
In Silico Modeling of Fabry Disease Pathophysiology for the Identification of Early Cellular Damage Biomarker Candidates

 , , , , , and
, , , , , and
Abstract
1. Introduction
2. Results
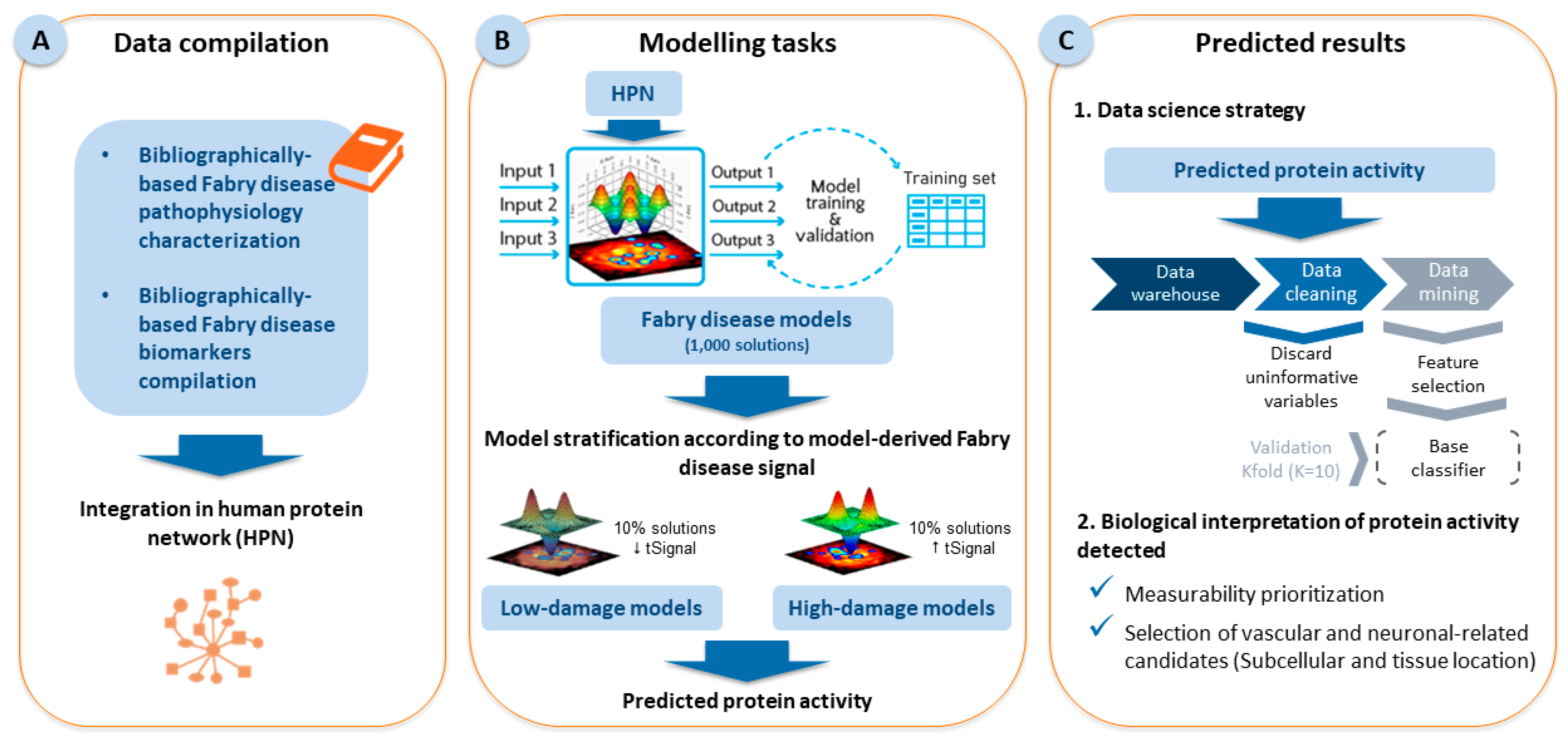
2.1. Fabry Disease Characterization and Model Creation
2.2. Results of the Data Science Strategy
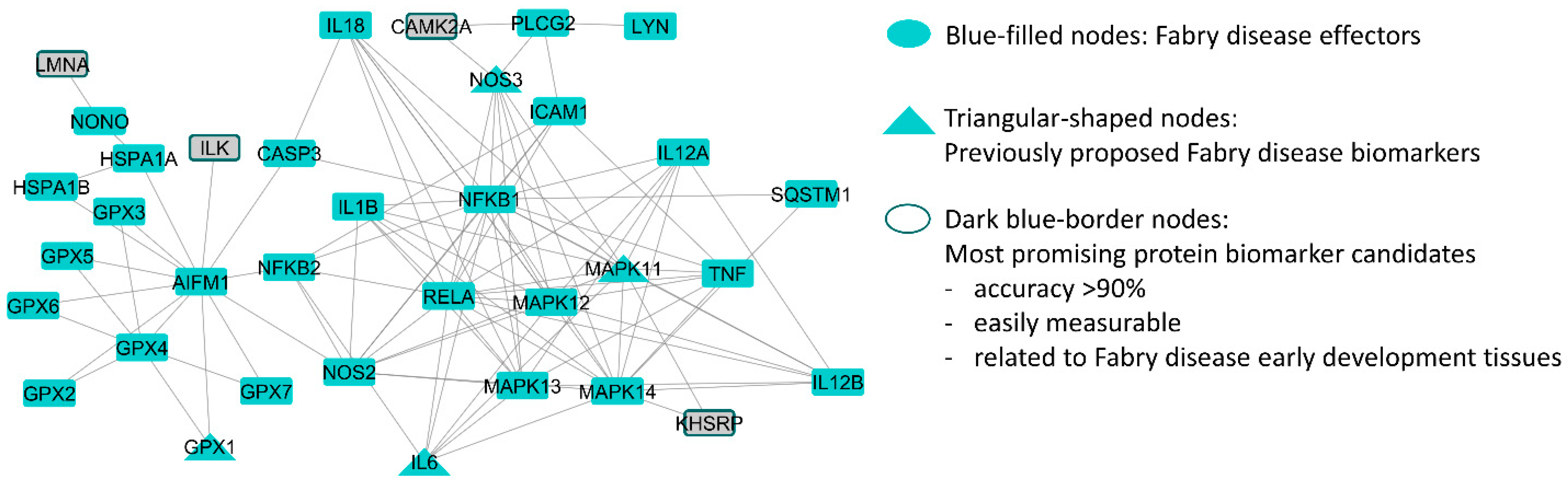
2.3. Biological and Clinical Filtering of the Candidates
3. Discussion
4. Materials and Methods
4.1. Generation of Mathematical Models
4.2. Statistical Analysis: Classifier Identification
4.3. Biological Data Compilation and Filter Application for the Selection of Risk Biomarkers
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Germain, D.P. Fabry disease. Orphanet J. Rare Dis. 2010, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Germain, D.P.; Oliveira, J.P.; Bichet, D.G.; Yoo, H.W.; Hopkin, R.J.; Lemay, R.; Politei, J.; Wanner, C.; Wilcox, W.R.; Warnock, D.G. Use of a rare disease registry for establishing phenotypic classification of previously unassigned GLA variants: A consensus classification system by a multispecialty Fabry disease genotype-phenotype workgroup. J. Med. Genet. 2020, 57, 542–551. [Google Scholar] [CrossRef]
- Echevarria, L.; Benistan, K.; Toussaint, A.; Dubourg, O.; Hagege, A.A.; Eladari, D.; Jabbour, F.; Beldjord, C.; De Mazancourt, P.; Germain, D.P. X-chromosome inactivation in female patients with Fabry disease. Clin. Genet. 