- Wang, J.C. Cellular roles of DNA topoisomerases: A molecular perspective. Nat. Rev. Mol. Cell Biol. 2002, 3, 430–440. [Google Scholar] [CrossRef]
- Wang, J.C. Moving one DNA double helix through another by a type II DNA topoisomerase: The story of a simple molecular machine. Q Rev Biophys 1998, 31, 107–144. [Google Scholar] [CrossRef]
- Nitiss, K.C.; Nitiss, J.L.; Hanakahi, L.A. DNA Damage by an essential enzyme: A delicate balance act on the tightrope. DNA Repair 2019, 82, 102639. [Google Scholar] [CrossRef]
- Pommier, Y.; Nussenzweig, A.; Takeda, S.; Austin, C. Human topoisomerases and their roles in genome stability and organization. Nat. Rev. Mol. Cell Biol. 2022, 23, 407–427. [Google Scholar] [CrossRef] [PubMed]
- Leppard, J.B.; Champoux, J.J. Human DNA topoisomerase I: Relaxation, roles, and damage control. Chromosoma 2005, 114, 75–85. [Google Scholar] [CrossRef]
- Austin, C.A.; Lee, K.C.; Swan, R.L.; Khazeem, M.M.; Manville, C.M.; Cridland, P.; Treumann, A.; Porter, A.; Morris, N.J.; Cowell, I.G. TOP2B: The First Thirty Years. Int. J. Mol. Sci. 2018, 19, 2765. [Google Scholar] [CrossRef]
- Cho, J.E.; Jinks-Robertson, S. Topoisomerase I and Genome Stability: The Good and the Bad. Methods Mol. Biol. 2018, 1703, 21–45. [Google Scholar] [PubMed]
- Uuskula-Reimand, L.; Wilson, M.D. Untangling the roles of TOP2A and TOP2B in transcription and cancer. Sci. Adv. 2022, 8, eadd4920. [Google Scholar] [CrossRef] [PubMed]
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Barcelo, J.M.; Rao, V.A.; Sordet, O.; Jobson, A.G.; Thibaut, L.; Miao, Z.H.; Seiler, J.A.; Zhang, H.; Marchand, C.; et al. Repair of topoisomerase I-mediated DNA damage. Prog. Nucleic Acid. Res. Mol. Biol. 2006, 81, 179–229. [Google Scholar]
- Pommier, Y. Drugging topoisomerases: Lessons and challenges. ACS Chem. Biol. 2013, 8, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Okoro, C.O.; Fatoki, T.H. A Mini Review of Novel Topoisomerase II Inhibitors as Future Anticancer Agents. Int. J. Mol. Sci. 2023, 24, 2532. [Google Scholar] [CrossRef]
- Pommier, Y.; Marchand, C. Interfacial inhibitors: Targeting macromolecular complexes. Nat. Rev. Drug Discov. 2011, 11, 25–36. [Google Scholar] [CrossRef]
- Wu, C.C.; Li, T.K.; Farh, L.; Lin, L.Y.; Lin, T.S.; Yu, Y.J.; Yen, T.J.; Chiang, C.W.; Chan, N.L. Structural basis of type II topoisomerase inhibition by the anticancer drug etoposide. Science 2011, 333, 459–462. [Google Scholar] [CrossRef]
- Staker, B.L.; Hjerrild, K.; Feese, M.D.; Behnke, C.A.; Burgin, A.B., Jr.; Stewart, L. The mechanism of topoisomerase I poisoning by a camptothecin analog. Proc. Natl. Acad. Sci. USA 2002, 99, 15387–15392. [Google Scholar] [CrossRef]
- Blower, T.R.; Bandak, A.; Lee, A.S.Y.; Austin, C.A.; Nitiss, J.L.; Berger, J.M. A complex suite of loci and elements in eukaryotic type II topoisomerases determine selective sensitivity to distinct poisoning agents. Nucleic Acids Res. 2019, 47, 8163–8179. [Google Scholar] [CrossRef]
- Jinks-Robertson, S.; Klein, H.L. Ribonucleotides in DNA: Hidden in plain sight. Nat. Struct. Mol. Biol. 2015, 22, 176–178. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.S.; Kunkel, T.A. Ribonucleotide Incorporation by Eukaryotic B-Family Replicases and Its Implications for Genome Stability. Annu. Rev. Biochem. 2022, 91, 133–155. [Google Scholar] [CrossRef] [PubMed]
- Bossaert, M.; Pipier, A.; Riou, J.F.; Noirot, C.; Nguyen, L.T.; Serre, R.F.; Bouchez, O.; Defrancq, E.; Calsou, P.; Britton, S.; et al. Transcription-associated topoisomerase 2alpha (TOP2A) activity is a major effector of cytotoxicity induced by G-quadruplex ligands. eLife 2021, 10, e65184. [Google Scholar] [CrossRef]
