

Taldefgrobep Alfa and the Phase 3 RESILIENT Trial in Spinal Muscular Atrophy

 , ,
, ,
Abstract
:1. Introduction
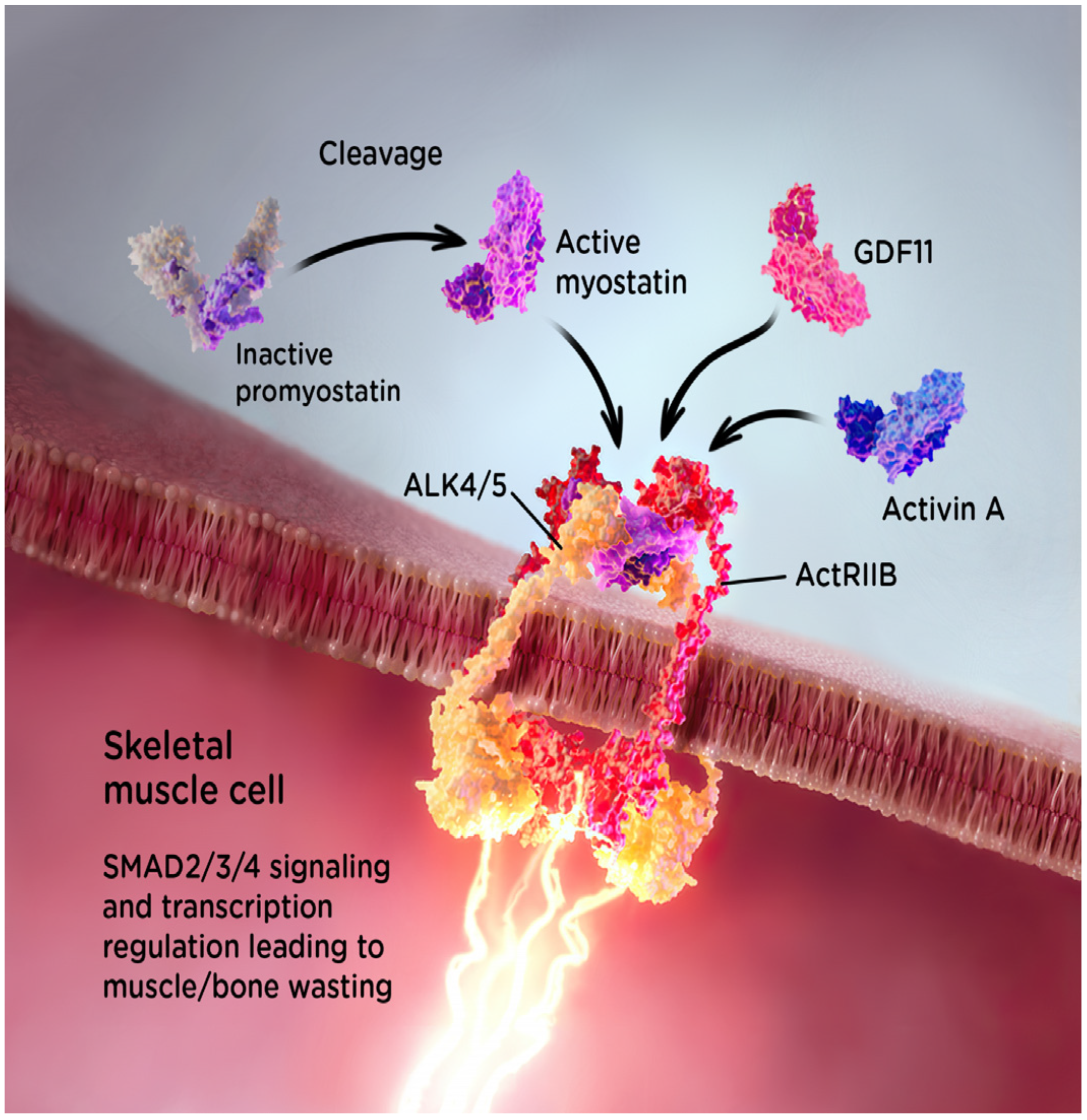
2. Myostatin and Muscle Growth
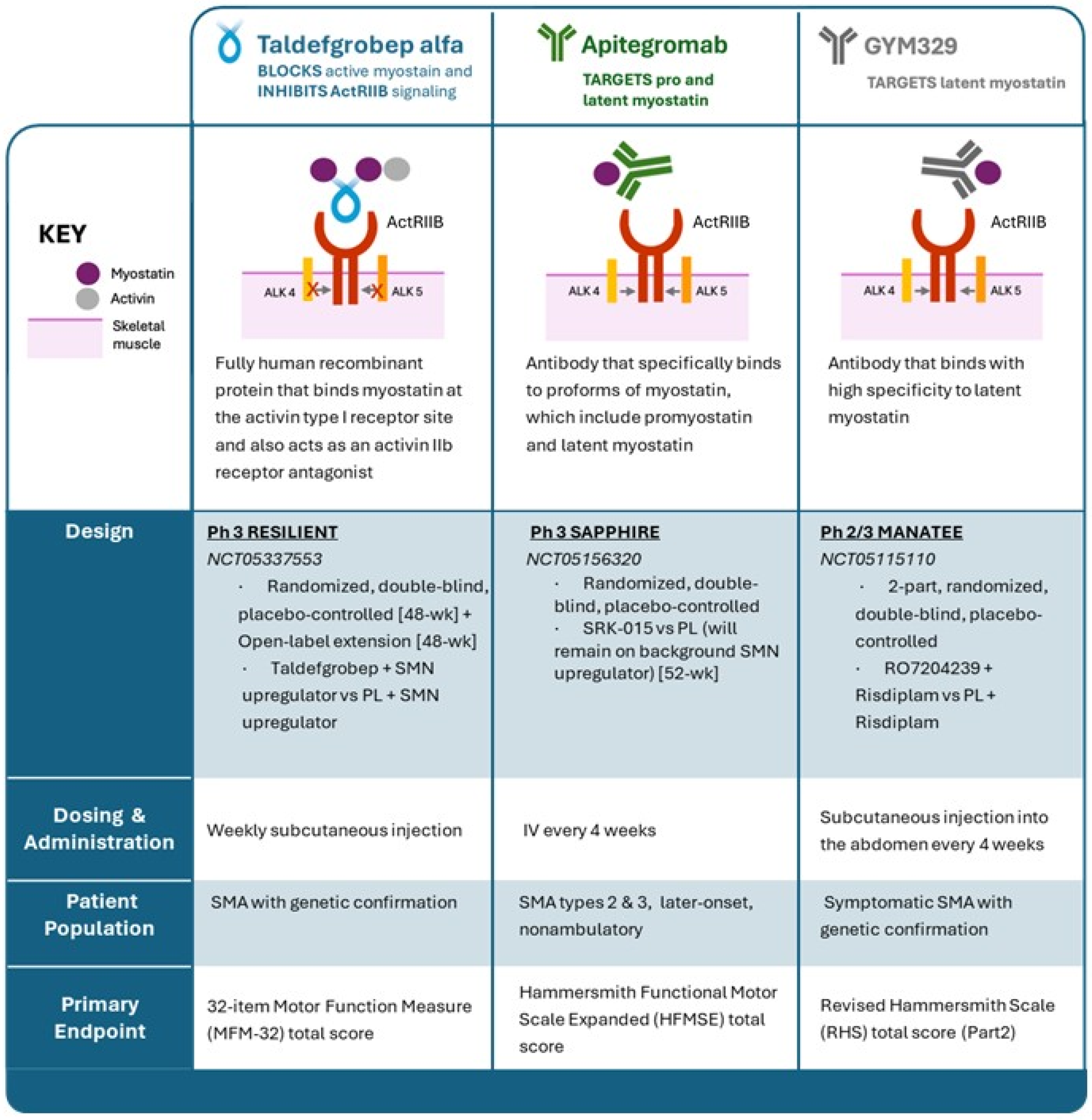
3. Taldefgrobep Alfa
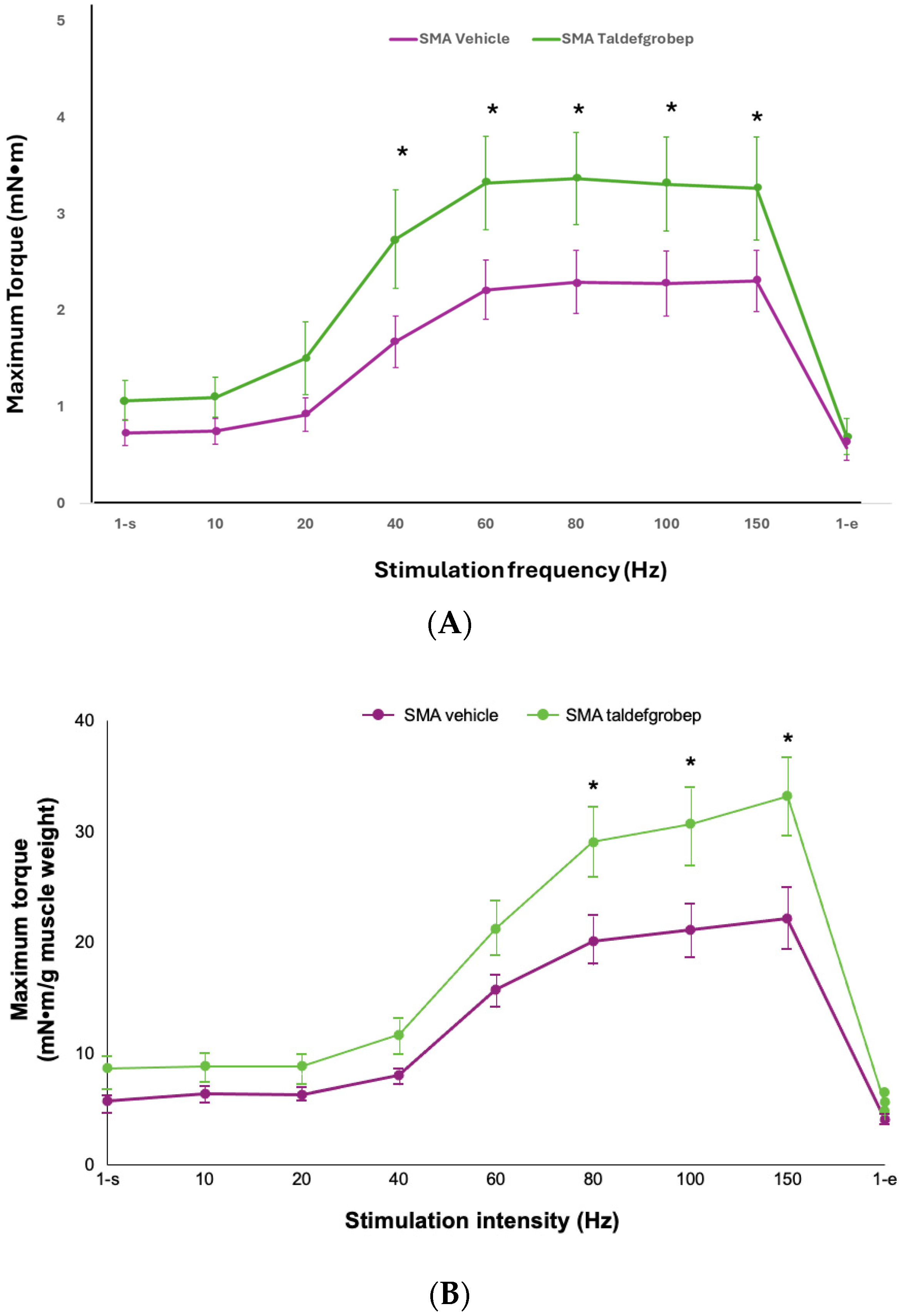
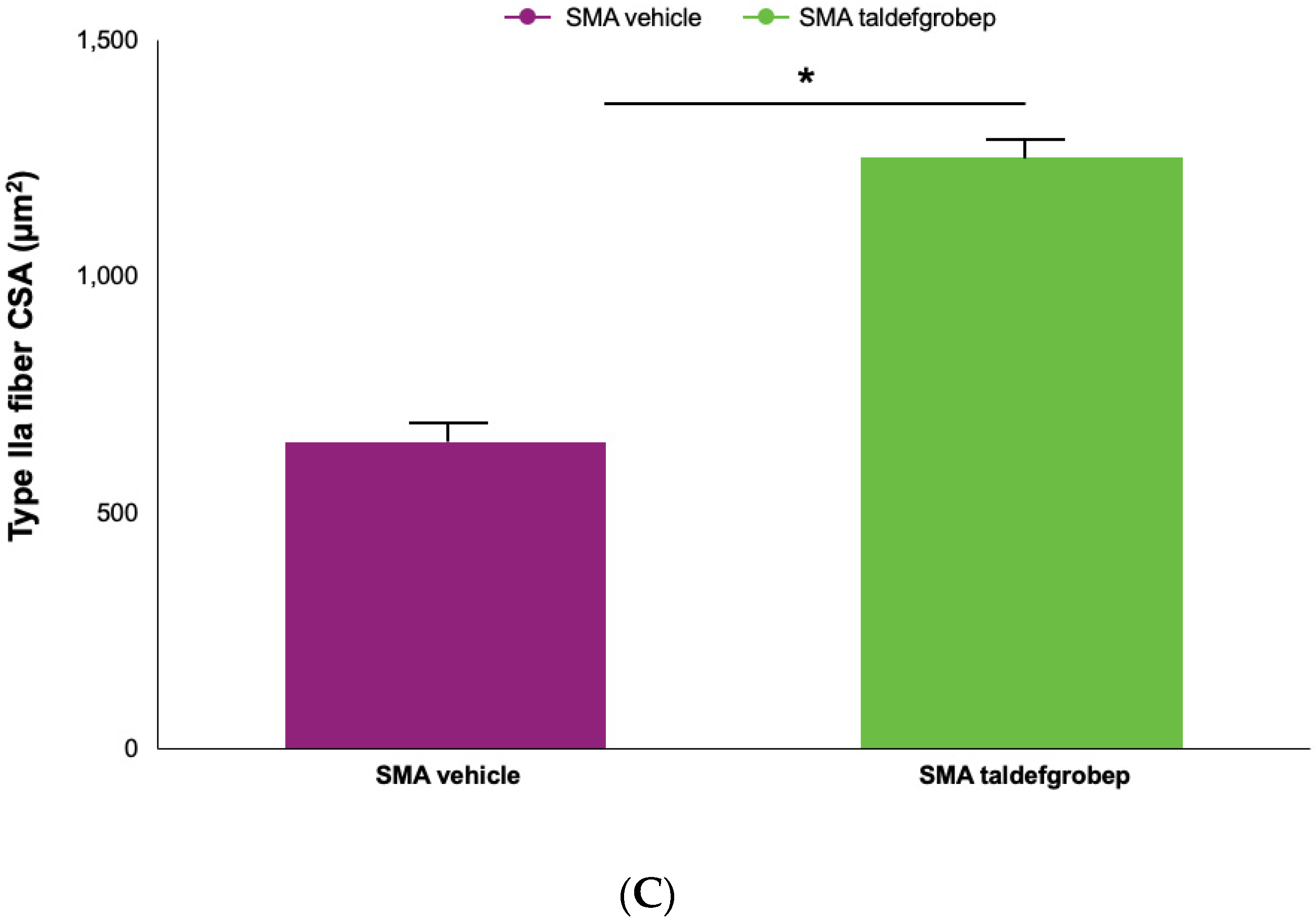
3.1. Preclinical Studies of Taldefgrobep in SMA
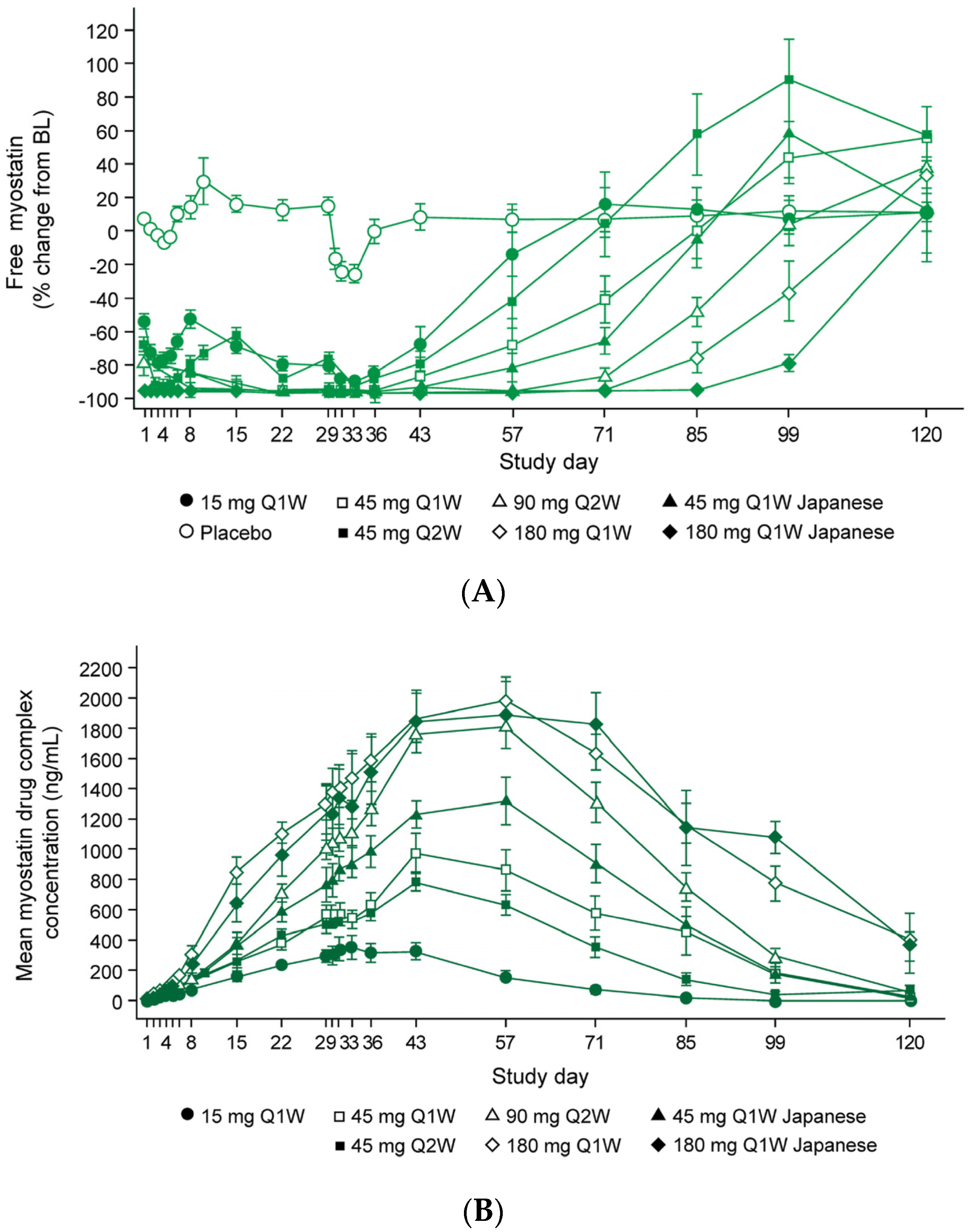
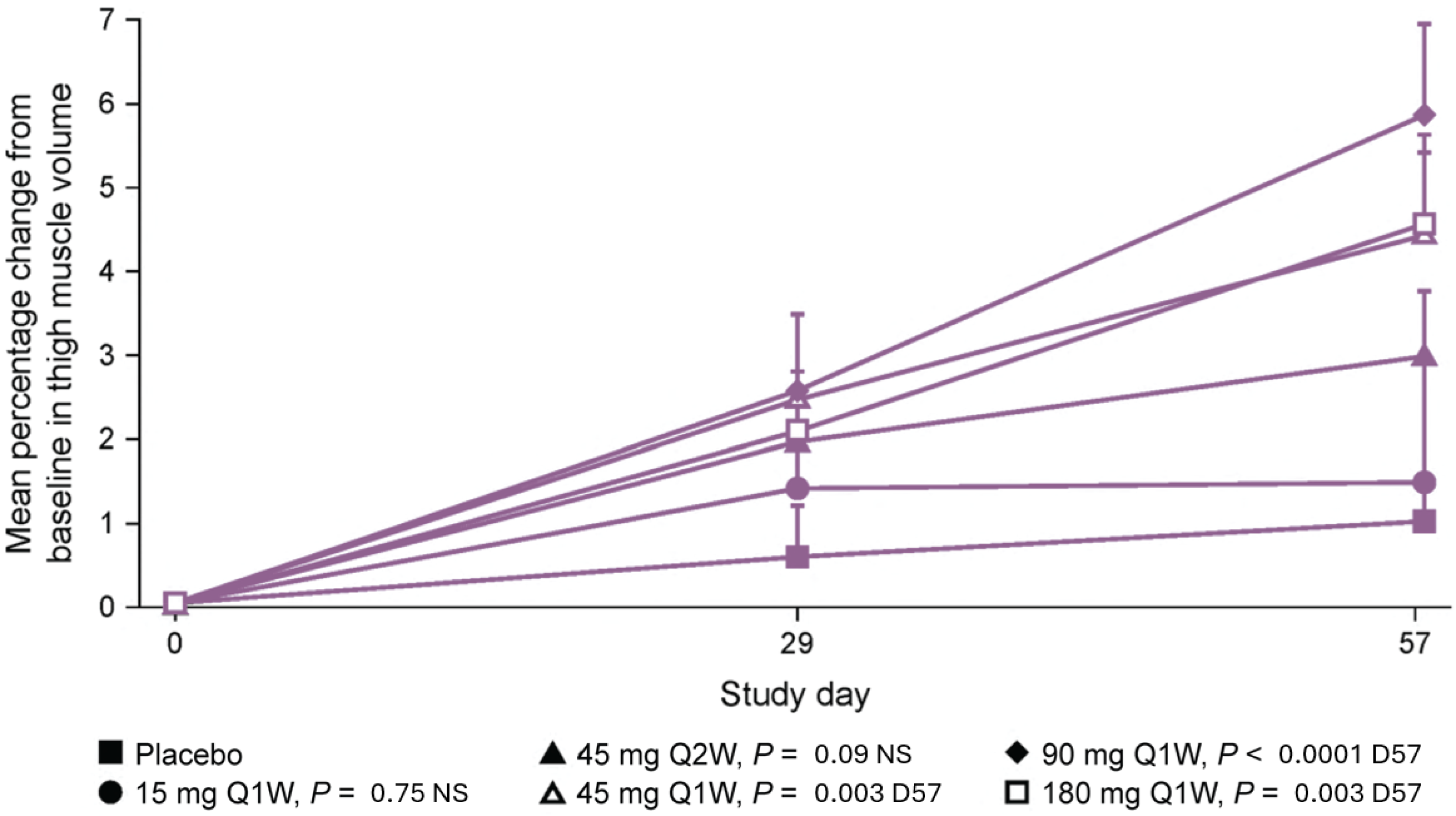
3.2. Taldefgrobep in the Clinical Setting
Healthy Adults
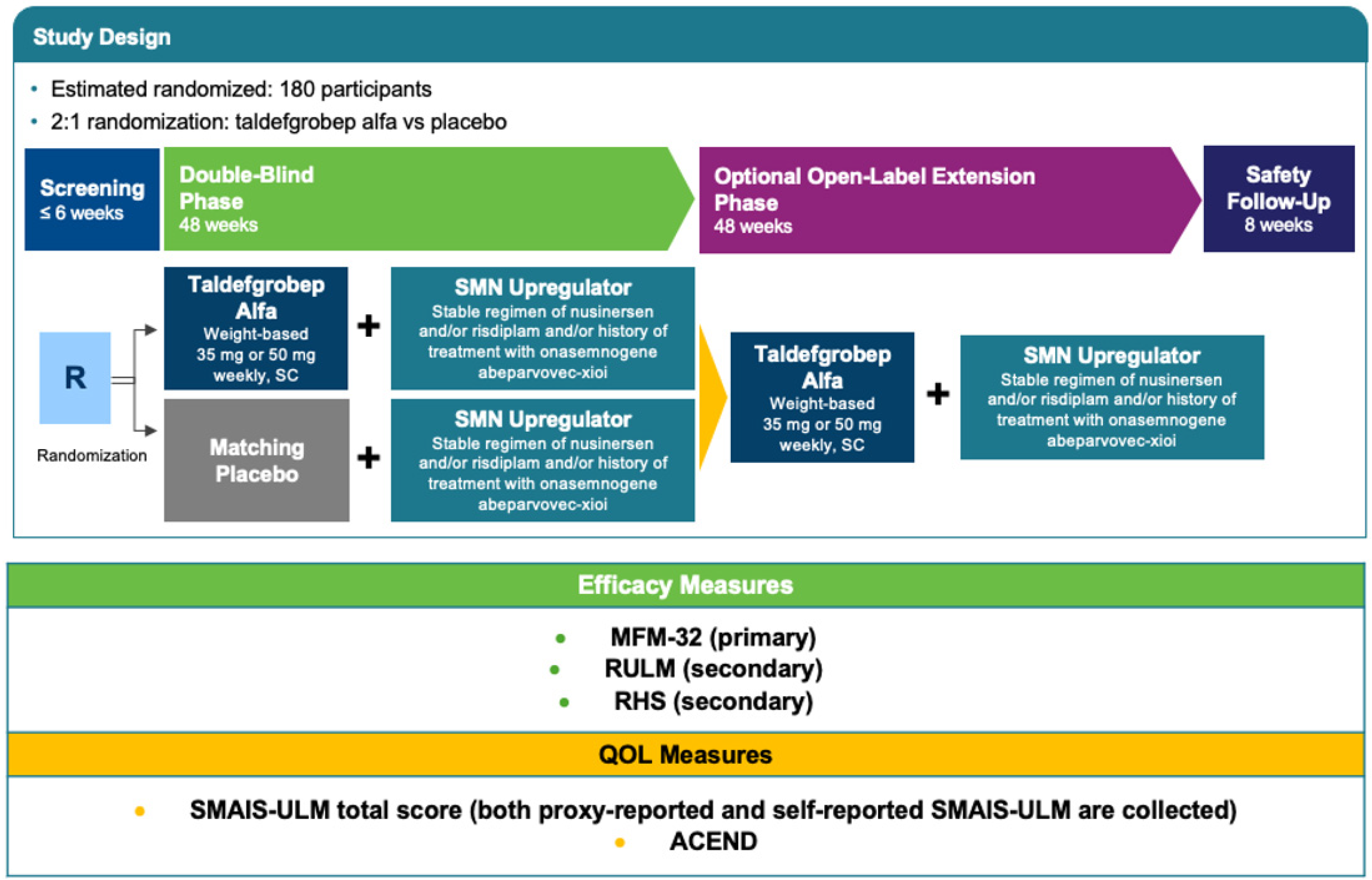
4. RESILIENT
4.1. Methods
4.1.1. Study Setting and Study Population
4.1.2. Interventions
4.1.3. Efficacy Outcomes and Safety Endpoints
4.1.4. Participant Timeline
4.1.5. Study Procedures