2016, 89, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Meikle, P.J.; Hopwood, J.J.; Clague, A.E.; Carey, W.F. Prevalence of lysosomal storage disorders. JAMA 1999, 281, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Burlina, A.B.; Polo, G.; Salviati, L.; Duro, G.; Zizzo, C.; Dardis, A.; Bembi, B.; Cazzorla, C.; Rubert, L.; Zordan, R.; et al. Newborn screening for lysosomal storage disorders by tandem mass spectrometry in North East Italy. J. Inherit. Metab. Dis. 2018, 41, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, P.V.; Campbell, C.; Klug, T.; Rogers, S.; Raburn-Miller, J.; Kiesling, J. Lysosomal storage disorder screening implementation: Findings from the first six months of full population pilot testing in Missouri. J. Pediatr. 2015, 166, 172–177. [Google Scholar] [CrossRef]
- Colon, C.; Ortolano, S.; Melcon-Crespo, C.; Alvarez, J.V.; Lopez-Suarez, O.E.; Couce, M.L.; Fernández-Lorenzo, J.R. Newborn screening for Fabry disease in the north-west of Spain. Eur. J. Pediatr. 2017, 176, 1075–1081. [Google Scholar] [CrossRef]
- Sawada, T.; Kido, J.; Yoshida, S.; Sugawara, K.; Momosaki, K.; Inoue, T.; Tajima, G.; Sawada, H.; Mastumoto, S.; Endo, F.; et al. Newborn screening for Fabry disease in the western region of Japan. Mol. Genet. Metab. Rep. 2020, 22, 100562. [Google Scholar] [CrossRef]
- Arends, M.; Wanner, C.; Hughes, D.; Mehta, A.; Oder, D.; Watkinson, O.T.; Elliott, P.M.; Linthorst, G.E.; Wijburg, F.A.; Biegstraaten, M.; et al. Characterization of Classical and Nonclassical Fabry Disease: A Multicenter Study. J. Am. Soc. Nephrol. 2017, 28, 1631–1641. [Google Scholar] [CrossRef]