- Stantial, N.; Rogojina, A.; Gilbertson, M.; Sun, Y.; Miles, H.; Shaltz, S.; Berger, J.; Nitiss, K.C.; Jinks-Robertson, S.; Nitiss, J.L. Trapped topoisomerase II initiates formation of de novo duplications via the nonhomologous end-joining pathway in yeast. Proc. Natl. Acad. Sci. USA 2020, 117, 26876–26884. [Google Scholar] [CrossRef]
- Boot, A.; Liu, M.; Stantial, N.; Shah, V.; Yu, W.; Nitiss, K.C.; Nitiss, J.L.; Jinks-Robertson, S.; Rozen, S.G. Recurrent mutations in topoisomerase IIalpha cause a previously undescribed mutator phenotype in human cancers. Proc. Natl. Acad. Sci. USA 2022, 119, e2114024119. [Google Scholar] [CrossRef] [PubMed]
- Levin, N.A.; Bjornsti, M.A.; Fink, G.R. A novel mutation in DNA topoisomerase I of yeast causes DNA damage and RAD9-dependent cell cycle arrest. Genetics 1993, 133, 799–814. [Google Scholar] [CrossRef]
- Ju, B.G.; Lunyak, V.V.; Perissi, V.; Garcia-Bassets, I.; Rose, D.W.; Glass, C.K.; Rosenfeld, M.G. A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science 2006, 312, 1798–1802. [Google Scholar] [CrossRef]
- Haffner, M.C.; De Marzo, A.M.; Meeker, A.K.; Nelson, W.G.; Yegnasubramanian, S. Transcription-induced DNA double strand breaks: Both oncogenic force and potential therapeutic target? Clin. Cancer Res. 2011, 17, 3858–3864. [Google Scholar] [CrossRef]
- Trotter, K.W.; King, H.A.; Archer, T.K. Glucocorticoid Receptor Transcriptional Activation via the BRG1-Dependent Recruitment of TOP2beta and Ku70/86. Mol. Cell Biol. 2015, 35, 2799–2817. [Google Scholar] [CrossRef]
- Madabhushi, R. The Roles of DNA Topoisomerase IIbeta in Transcription. Int. J. Mol. Sci. 2018, 19, 1917. [Google Scholar] [CrossRef]
- Ezoe, S. Secondary leukemia associated with the anti-cancer agent, etoposide, a topoisomerase II inhibitor. Int. J. Env. Environ. Res. Public. Health 2012, 9, 2444–2453. [Google Scholar] [CrossRef] [PubMed]
- Buttmann, M.; Seuffert, L.; Mader, U.; Toyka, K.V. Malignancies after mitoxantrone for multiple sclerosis: A retrospective cohort study. Neurology 2016, 86, 2203–2207. [Google Scholar] [CrossRef] [PubMed]
- Pendleton, M.; Lindsey, R.H., Jr.; Felix, C.A.; Grimwade, D.; Osheroff, N. Topoisomerase II and leukemia. Ann. N. Y. Acad. Sci. 2014, 1310, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Thrash, C.; Voelkel, K.; DiNardo, S.; Sternglanz, R. Identification of Saccharomyces cerevisiae mutants deficient in DNA topoisomerase I activity. J. Biol. Chem. 1984, 259, 1375–1377. [Google Scholar] [CrossRef] [PubMed]
- Uemura, T.; Yanagida, M. Isolation of type I and II DNA topoisomerase mutants from fission yeast: Single and double mutants show different phenotypes in cell growth and chromatin organization. EMBO J. 1984, 3, 1737–1744. [Google Scholar] [CrossRef]
- Manzo, S.G.; Hartono, S.R.; Sanz, L.A.; Marinello, J.; De Biasi, S.; Cossarizza, A.; Capranico, G.; Chedin, F. DNA Topoisomerase I differentially modulates R-loops across the human genome. Genome Biol. 2018, 19, 100. [Google Scholar] [CrossRef]
- Brickner, J.R.; Garzon, J.L.; Cimprich, K.A. Walking a tightrope: The complex balancing act of R-loops in genome stability. Mol. Cell 2022, 82, 2267–2297. [Google Scholar] [CrossRef]
- Robinson, J.; Raguseo, F.; Nuccio, S.P.; Liano, D.; Di Antonio, M. DNA G-quadruplex structures: More than simple roadblocks to transcription? Nucleic Acids Res. 2021, 49, 8419–8431. [Google Scholar] [CrossRef]
- Ghilain, C.; Vidal-Cruchez, O.; Joly, A.; Debatisse, M.; Gilson, E.; Giraud-Panis, M.J. Innovative Tools for DNA Topology Probing in Human Cells Reveal a Build-Up of Positive Supercoils Following Replication Stress at Telomeres and at the FRA3B Fragile Site. Cells 2024, 13, 1361. [Google Scholar] [CrossRef]