4.1.6. Statistical Methods and Sample Size
4.1.7. Ethics
5. Discussion
6. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aragon-Gawinska, K.; Mouraux, C.; Dangouloff, T.; Servais, L. Spinal Muscular Atrophy Treatment in Patients Identified by Newborn Screening—A Systematic Review. Genes 2023, 14, 1377. [Google Scholar] [CrossRef] [PubMed]
- Burr, P.; Reddivari, A.K.R. National Library of Medicine. Available online: http://www.ncbi.nlm.nih.gov/books/NBK560687/ (accessed on 14 May 2024).
- Mercuri, E.; Sumner, C.J.; Muntoni, F.; Darras, B.T.; Finkel, R.S. Spinal muscular atrophy. Nat. Rev. Dis. Prim. 2022, 8, 52. [Google Scholar] [CrossRef] [PubMed]
- Calucho, M.; Bernal, S.; Alías, L.; March, F.; Venceslá, A.; Rodríguez-Álvarez, F.J.; Aller, E.; Fernández, R.M.; Borrego, S.; Millán, J.M.; et al. Correlation between SMA type and SMN2 copy number revisited: An analysis of 625 unrelated Spanish patients and a compilation of 2834 reported cases. Neuromuscul. Disord. 2018, 28, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Lair, L.; Qureshi, I.; Bechtold, C.; Heller, L.; Durham, S.; Campbell, D.; Marin, J.; Chen, K.; Coric, V. P04 Taldefgrobep alfa: Preclinical and clinical data supporting the phase 3 RESILIENT study in spinal muscular atrophy. Neuromuscul. Disord. 2023, 33, S163. [Google Scholar] [CrossRef]
- Cure SMA. Available online: https://www.curesma.org/testing-diagnosis/ (accessed on 30 May 2024).
- Annoussamy, M.; Seferian, A.M.; Daron, A.; Péréon, Y.; Cances, C.; Vuillerot, C.; De Waele, L.; Laugel, V.; Schara, U.; Gidaro, T.; et al. Natural history of Type 2 and 3 spinal muscular atrophy: 2-year NatHis-SMA study. Ann. Clin. Transl. Neurol. 2020, 8, 359–373. [Google Scholar] [CrossRef]
- Oskoui, M.; Servais, L. Spinal Muscular Atrophy. Contin. Lifelong Learn. Neurol. 2023, 29, 1564–1584. [Google Scholar] [CrossRef]
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1723–1732. [Google Scholar] [CrossRef]
- Spinraza. Available online: https://www.spinraza.com/content/dam/commercial/spinraza/caregiver/en_us/pdf/spinraza-prescribing-information.pdf (accessed on 30 May 2024).
- Novartis. Available online: https://www.novartis.com/us-en/sites/novartis_us/files/zolgensma.pdf (accessed on 30 May 2024).
- Mendell, J.R.; Al-Zaidy, S.; Shell, R.; Arnold, W.D.; Rodino-Klapac, L.R.; Prior, T.W.; Lowes, L.; Alfano, L.; Berry, K.; Church, K.; et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1713–1722. [Google Scholar] [CrossRef]
- Darras, B.T.; Masson, R.; Mazurkiewicz-Bełdzińska, M.; Rose, K.; Xiong, H.; Zanoteli, E.; Baranello, G.; Bruno, C.; Vlodavets, D.; Wang, Y.; et al. Risdiplam-Treated Infants with Type 1 Spinal Muscular Atrophy versus Historical Controls. N. Engl. J. Med. 2021, 385, 427–435. [Google Scholar] [CrossRef]
- Genentech. Available online: https://www.gene.com/download/pdf/evrysdi_prescribing.pdf (accessed on 5 June 2024).
- Mercuri, E.; Deconinck, N.; Mazzone, E.S.; Nascimento, A.; Oskoui, M.; Saito, K.; Vuillerot, C.; Baranello, G.; Boespflug-Tanguy, O.; Goemans, N.; et al. Safety and efficacy of once-daily risdiplam in type 2 and non-ambulant type 3 spinal muscular atrophy (SUNFISH part 2): A phase 3, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2022, 21, 42–52. [Google Scholar] [CrossRef]
- Mendell, J.R.; Al-Zaidy, S.A.; Lehman, K.J.; McColly, M.; Lowes, L.P.; Alfano, L.N.; Reash, N.F.; Iammarino, M.A.; Church, K.R.; Kleyn, A.; et al. Five-Year Extension Results of the Phase 1 START Trial of Onasemnogene Abeparvovec in Spinal Muscular Atrophy. JAMA Neurol. 2021, 78, 834–841. [Google Scholar] [CrossRef] [PubMed]
- Day, J.W.; Finkel, R.S.; A Chiriboga, C.; Connolly, A.M.; Crawford, T.O.; Darras, B.T.; Iannaccone, S.T.; Kuntz, N.L.; Peña, L.D.M.; Shieh, P.B.; et al. Onasemnogene abeparvovec gene therapy for symptomatic infantile-onset spinal muscular atrophy in patients with two copies of SMN2 (STR1VE): An open-label, single-arm, multicentre, phase 3 trial. Lancet Neurol. 2021, 20, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Coratti, G.; Cutrona, C.; Pera, M.C.; Bovis, F.; Ponzano, M.; Chieppa, F.; Antonaci, L.; Sansone, V.; Finkel, R.; Pane, M.; et al. Motor function in type 2 and 3 SMA patients treated with Nusinersen: A critical review and meta-analysis. Orphanet J. Rare Dis. 2021, 16, 430. [Google Scholar] [CrossRef]
- Mercuri, E.; Darras, B.T.; Chiriboga, C.A.; Day, J.W.; Campbell, C.; Connolly, A.M.; Iannaccone, S.T.; Kirschner, J.; Kuntz, N.L.; Saito, K.; et al. Nusinersen versus Sham Control in Later-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2018, 378, 625–635. [Google Scholar] [CrossRef]
- Finkel, R.S.; Chiriboga, C.A.; Vajsar, J.; Day, J.W.; Montes, J.; De Vivo, D.C.; Yamashita, M.; Rigo, F.; Hung, G.; Schneider, E.; et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: A phase 2, open-label, dose-escalation study. Lancet 2016, 388, 3017–3026. [Google Scholar] [CrossRef]
- Mercuri, E.; Baranello, G.; Boespflug-Tanguy, O.; De Waele, L.; Goemans, N.; Kirschner, J.; Masson, R.; Mazzone, E.S.; Pechmann, A.; Pera, M.C.; et al. Risdiplam in types 2 and 3 spinal muscular atrophy: A randomised, placebo-controlled, dose-finding trial followed by 24 months of treatment. Eur. J. Neurol. 2022, 30, 1945–1956. [Google Scholar] [CrossRef]
- Schorling, D.C.; Pechmann, A.; Kirschner, J. Advances in Treatment of Spinal Muscular Atrophy—New Phenotypes, New Challenges, New Implications for Care. J. Neuromuscul. Dis. 2020, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wirth, B.; Karakaya, M.; Kye, M.J.; Mendoza-Ferreira, N. Twenty-Five Years of Spinal Muscular Atrophy Research: From Phenotype to Genotype to Therapy, and What Comes Next. Annu. Rev. Genom. Hum. Genet. 2020, 21, 231–261. [Google Scholar] [CrossRef] [PubMed]
- Day, J.W.; Howell, K.; Place, A.; Long, K.; Rossello, J.; Kertesz, N.; Nomikos, G. Advances and limitations for the treatment of spinal muscular atrophy. BMC Pediatr. 2022, 22, 632. [Google Scholar] [CrossRef]
- Cure SMA. Available online: https://www.curesma.org/wp-content/uploads/2022/08/NIH_SMA_Research_FactSheet_vFnl.pdf (accessed on 31 May 2023).
- Günther, R.; Wurster, C.D.; Brakemeier, S.; Osmanovic, A.; Schreiber-Katz, O.; Petri, S.; Uzelac, Z.; Hiebeler, M.; Thiele, S.; Walter, M.C.; et al. Long-term efficacy and safety of nusinersen in adults with 5q spinal muscular atrophy: A prospective European multinational observational study. Lancet Reg. Health Eur. 2024, 39, 100862. [Google Scholar] [CrossRef]
- Gavriilaki, M.; Moschou, M.; Papaliagkas, V.; Notas, K.; Chatzikyriakou, E.; Papagiannopoulos, S.; Arnaoutoglou, M.; Kimiskidis, V.K. Nusinersen in Adults with 5q Spinal Muscular Atrophy: A Systematic Review and Meta-analysis. Neurotherapeutics 2022, 19, 464–475. [Google Scholar] [CrossRef] [PubMed]
- Schuelke, M.; Wagner, K.R.; Stolz, L.E.; Hübner, C.; Riebel, T.; Kömen, W.; Braun, T.; Tobin, J.F.; Lee, S.-J. Myostatin Mutation Associated with Gross Muscle Hypertrophy in a Child. N. Engl. J. Med. 2004, 350, 2682–2688. [Google Scholar] [CrossRef] [PubMed]
- Abati, E.; Manini, A.; Comi, G.P.; Corti, S. Inhibition of myostatin and related signaling pathways for the treatment of muscle atrophy in motor neuron diseases. Cell. Mol. Life Sci. 2022, 79, 1–21. [Google Scholar] [CrossRef]
- McPherron, A.C.; Lee, S.-J. Double muscling in cattle due to mutations in the myostatin gene. Proc. Natl. Acad. Sci. USA 1997, 94, 12457–12461. [Google Scholar] [CrossRef] [PubMed]
- National Library of Medicine. Available online: https://medlineplus.gov/genetics/gene/mstn/ (accessed on 30 May 2024).