- Christman, M.F.; Dietrich, F.S.; Fink, G.R. Mitotic recombination in the rDNA of S. cerevisiae is suppressed by the combined action of DNA topoisomerases I and II. Cell 1988, 55, 413–425. [Google Scholar] [CrossRef]
- D’Alfonso, A.; Micheli, G.; Camilloni, G. rDNA transcription, replication and stability in Saccharomyces cerevisiae. Semin. Cell Dev. Biol. 2024, 159, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.A.; Wang, J.C. A subthreshold level of DNA topoisomerases leads to the excision of yeast rDNA as extrachromosomal rings. Cell 1989, 57, 975–985. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, D.A.; Guarente, L. Extrachromosomal rDNA circles—A cause of aging in yeast. Cell 1997, 91, 1033–1042. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, T.; Kumar, P.; Koseoglu, M.M.; Dutta, A. Discoveries of Extrachromosomal Circles of DNA in Normal and Tumor Cells. Trends Genet. 2018, 34, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Kerrigan, D.; Covey, J.M.; Kao-Shan, C.S.; Whang-Peng, J. Sister chromatid exchanges, chromosomal aberrations, and cytotoxicity produced by antitumor topoisomerase II inhibitors in sensitive (DC3F) and resistant (DC3F/9-OHE) Chinese hamster cells. Cancer Res. 1988, 48, 512–516. [Google Scholar]
- Nitiss, J.; Wang, J.C. DNA topoisomerase-targeting antitumor drugs can be studied in yeast. Proc. Natl. Acad. Sci. USA 1988, 85, 7501–7505. [Google Scholar] [CrossRef]
- Westhorpe, R.; Roske, J.J.; Yeeles, J.T.P. Mechanisms controlling replication fork stalling and collapse at topoisomerase 1 cleavage complexes. Mol. Cell 2024, 84, 3469–3481. [Google Scholar] [CrossRef]
- Milano, L.; Gautam, A.; Caldecott, K.W. DNA damage and transcription stress. Mol. Cell 2024, 84, 70–79. [Google Scholar] [CrossRef]
- Pourquier, P.; Pilon, A.A.; Kohlhagen, G.; Mazumder, A.; Sharma, A.; Pommier, Y. Trapping of mammalian topoisomerase I and recombinations induced by damaged DNA containing nicks or gaps. Importance of DNA end phosphorylation and camptothecin effects. J. Biol. Chem. 1997, 272, 26441–26447. [Google Scholar] [CrossRef]
- Lippert, M.J.; Kim, N.; Cho, J.E.; Larson, R.P.; Schoenly, N.E.; O’Shea, S.H.; Jinks-Robertson, S. Role for topoisomerase 1 in transcription-associated mutagenesis in yeast. Proc. Natl. Acad. Sci. USA 2011, 108, 698–703. [Google Scholar] [CrossRef]
- Takahashi, T.; Burguiere-Slezak, G.; Van der Kemp, P.A.; Boiteux, S. Topoisomerase 1 provokes the formation of short deletions in repeated sequences upon high transcription in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2011, 108, 692–697. [Google Scholar] [CrossRef]
- Kim, N.; Huang, S.N.; Williams, J.S.; Li, Y.C.; Clark, A.B.; Cho, J.E.; Kunkel, T.A.; Pommier, Y.; Jinks-Robertson, S. Mutagenic processing of ribonucleotides in DNA by yeast topoisomerase I. Science 2011, 332, 1561–1564. [Google Scholar] [CrossRef]
- Zhou, Z.X.; Williams, J.S.; Lujan, S.A.; Kunkel, T.A. Ribonucleotide incorporation into DNA during DNA replication and its consequences. Crit. Rev. Biochem. Mol. Biol. 2021, 56, 109–124. [Google Scholar] [CrossRef]
- Sekiguchi, J.; Shuman, S. Site-specific ribonuclease activity of eukaryotic DNA topoisomerase I. Mol. Cell 1997, 1, 89–97. [Google Scholar] [CrossRef]
- Sparks, J.L.; Burgers, P.M. Error-free and mutagenic processing of topoisomerase 1-provoked damage at genomic ribonucleotides. EMBO J. 2015, 34, 1259–1269. [Google Scholar] [CrossRef]