- McPherron, A.C.; Lawler, A.M.; Lee, S.-J. Regulation of skeletal muscle mass in mice by a new TGF-p superfamily member. Nature 1997, 387, 83–90. [Google Scholar] [CrossRef]
- Lodberg, A. Principles of the activin receptor signaling pathway and its inhibition. Cytokine Growth Factor Rev. 2021, 60, 1–17. [Google Scholar] [CrossRef]
- Clézardin, P.; Sousa, S.; Croset, M.; Pantano, F.; Confavreux, C. Current and Emerging Bone-Targeted Therapies for the Treatment of Bone Metastases from Solid Tumors. In Encyclopedia of Bone Biology, 1st ed.; Zaidi, M., Ed.; Academic Press: Cambridge, MA, USA; Elsevier: Amsterdam, The Netherlands, 2020; pp. 403–420. [Google Scholar] [CrossRef]
- Esposito, P.; Picciotto, D.; Battaglia, Y.; Costigliolo, F.; Viazzi, F.; Verzola, D. Myostatin: Basic biology to clinical application. In Advances in Clinical Chemistry, 1st ed.; Makowski, G.S., Ed.; Academic Press: Cambridge, MA, USA; Elsevier: Amsterdam, The Netherlands, 2022; Volume 106, pp. 181–234. [Google Scholar] [CrossRef]
- Baig, M.H.; Ahmad, K.; Moon, J.S.; Park, S.-Y.; Lim, J.H.; Chun, H.J.; Qadri, A.F.; Hwang, Y.C.; Jan, A.T.; Ahmad, S.S.; et al. Myostatin and its Regulation: A Comprehensive Review of Myostatin Inhibiting Strategies. Front. Physiol. 2022, 13, 876078. [Google Scholar] [CrossRef]
- Mariot, V.; Joubert, R.; Hourdé, C.; Féasson, L.; Hanna, M.; Muntoni, F.; Maisonobe, T.; Servais, L.; Bogni, C.; Le Panse, R.; et al. Downregulation of myostatin pathway in neuromuscular diseases may explain challenges of anti-myostatin therapeutic approaches. Nat. Commun. 2017, 8, 1859. [Google Scholar] [CrossRef]
- Rodgers, B.D.; Ward, C.W. Myostatin/Activin Receptor Ligands in Muscle and the Development Status of Attenuating Drugs. Endocr. Rev. 2021, 43, 329–365. [Google Scholar] [CrossRef]
- Statland, J.M.; Campbell, C.; Desai, U.; Karam, C.; Díaz-Manera, J.; Guptill, J.T.; Korngut, L.; Genge, A.; Tawil, R.N.; Elman, L.; et al. Randomized phase 2 study of ACE-083, a muscle-promoting agent, in facioscapulohumeral muscular dystrophy. Muscle Nerve 2022, 66, 50–62. [Google Scholar] [CrossRef]
- Wagner, K.R.; Fleckenstein, J.L.; Amato, A.A.; Barohn, R.J.; Bushby, K.; Escolar, D.M.; Flanigan, K.M.; Pestronk, A.; Tawil, R.; Wolfe, G.I.; et al. A phase I/IItrial of MYO-029 in adult subjects with muscular dystrophy. Ann. Neurol. 2008, 63, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Muntoni, F.; Byrne, B.J.; McMillan, H.J.; Ryan, M.M.; Wong, B.L.; Dukart, J.; Bansal, A.; Cosson, V.; Dreghici, R.; Guridi, M.; et al. The Clinical Development of Taldefgrobep Alfa: An Anti-Myostatin Adnectin for the Treatment of Duchenne Muscular Dystrophy. Neurol. Ther. 2024, 13, 183–219. [Google Scholar] [CrossRef]
- National Library of Medicine. Available online: https://clinicaltrials.gov/study/NCT03710941 (accessed on 30 May 2024).
- Jha, N.N.; Kim, J.-K.; Her, Y.-R.; Monani, U.R. Muscle: To the neuromuscular disease phenotype in spinal muscular atrophy. J. Clin. Investig. 2023, 8. [Google Scholar] [CrossRef] [PubMed]
- Bricceno, K.V.; Martinez, T.; Leikina, E.; Duguez, S.; Partridge, T.A.; Chernomordik, L.V.; Fischbeck, K.H.; Sumner, C.J.; Burnett, B.G. Survival motor neuron protein deficiency impairs myotube formation by altering myogenic gene expression and focal adhesion dynamics. Hum. Mol. Genet. 2014, 23, 4745–4757. [Google Scholar] [CrossRef] [PubMed]
- Hayhurst, M.; Wagner, A.K.; Cerletti, M.; Wagers, A.J.; Rubin, L.L. A cell-autonomous defect in skeletal muscle satellite cells expressing low levels of survival of motor neuron protein. Dev. Biol. 2012, 368, 323–334. [Google Scholar] [CrossRef]
- Long, K.K.; O’shea, K.M.; Khairallah, R.J.; Howell, K.; Paushkin, S.; Chen, K.S.; Cote, S.M.; Webster, M.T.; Stains, J.P.; Treece, E.; et al. Specific inhibition of myostatin activation is beneficial in mouse models of SMA therapy. Hum. Mol. Genet. 2018, 28, 1076–1089. [Google Scholar] [CrossRef]
- Mackels, L.; Mariot, V.; Buscemi, L.; Servais, L.; Dumonceaux, J. Impact of Disease Severity and Disease-Modifying Therapies on Myostatin Levels in SMA Patients. Int. J. Mol. Sci. 2024, 25, 8763. [Google Scholar] [CrossRef]
- Zhou, H.; Meng, J.; Malerba, A.; Catapano, F.; Sintusek, P.; Jarmin, S.; Feng, L.; Lu-Nguyen, N.; Sun, L.; Mariot, V.; et al. Myostatin inhibition in combination with antisense oligonucleotide therapy improves outcomes in spinal muscular atrophy. J. Cachex-Sarcopenia Muscle 2020, 11, 768–782. [Google Scholar] [CrossRef]
- Nielsen, T.L.; Vissing, J.; Krag, T.O. Antimyostatin Treatment in Health and Disease: The Story of Great Expectations and Limited Success. Cells 2021, 10, 533. [Google Scholar] [CrossRef]
- ScienceDirect. Available online: https://www.sciencedirect.com/topics/medicine-and-dentistry/activin-receptor-1 (accessed on 6 June 2024).
- Lair, L.; Qureshi, I.; Bechtold, C.; Durham, S.; Campbell, D.; Marin, J.; Coric, V. The Phase 3 RESILIENT Study: Taldefgrobep Alfa in Spinal Muscular Atrophy (P2-11.008). Neurology 2024, 102, 3255. [Google Scholar] [CrossRef]
- Latres, E.; Mastaitis, J.; Fury, W.; Miloscio, L.; Trejos, J.; Pangilinan, J.; Okamoto, H.; Cavino, K.; Na, E.; Papatheodorou, A.; et al. Activin A more prominently regulates muscle mass in primates than does GDF8. Nat. Commun. 2017, 8, 15153. [Google Scholar] [CrossRef] [PubMed]
- National Library of Medicine. Available online: https://clinicaltrials.gov/study/NCT03100630 (accessed on 30 May 2024).
- National Library of Medicine. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT02145234 (accessed on 30 May 2024).
- Lair, L.; Qureshi, I.; Bechtold, C.; Heller, L.; Durham, S.; Campbell, D.; Marin, J.; Chen, K.; Coric, V.I. Taldefgrobep alfa Preclinical and Clinical Data Supporting the Phase 3 RESILIENT Study in SMA. In Proceedings of the 4th Scientific International Congress of Spinal Muscular Atrophy, Ghent, Belgium, 16 March 2024. [Google Scholar]
- National Library of Medicine. Available online: https://clinicaltrials.gov/study/NCT05337553 (accessed on 31 December 2023).
- Biohaven, Ltd. Available online: https://ir.biohaven.com/news-releases/news-release-details/biohaven-completes-enrollment-pivotal-phase-3-study-taldefgrobep/ (accessed on 6 June 2024).