- Huang, S.Y.; Ghosh, S.; Pommier, Y. Topoisomerase I alone is sufficient to produce short DNA deletions and can also reverse nicks at ribonucleotide sites. J. Biol. Chem. 2015, 290, 14068–14076. [Google Scholar] [CrossRef]
- Cerritelli, S.M.; Crouch, R.J. RNase H2-RED carpets the path to eukaryotic RNase H2 functions. DNA Repair 2019, 84, 102736. [Google Scholar] [CrossRef]
- Colley, W.C.; van der Merwe, M.; Vance, J.R.; Burgin, A.B., Jr.; Bjornsti, M.A. Substitution of conserved residues within the active site alters the cleavage religation equilibrium of DNA topoisomerase I. J. Biol. Chem. 2004, 279, 54069–54078. [Google Scholar] [CrossRef]
- Fertala, J.; Vance, J.R.; Pourquier, P.; Pommier, Y.; Bjornsti, M.A. Substitutions of Asn-726 in the active site of yeast DNA topoisomerase I define novel mechanisms of stabilizing the covalent enzyme-DNA intermediate. J. Biol. Chem. 2000, 275, 15246–15253. [Google Scholar] [CrossRef]
- Cho, J.E.; Jinks-Robertson, S. Deletions associated with stabilization of the Top1 cleavage complex in yeast are products of the nonhomologous end-joining pathway. Proc. Natl. Acad. Sci. USA 2019, 116, 22683–22691. [Google Scholar] [CrossRef]
- Cristini, A.; Ricci, G.; Britton, S.; Salimbeni, S.; Huang, S.N.; Marinello, J.; Calsou, P.; Pommier, Y.; Favre, G.; Capranico, G.; et al. Dual Processing of R-Loops and Topoisomerase I Induces Transcription-Dependent DNA Double-Strand Breaks. Cell Rep. 2019, 28, 3167–3181. [Google Scholar] [CrossRef]
- Rubio-Contreras, D.; Gomez-Herreros, F. TDP1 suppresses chromosomal translocations and cell death induced by abortive TOP1 activity during gene transcription. Nat. Commun. 2023, 14, 6940. [Google Scholar] [CrossRef] [PubMed]
- Lebedeva, N.; Auffret Vander Kemp, P.; Bjornsti, M.A.; Lavrik, O.; Boiteux, S. Trapping of DNA topoisomerase I on nick-containing DNA in cell free extracts of Saccharomyces cerevisiae. DNA Repair 2006, 5, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Reijns, M.A.M.; Parry, D.A.; Williams, T.C.; Nadeu, F.; Hindshaw, R.L.; Rios Szwed, D.O.; Nicholson, M.D.; Carroll, P.; Boyle, S.; Royo, R.; et al. Signatures of TOP1 transcription-associated mutagenesis in cancer and germline. Nature 2022, 602, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Sun, Y.; Huang, S.N.; Nitiss, J.L. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell Biol. 2016, 17, 703–721. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, K.; Ikegami, Y.; Ishida, R.; Andoh, T. Inhibition of topoisomerase II by antitumor agents bis (2.6-dioxopiperazine) derivatives. Cancer Res. 1991, 51, 4903–4908. [Google Scholar]
- Drake, F.H.; Hofmann, G.A.; Mong, S.M.; Bartus, J.O.; Hertzberg, R.P.; Johnson, R.K.; Mattern, M.R.; Mirabelli, C.K. In vitro and intracellular inhibition of topoisomerase II by the antitumor agent merbarone. Cancer Res. 1989, 49, 2578–2583. [Google Scholar]
- Jensen, L.H.; Nitiss, K.C.; Rose, A.; Dong, J.; Zhou, J.; Hu, T.; Osheroff, N.; Jensen, P.B.; Sehested, M.; Nitiss, J.L. A novel mechanism of cell killing by anti-topoisomerase II bisdioxopiperazines. J. Biol. Chem. 2000, 275, 2137–2146. [Google Scholar] [CrossRef]
- Amoiridis, M.; Verigos, J.; Meaburn, K.; Gittens, W.H.; Ye, T.; Neale, M.J.; Soutoglou, E. Inhibition of topoisomerase 2 catalytic activity impacts the integrity of heterochromatin and repetitive DNA and leads to interlinks between clustered repeats. Nat. Commun. 2024, 15, 5727. [Google Scholar] [CrossRef]
- Fortune, J.M.; Osheroff, N. Merbarone inhibits the catalytic activity of human topoisomerase IIalpha by blocking DNA cleavage. J. Biol. Chem. 1998, 273, 17643–17650. [Google Scholar] [CrossRef]
- Spell, R.M.; Holm, C. Nature and distribution of chromosomal intertwinings in Saccharomyces cerevisiae. Mol. Cell Biol. 1994, 14, 1465–1476. [Google Scholar]
- Jeggo, P.A.; Caldecott, K.; Pidsley, S.; Banks, G.R. Sensitivity of Chinese hamster ovary mutants defective in DNA double strand break repair to topoisomerase II inhibitors. Cancer Res. 1989, 49, 7057–7063. [Google Scholar]
- Kerrigan, D.; Pommier, Y.; Kohn, K.W. Protein-linked DNA strand breaks produced by etoposide and teniposide in mouse L1210 and human VA-13 and HT-29 cell lines: Relationship to cytotoxicity. NCI Monogr. 1987, 4, 117–121. [Google Scholar]
- Anderson, R.D.; Berger, N.A. International Commission for Protection Against Environmental Mutagens and Carcinogens. Mutagenicity and carcinogenicity of topoisomerase-interactive agents. Mutat. Res. 1994, 309, 109–142. [Google Scholar] [CrossRef]
- Berger, N.A.; Chatterjee, S.; Schmotzer, J.A.; Helms, S.R. Etoposide (VP-16-213)-induced gene alterations: Potential contribution to cell death. Proc. Natl. Acad. Sci. USA 1991, 88, 8740–8743. [Google Scholar] [CrossRef]
- Masurekar, M.; Krueuzer, K.N.; Ripley, L.S. The specificity of topoisomerase-mediated DNA cleavage defines acridine-induced frameshift specificity within a hotspot in bacteriophage T4. Genetics 1991, 128, 193. [Google Scholar] [CrossRef]
- Helbig, R.; Gerland, E.; Speit, G. The molecular nature of mutations induced by adriamycin at the hprt locus of V79 cells. Mutagenesis 1994, 9, 113–116. [Google Scholar] [CrossRef]
- Zhang, W.; Gou, P.; Dupret, J.M.; Chomienne, C.; Rodrigues-Lima, F. Etoposide, an anticancer drug involved in therapy-related secondary leukemia: Enzymes at play. Transl. Oncol. 2021, 14, 101169. [Google Scholar] [CrossRef]
- Lovett, B.D.; Lo Nigro, L.; Rappaport, E.F.; Blair, I.A.; Osheroff, N.; Zheng, N.; Megonigal, M.D.; Williams, W.R.; Nowell, P.C.; Felix, C.A. Near-precise interchromosomal recombination and functional DNA topoisomerase II cleavage sites at MLL and AF-4 genomic breakpoints in treatment-related acute lymphoblastic leukemia with t (4;11) translocation. Proc. Natl. Acad. Sci. USA 2001, 98, 9802–9807. [Google Scholar] [CrossRef]
- Lovett, B.D.; Strumberg, D.; Blair, I.A.; Pang, S.; Burden, D.A.; Megonigal, M.D.; Rappaport, E.F.; Rebbeck, T.R.; Osheroff, N.; Pommier, Y.G.; et al. Etoposide metabolites enhance DNA topoisomerase II cleavage near leukemia-associated MLL translocation breakpoints. Biochemistry 2001, 40, 1159–1170. [Google Scholar] [CrossRef]
- Deweese, J.E.; Osheroff, N. The DNA cleavage reaction of topoisomerase II: Wolf in sheep’s clothing. Nucleic Acids Res. 2009, 37, 738–748. [Google Scholar] [CrossRef]
- Canela, A.; Maman, Y.; Huang, S.N.; Wutz, G.; Tang, W.; Zagnoli-Vieira, G.; Callen, E.; Wong, N.; Day, A.; Peters, J.M.; et al. Topoisomerase II-Induced Chromosome Breakage and Translocation Is Determined by Chromosome Architecture and Transcriptional Activity. Mol. Cell 2019, 75, 252–266. [Google Scholar] [CrossRef]
- Sciascia, N.; Wu, W.; Zong, D.; Sun, Y.; Wong, N.; John, S.; Wangsa, D.; Ried, T.; Bunting, S.F.; Pommier, Y.; et al. Suppressing proteasome mediated processing of topoisomerase II DNA-protein complexes preserves genome integrity. eLife 2020, 9, e53447. [Google Scholar] [CrossRef]
- Azarova, A.M.; Lyu, Y.L.; Lin, C.P.; Tsai, Y.C.; Lau, J.Y.; Wang, J.C.; Liu, L.F. Roles of DNA topoisomerase II isozymes in chemotherapy and secondary malignancies. Proc. Natl. Acad. Sci. USA 2007, 104, 11014–11019. [Google Scholar] [CrossRef]
- Kingma, P.S.; Corbett, A.H.; Burcham, P.C.; Marnett, L.J.; Osheroff, N. Abasic sites stimulate double-stranded DNA cleavage mediated by topoisomerase II. DNA lesions as endogenous topoisomerase II poisons. J. Biol. Chem. 1995, 270, 21441–21444. [Google Scholar] [CrossRef]