- Lair, L.; Qureshi, I.; Bechtold, C.; Durham, S.; Campbell, D.; Marin, J.; Coric, V. The Phase 3 RESILIENT Study in Spinal Muscular Atrophy. In Proceedings of the 9th Congress of the European Academy of Neurology, Budapest, Hungary, 14 July 2023. [Google Scholar]
- Bertini, E.; Dessaud, E.; Mercuri, E.; Muntoni, F.; Kirschner, J.; Reid, C.; Lusakowska, A.; Comi, G.P.; Cuisset, J.-M.; Abitbol, J.-L.; et al. Safety and efficacy of olesoxime in patients with type 2 or non-ambulatory type 3 spinal muscular atrophy: A randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol. 2017, 16, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Bérard, C.; Payan, C.; Hodgkinson, I.; Fermanian, J.; The MFM Collaborative Study Group. A motor function measure scale for neuromuscular diseases. Construction and validation study. Neuromuscul. Disord. 2005, 15, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Trundell, D.; Le Scouiller, S.; Le Goff, L.; Gorni, K.; Vuillerot, C. Assessment of the validity and reliability of the 32-item Motor Function Measure in individuals with Type 2 or non-ambulant Type 3 spinal muscular atrophy. PLoS ONE 2020, 15, e0238786. [Google Scholar] [CrossRef] [PubMed]
- Trundell, D.; Le Scouiller, S.; Gorni, K.; Seabrook, T.; Vuillerot, C.; the SMA MFM Study Group. Validity and Reliability of the 32-Item Motor Function Measure in 2-to 5-Year-Olds with Neuromuscular Disorders and 2-to 25-Year-Olds with Spinal Muscular Atrophy. Neurol. Ther. 2020, 9, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Duong, T.; Staunton, H.; Braid, J.; Barriere, A.; Trzaskoma, B.; Gao, L.; Willgoss, T.; Cruz, R.; Gusset, N.; Gorni, K.; et al. A Patient-Centered Evaluation of Meaningful Change on the 32-Item Motor Function Measure in Spinal Muscular Atrophy Using Qualitative and Quantitative Data. Front. Neurol. 2022, 12, 770423. [Google Scholar] [CrossRef]
- Vuillerot, C.; Payan, C.; Iwaz, J.; Ecochard, R.; Bérard, C. Responsiveness of the Motor Function Measure in Patients with Spinal Muscular Atrophy. Arch. Phys. Med. Rehabilitation 2013, 94, 1555–1561. [Google Scholar] [CrossRef]
- Seferian, A.M.; Moraux, A.; Canal, A.; Decostre, V.; Diebate, O.; Le Moing, A.G.; Gidaro, T.; Deconinck, N.; Van Parys, F.; Vereecke, W.; et al. Upper Limb Evaluation and One-Year Follow Up of Non-Ambulant Patients with Spinal Muscular Atrophy: An Observational Multicenter Trial. PLoS ONE 2015, 10, e0121799. [Google Scholar] [CrossRef]
- Wu, J.W.; Pepler, L.; Maturi, B.M.; Afonso, A.C.F.; Sarmiento, J.; Haldenby, R.M. Systematic Review of Motor Function Scales and Patient-Reported Outcomes in Spinal Muscular Atrophy. Am. J. Phys. Med. Rehabil. 2021, 101, 590–608. [Google Scholar] [CrossRef]
- Pechmann, A.; Behrens, M.; Dörnbrack, K.; Tassoni, A.; Wenzel, F.; Stein, S.; Vogt, S.; Zöller, D.; Bernert, G.; Hagenacker, T.; et al. Improved upper limb function in non-ambulant children with SMA type 2 and 3 during nusinersen treatment: A prospective 3-years SMArtCARE registry study. Orphanet J. Rare Dis. 2022, 17, 384. [Google Scholar] [CrossRef]
- National Library of Medicine. Available online: https://clinicaltrials.gov/study/NCT02908685 (accessed on 31 December 2023).
- Pera, M.C.; Coratti, G.; Mazzone, E.S.; Montes, J.; Scoto, M.; De Sanctis, R.; Main, M.; Mayhew, A.; Lofra, R.M.; Young, S.D.; et al. Revised upper limb module for spinal muscular atrophy: 12 month changes. Muscle Nerve 2019, 59, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Maggi, L.; Bello, L.; Bonanno, S.; Govoni, A.; Caponnetto, C.; Passamano, L.; Grandis, M.; Trojsi, F.; Cerri, F.; Ferraro, M.; et al. Nusinersen safety and effects on motor function in adult spinal muscular atrophy type 2 and 3. J. Neurol. Neurosurg. Psychiatry 2020, 91, 1166–1174. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, D.; Scoto, M.; Mayhew, A.; Main, M.; Mazzone, E.S.; Montes, J.; de Sanctis, R.; Young, S.D.; Salazar, R.; Glanzman, A.M.; et al. Revised Hammersmith Scale for spinal muscular atrophy: A SMA specific clinical outcome assessment tool. PLoS ONE 2017, 12, e0172346. [Google Scholar] [CrossRef] [PubMed]
- Stimpson, G.; Ramsey, D.; Wolfe, A.; Mayhew, A.; Scoto, M.; Baranello, G.; Lofra, R.M.; Main, M.; Milev, E.; Coratti, G.; et al. 2-Year Change in Revised Hammersmith Scale Scores in a Large Cohort of Untreated Paediatric Type 2 and 3 SMA Participants. J. Clin. Med. 2023, 12, 1920. [Google Scholar] [CrossRef]
- Ramsey, D.; Ramdharry, G.; Scoto, M.; Muntoni, F.; Wallace, A.; on behalf of the SMA REACH UK Network. Revised Hammersmith Scale for spinal muscular atrophy: Inter and intra-rater reliability and agreement. PLoS ONE 2022, 17, e0278996. [Google Scholar] [CrossRef]
- Mazzone, E.S.; Mayhew, A.; Montes, J.; Ramsey, D.; Fanelli, L.; Young, S.D.; Salazar, R.; De Sanctis, R.; Pasternak, A.; Glanzman, A.; et al. Revised upper limb module for spinal muscular atrophy: Development of a new module. Muscle Nerve 2017, 55, 869–874. [Google Scholar] [CrossRef]
- Coratti, G.; Pera, M.C.; Montes, J.; Scoto, M.; Pasternak, A.; Bovis, F.; Sframeli, M.; D’Amico, A.; Pane, M.; Albamonte, E.; et al. Revised upper limb module in type II and III spinal muscular atrophy: 24-month changes. Neuromuscul. Disord. 2021, 32, 36–42. [Google Scholar] [CrossRef]
- Trundell, D.; Skalicky, A.; Staunton, H.; Hareendran, A.; Le Scouiller, S.; Barrett, L.; Cooper, O.; Gorni, K.; Seabrook, T.; Jethwa, S.; et al. Development of the SMA independence scale–upper limb module (SMAIS–ULM): A novel scale for individuals with Type 2 and non-ambulant Type 3 SMA. J. Neurol. Sci. 2021, 432, 120059. [Google Scholar] [CrossRef]
- Matsumoto, H.; Clayton-Krasinski, D.A.; Klinge, S.A.; Gomez, J.A.; Booker, W.A.; Hyman, J.E.; Roye, D.P.; Vitale, M.G. Development and Initial Validation of the Assessment of Caregiver Experience with Neuromuscular Disease. J. Pediatr. Orthop. 2011, 31, 284–292. [Google Scholar] [CrossRef]
- SMAtrial. Available online: https://www.smatrial.com (accessed on 6 June 2024).