- Sabourin, M.; Osheroff, N. Sensitivity of human type II topoisomerases to DNA damage: Stimulation of enzyme-mediated DNA cleavage by abasic, oxidized and alkylated lesions. Nucleic Acids Res. 2000, 28, 1947–1954. [Google Scholar] [CrossRef]
- Wang, Y.; Knudsen, B.R.; Bjergbaek, L.; Westergaard, O.; Andersen, A.H. Stimulated activity of human topoisomerases IIalpha and IIbeta on RNA-containing substrates. J. Biol. Chem. 1999, 274, 22839–22846. [Google Scholar] [CrossRef]
- Pourquier, P.; Waltman, J.L.; Urasaki, Y.; Loktionova, N.A.; Pegg, A.E.; Nitiss, J.L.; Pommier, Y. Topoisomerase I-mediated cytotoxicity of N-methyl-N’-nitro-N-nitrosoguanidine: Trapping of topoisomerase I by the O6-methylguanine. Cancer Res. 2001, 61, 53–58. [Google Scholar]
- Nitiss, J.L.; Nitiss, K.C.; Rose, A.; Waltman, J.L. Overexpression of type I topoisomerases sensitizes yeast cells to DNA damage. J. Biol. Chem. 2001, 276, 26708–26714. [Google Scholar] [CrossRef]
- Nitiss, J.L. DNA Topoisomerases in DNA Repair and DNA Damage Tolerance. In DNA Damage and Repair: DNA Repair in Higher Eukaryotes; Nickoloff, J.A., Hoekstra, M.F., Eds.; Humana Press: Totowa, NJ, USA, 1998; Volume 2, pp. 517–537. [Google Scholar]
- Walker, J.V.; Nitiss, K.C.; Jensen, L.H.; Mayne, C.; Hu, T.; Jensen, P.B.; Sehested, M.; Hsieh, T.; Nitiss, J.L. A mutation in human topoisomerase II alpha whose expression is lethal in DNA repair-deficient yeast cells. J. Biol. Chem. 2004, 279, 25947–25954. [Google Scholar] [CrossRef]
- Whelan, W.L.; Gocke, E.; Manney, T.R. The CAN1 locus of Saccharomyces cerevisiae: Fine-structure analysis and forward mutation rates. Genetics 1979, 91, 35–51. [Google Scholar] [CrossRef]
- Lang, G.I.; Murray, A.W. Estimating the per-base-pair mutation rate in the yeast Saccharomyces cerevisiae. Genetics 2008, 178, 67–82. [Google Scholar] [CrossRef]
- Nitiss, K.C.; Malik, M.; He, X.; White, S.W.; Nitiss, J.L. Tyrosyl-DNA phosphodiesterase (Tdp1) participates in the repair of Top2-mediated DNA damage. Proc. Natl. Acad. Sci. USA 2006, 103, 8953–8958. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef]
- Bandak, A.F.; Blower, T.R.; Nitiss, K.C.; Gupta, R.; Lau, A.Y.; Guha, R.; Nitiss, J.L.; Berger, J.M. Naturally mutagenic sequence diversity in a human type II topoisomerase. Proc. Natl. Acad. Sci. USA 2023, 120, e2302064120. [Google Scholar] [CrossRef]
- Chen, S.H.; Chan, N.L.; Hsieh, T.S. New mechanistic and functional insights into DNA topoisomerases. Annu. Rev. Biochem. 2013, 82, 139–170. [Google Scholar] [CrossRef]
- Vidmar, V.; Vayssieres, M.; Lamour, V. What’s on the Other Side of the Gate: A Structural Perspective on DNA Gate Opening of Type IA and IIA DNA Topoisomerases. Int. J. Mol. Sci. 2023, 24, 3986. [Google Scholar] [CrossRef]
- Evoli, S.; Kariyawasam, N.L.; Nitiss, K.C.; Nitiss, J.L.; Wereszczynski, J. Modeling allosteric mechanisms of eukaryotic type II topoisomerases. Biophys. J. 2024, 123, 1620–1634. [Google Scholar] [CrossRef]
- Bandak, A.F.; Blower, T.R.; Nitiss, K.C.; Shah, V.; Nitiss, J.L.; Berger, J.M. Using energy to go downhill-a genoprotective role for ATPase activity in DNA topoisomerase II. Nucleic Acids Res. 2024, 52, 1313–1324. [Google Scholar] [CrossRef]
- Sun, Y.; Saha, L.K.; Saha, S.; Jo, U.; Pommier, Y. Debulking of topoisomerase DNA-protein crosslinks (TOP-DPC) by the proteasome, non-proteasomal and non-proteolytic pathways. DNA Repair 2020, 94, 102926. [Google Scholar] [CrossRef]
- Riccio, A.A.; Schellenberg, M.J.; Williams, R.S. Molecular mechanisms of topoisomerase 2 DNA-protein crosslink resolution. Cell Mol. Life Sci. 2020, 77, 81–91. [Google Scholar] [CrossRef]
- Zagnoli-Vieira, G.; Caldecott, K.W. Untangling trapped topoisomerases with tyrosyl-DNA phosphodiesterases. DNA Repair 2020, 94, 102900. [Google Scholar] [CrossRef]
- Mao, Y.; Desai, S.D.; Ting, C.Y.; Hwang, J.; Liu, L.F. 26 S proteasome-mediated degradation of topoisomerase II cleavable complexes. J. Biol. Chem. 2001, 276, 40652–40658. [Google Scholar] [CrossRef]
- Tammaro, M.; Barr, P.; Ricci, B.; Yan, H. Replication-dependent and transcription-dependent mechanisms of DNA double-strand break induction by the topoisomerase 2-targeting drug etoposide. PLoS ONE 2013, 8, e79202. [Google Scholar] [CrossRef]
- Vaz, B.; Popovic, M.; Newman, J.A.; Fielden, J.; Aitkenhead, H.; Halder, S.; Singh, A.N.; Vendrell, I.; Fischer, R.; Torrecilla, I.; et al. Metalloprotease SPRTN/DVC1 Orchestrates Replication-Coupled DNA-Protein Crosslink Repair. Mol. Cell 2016, 64, 704–719. [Google Scholar] [CrossRef]
- Huang, M.; Yao, F.; Nie, L.; Wang, C.; Su, D.; Zhang, H.; Li, S.; Tang, M.; Feng, X.; Yu, B.; et al. FACS-based genome-wide CRISPR screens define key regulators of DNA damage signaling pathways. Mol. Cell 2023, 83, 2810–2828.e6. [Google Scholar] [CrossRef]
- Maede, Y.; Shimizu, H.; Fukushima, T.; Kogame, T.; Nakamura, T.; Miki, T.; Takeda, S.; Pommier, Y.; Murai, J. Differential and common DNA repair pathways for topoisomerase I- and II-targeted drugs in a genetic DT40 repair cell screen panel. Mol. Cancer Ther. 2014, 13, 214–220. [Google Scholar] [CrossRef]
- Borda, M.A.; Palmitelli, M.; Veron, G.; Gonzalez-Cid, M.; de Campos, M.N. Tyrosyl-DNA-phosphodiesterase I (TDP1) participates in the removal and repair of stabilized-Top2alpha cleavage complexes in human cells. Mutat. Res. 2015, 781, 37–48. [Google Scholar] [CrossRef]
- Hartsuiker, E.; Neale, M.J.; Carr, A.M. Distinct requirements for the Rad32(Mre11) nuclease and Ctp1(CtIP) in the removal of covalently bound topoisomerase I and II from DNA. Mol. Cell 2009, 33, 117–123. [Google Scholar] [CrossRef]
- Lee, K.C.; Padget, K.; Curtis, H.; Cowell, I.G.; Moiani, D.; Sondka, Z.; Morris, N.J.; Jackson, G.H.; Cockell, S.J.; Tainer, J.A.; et al. MRE11 facilitates the removal of human topoisomerase II complexes from genomic DNA. Biol. Open 2012, 1, 863–873. [Google Scholar] [CrossRef]
- Hoa, N.N.; Shimizu, T.; Zhou, Z.W.; Wang, Z.Q.; Deshpande, R.A.; Paull, T.T.; Akter, S.; Tsuda, M.; Furuta, R.; Tsutsui, K.; et al. Mre11 Is Essential for the Removal of Lethal Topoisomerase 2 Covalent Cleavage Complexes. Mol. Cell 2016, 64, 1010. [Google Scholar] [CrossRef]
- Sun, Y.; Soans, E.; Mishina, M.; Petricci, E.; Pommier, Y.; Nitiss, K.C.; Nitiss, J.L. Requirements for MRN endonuclease processing of topoisomerase II-mediated DNA damage in mammalian cells. Front. Mol. Biosci. 2022, 9, 1007064. [Google Scholar] [CrossRef]
- Schellenberg, M.J.; Lieberman, J.A.; Herrero-Ruiz, A.; Butler, L.R.; Williams, J.G.; Munoz-Cabello, A.M.; Mueller, G.A.; London, R.E.; Cortes-Ledesma, F.; Williams, R.S. ZATT (ZNF451)-mediated resolution of topoisomerase 2 DNA-protein cross-links. Science 2017, 357, 1412–1416. [Google Scholar] [CrossRef]
- D’Alessandro, G.; Morales-Juarez, D.A.; Richards, S.L.; Nitiss, K.C.; Serrano-Benitez, A.; Wang, J.; Thomas, J.C.; Gupta, V.; Voigt, A.; Belotserkovskaya, R.; et al. RAD54L2 counters TOP2-DNA adducts to promote genome stability. Sci. Adv. 2023, 9, eadl2108. [Google Scholar] [CrossRef]
- Zhang, H.; Xiong, Y.; Sun, Y.; Park, J.M.; Su, D.; Feng, X.; Keast, S.; Tang, M.; Huang, M.; Wang, C.; et al. RAD54L2-mediated DNA damage avoidance pathway specifically preserves genome integrity in response to topoisomerase 2 poisons. Sci. Adv. 2023, 9, eadi6681. [Google Scholar] [CrossRef]