- Kourakis, S.; Timpani, C.A.; Campelj, D.G.; Hafner, P.; Gueven, N.; Fischer, D.; Rybalka, E. Standard of care versus new-wave corticosteroids in the treatment of Duchenne muscular dystrophy: Can we do better? Orphanet J. Rare Dis. 2021, 16, 117. [Google Scholar] [CrossRef]
- Kharraz, Y.; Guerra, J.; Pessina, P.; Serrano, A.L.; Muñoz-Cánoves, P. Understanding the Process of Fibrosis in Duchenne Muscular Dystrophy. BioMed Res. Int. 2014, 2014, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Quattrocelli, M.; Zelikovich, A.S.; Salamone, I.M.; Fischer, J.A.; McNally, E.M. Mechanisms and Clinical Applications of Glucocorticoid Steroids in Muscular Dystrophy. J. Neuromuscul. Dis. 2021, 8, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Apkon, S.D.; Blackwell, A.; Brumbaugh, D.; Case, L.E.; Clemens, P.R.; Hadjiyannakis, S.; Pandya, S.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018, 17, 251–267. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. Available online: https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-first-targeted-treatment-rare-duchenne-muscular-dystrophy-mutation (accessed on 6 June 2024).
- Dangouloff, T.; Burghes, A.; Tizzano, E.F.; Servais, L. 244th ENMC international workshop: Newborn screening in spinal muscular atrophy May 10–12, 2019, Hoofdorp, The Netherlands. Neuromuscul. Disord. 2019, 30, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Nishio, H.; Niba, E.T.E.; Saito, T.; Okamoto, K.; Takeshima, Y.; Awano, H. Spinal Muscular Atrophy: The Past, Present, and Future of Diagnosis and Treatment. Int. J. Mol. Sci. 2023, 24, 11939. [Google Scholar] [CrossRef] [PubMed]
- Servais, L.; Baranello, G.; Scoto, M.; Daron, A.; Oskoui, M. Therapeutic interventions for spinal muscular atrophy: Preclinical and early clinical development opportunities. Expert Opin. Investig. Drugs 2021, 30, 519–527. [Google Scholar] [CrossRef]
- Cano, S.J.; Mayhew, A.; Glanzman, A.M.; Krosschell, K.J.; Swoboda, K.J.; Main, M.; Steffensen, B.F.; Bérard, C.; Girardot, F.; Payan, C.A.; et al. Rasch analysis of clinical outcome measures in spinal muscular atrophy. Muscle Nerve 2013, 49, 422–430. [Google Scholar] [CrossRef]
- Oskoui, M.; Day, J.W.; Deconinck, N.; Mazzone, E.S.; Nascimento, A.; Saito, K.; Vuillerot, C.; Baranello, G.; Goemans, N.; Kirschner, J.; et al. Two-year efficacy and safety of risdiplam in patients with type 2 or non-ambulant type 3 spinal muscular atrophy (SMA). J. Neurol. 2023, 270, 2531–2546. [Google Scholar] [CrossRef]
- Vincent-Genod, D.P.; Rippert, P.; Coton, J.; Le Goff, L.; Barriere, A.O.; Berruyer, A.; Bernard, M.; Garde, C.; Gutierrez-Garcia, M.; Gilabert, S.; et al. Scoring People with Spinal Muscular Atrophy on the Motor Function Measure Using the Microsoft Kinect. Pediatr. Phys. Ther. 2023, 35, 36–41. [Google Scholar] [CrossRef]
- Panero, E.; D’alessandro, R.; Cavallina, I.; Davico, C.; Mongini, T.; Gastaldi, L.; Ricci, F. Wearable Inertial Devices in Duchenne Muscular Dystrophy: A Scoping Review. Appl. Sci. 2023, 13, 1268. [Google Scholar] [CrossRef]
- Ricotti, V.; Kadirvelu, B.; Selby, V.; Festenstein, R.; Mercuri, E.; Voit, T.; Faisal, A.A. Wearable full-body motion tracking of activities of daily living predicts disease trajectory in Duchenne muscular dystrophy. Nat. Med. 2023, 29, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Servais, L.; Camino, E.; Clement, A.; McDonald, C.M.; Lukawy, J.; Lowes, L.P.; Eggenspieler, D.; Cerreta, F.; Strijbos, P. First Regulatory Qualification of a Novel Digital Endpoint in Duchenne Muscular Dystrophy: A Multi-Stakeholder Perspective on the Impact for Patients and for Drug Development in Neuromuscular Diseases. Digit. Biomarkers 2021, 5, 183–190. [Google Scholar] [CrossRef]
- Servais, L.; Eggenspieler, D.; Poleur, M.; Grelet, M.; Muntoni, F.; Strijbos, P.; Annoussamy, M. First regulatory qualification of a digital primary endpoint to measure treatment efficacy in DMD. Nat. Med. 2023, 29, 2391–2392. [Google Scholar] [CrossRef] [PubMed]
- Ohio State News. Available online: https://news.osu.edu/first-wearable-health-sensor-for-monitoring-muscle-atrophy/ (accessed on 30 May 2024).
- Iijima, K.; Watanabe, H.; Nakashiro, Y.; Iida, Y.; Nonaka, M.; Moriwaka, F.; Hamada, S. Long-term effects of the gait treatment using a wearable cyborg hybrid assistive limb in a patient with spinal and bulbar muscular atrophy: A case report with 5 years of follow-up. Front. Neurol. 2023, 14, 1143820. [Google Scholar] [CrossRef]
- Dangouloff, T.; Hiligsmann, M.; Deconinck, N.; D’Amico, A.; Seferian, A.M.; Boemer, F.; Servais, L. Financial cost and quality of life of patients with spinal muscular atrophy identified by symptoms or newborn screening. Dev. Med. Child Neurol. 2022, 65, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Oskoui, M.; Dangouloff, T.; Servais, L. Universal Newborn Screening for Spinal Muscular Atrophy. JAMA Pediatr. 2024, 178, 520. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, O.; Vill, K.; Pfaffenlehner, M.; Behrens, M.; Weiß, C.; Johannsen, J.; Friese, J.; Hahn, A.; Ziegler, A.; Illsinger, S.; et al. Clinical Effectiveness of Newborn Screening for Spinal Muscular Atrophy. JAMA Pediatr. 2024, 178, 540. [Google Scholar] [CrossRef]
- Baranello, G.; Gorni, K.; Daigl, M.; Kotzeva, A.; Evans, R.; Hawkins, N.; Scott, D.A.; Mahajan, A.; Muntoni, F.; Servais, L. Prognostic Factors and Treatment-Effect Modifiers in Spinal Muscular Atrophy. Clin. Pharmacol. Ther. 2021, 110, 1435–1454. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Key Inclusion Criteria [5] | Key Exclusion Criteria [5] |
|---|---|
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Servais, L.; Lair, L.L.; Connolly, A.M.; Byrne, B.J.; Chen, K.S.; Coric, V.; Qureshi, I.; Durham, S.; Campbell, D.J.; Maclaine, G.; et al. Taldefgrobep Alfa and the Phase 3 RESILIENT Trial in Spinal Muscular Atrophy. Int. J. Mol. Sci. 2024, 25, 10273. https://doi.org/10.3390/ijms251910273
Servais L, Lair LL, Connolly AM, Byrne BJ, Chen KS, Coric V, Qureshi I, Durham S, Campbell DJ, Maclaine G, et al. Taldefgrobep Alfa and the Phase 3 RESILIENT Trial in Spinal Muscular Atrophy. International Journal of Molecular Sciences. 2024; 25(19):10273. https://doi.org/10.3390/ijms251910273
Chicago/Turabian StyleServais, Laurent, Lindsey Lee Lair, Anne M. Connolly, Barry J. Byrne, Karen S. Chen, Vlad Coric, Irfan Qureshi, Susan Durham, Daniel J. Campbell, Grant Maclaine, and et al. 2024. "Taldefgrobep Alfa and the Phase 3 RESILIENT Trial in Spinal Muscular Atrophy" International Journal of Molecular Sciences 25, no. 19: 10273. https://doi.org/10.3390/ijms251910273