- Malik, M.; Nitiss, K.C.; Enriquez-Rios, V.; Nitiss, J.L. Roles of nonhomologous end-joining pathways in surviving topoisomerase II-mediated DNA damage. Mol. Cancer Ther. 2006, 5, 1405–1414. [Google Scholar] [CrossRef]
- Mistry, A.R.; Felix, C.A.; Whitmarsh, R.J.; Mason, A.; Reiter, A.; Cassinat, B.; Parry, A.; Walz, C.; Wiemels, J.L.; Segal, M.R.; et al. DNA topoisomerase II in therapy-related acute promyelocytic leukemia. N. Engl. J. Med. 2005, 352, 1529–1538. [Google Scholar] [CrossRef]
- Negrini, M.; Felix, C.A.; Martin, C.; Lange, B.J.; Nakamura, T.; Canaani, E.; Croce, C.M. Potential topoisomerase II DNA-binding sites at the breakpoints of a t (9;11) chromosome translocation in acute myeloid leukemia. Cancer Res. 1993, 53, 4489–4492. [Google Scholar]
- Ellis, R.; Brown, S.; Boggild, M. Therapy-related acute leukaemia with mitoxantrone: Four years on, what is the risk and can it be limited? Mult. Scler. 2015, 21, 642–645. [Google Scholar] [CrossRef]
- Merrouche, Y.; Mugneret, F.; Cahn, J.Y. Secondary acute promyelocytic leukemia following irinotecan and oxaliplatin for advanced colon cancer. Ann. Oncol. 2006, 17, 1025–1026. [Google Scholar] [CrossRef]
- Xue, Y.; Lu, D.; Guo, Y.; Lin, B. Specific chromosomal translocations and therapy-related leukemia induced by bimolane therapy for psoriasis. Leuk. Res. 1992, 16, 1113–1123. [Google Scholar] [CrossRef]
- Vuong, M.C.; Hasegawa, L.S.; Eastmond, D.A. A comparative study of the cytotoxic and genotoxic effects of ICRF-154 and bimolane, two catalytic inhibitors of topoisomerase II. Mutat. Res. 2013, 750, 63–71. [Google Scholar] [CrossRef]
- Jin, H.; Gulhan, D.C.; Geiger, B.; Ben-Isvy, D.; Geng, D.; Ljungstrom, V.; Park, P.J. Accurate and sensitive mutational signature analysis with MuSiCal. Nat. Genet. 2024, 56, 541–552. [Google Scholar] [CrossRef]
- Broderick, L.; Clay, G.M.; Blum, R.H.; Liu, Y.; McVicar, R.; Papes, F.; Booshehri, L.M.; Cowell, I.G.; Austin, C.A.; Putnam, C.D.; et al. Disease-associated mutations in topoisomerase IIbeta result in defective NK cells. J. Allergy Clin. Immunol. 2022, 149, 2171–2176.e3. [Google Scholar] [CrossRef]
- Lam, C.W.; Yeung, W.L.; Law, C.Y. Global developmental delay and intellectual disability associated with a de novo TOP2B mutation. Clin Chim Acta 2017, 469, 63–68. [Google Scholar] [CrossRef]
- Scott, P.; Al Kindi, A.; Al Fahdi, A.; Al Yarubi, N.; Bruwer, Z.; Al Adawi, S.; Nandhagopal, R. Spinocerebellar ataxia with axonal neuropathy type 1 revisited. J. Clin. Neurosci. 2019, 67, 139–144. [Google Scholar] [CrossRef]
- Zagnoli-Vieira, G.; Bruni, F.; Thompson, K.; He, L.; Walker, S.; de Brouwer, A.P.M.; Taylor, R.W.; Niyazov, D.; Caldecott, K.W. Confirming TDP2 mutation in spinocerebellar ataxia autosomal recessive 23 (SCAR23). Neurol. Genet. 2018, 4, e262. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nitiss, K.C.; Bandak, A.; Berger, J.M.; Nitiss, J.L. Genome Instability Induced by Topoisomerase Misfunction. Int. J. Mol. Sci. 2024, 25, 10247. https://doi.org/10.3390/ijms251910247
Nitiss KC, Bandak A, Berger JM, Nitiss JL. Genome Instability Induced by Topoisomerase Misfunction. International Journal of Molecular Sciences. 2024; 25(19):10247. https://doi.org/10.3390/ijms251910247
Chicago/Turabian StyleNitiss, Karin C., Afif Bandak, James M. Berger, and John L. Nitiss. 2024. "Genome Instability Induced by Topoisomerase Misfunction" International Journal of Molecular Sciences 25, no. 19: 10247. https://doi.org/10.3390/ijms251910247