- Michaud, M.; Mauhin, W.; Belmatoug, N.; Garnotel, R.; Bedreddine, N.; Catros, F.; Ancellin, S.; Lidove, O.; Gaches, F. When and How to Diagnose Fabry Disease in Clinical Pratice. Am. J. Med. Sci. 2020, 360, 641–649. [Google Scholar] [CrossRef]
- Stankowski, K.; Figliozzi, S.; Battaglia, V.; Catapano, F.; Francone, M.; Monti, L. Fabry Disease: More than a Phenocopy of Hypertrophic Cardiomyopathy. J. Clin. Med. 2023, 12, 7061. [Google Scholar] [CrossRef] [PubMed]
- Esposito, P.; Caputo, C.; Repetto, M.; Somaschini, A.; Pietro, B.; Colomba, P.; Zizzo, C.; Parodi, A.; Zanetti, V.; Canepa, M.; et al. Diagnosing Fabry nephropathy: The challenge of multiple kidney disease. BMC Nephrol. 2023, 24, 344. [Google Scholar] [CrossRef] [PubMed]
- Blasco, M.; Quiroga, B.; García-Aznar, J.M.; Castro-Alonso, C.; Fernández-Granados, S.J.; Luna, E.; Fernández Fresnedo, G.; Ossorio, M.; Izquierdo, M.J.; Sanchez-Ospina, D.; et al. Genetic Characterization of Kidney Failure of Unknown Etiology in Spain: Findings from the GENSEN Study. Am. J. Kidney Dis. 2024. [Google Scholar] [CrossRef] [PubMed]
- Buechner, S.; Moretti, M.; Burlina, A.P.; Cei, G.; Manara, R.; Ricci, R.; Mignani, R.; Parini, R.; Di Vito, R.; Giordano, G.P.; et al. Central nervous system involvement in Anderson-Fabry disease: A clinical and MRI retrospective study. J. Neurol. Neurosurg. Psychiatry 2008, 79, 1249–1254. [Google Scholar] [CrossRef] [PubMed]
- Alcalay, R.N.; Wolf, P.; Levy, O.A.; Kang, U.J.; Waters, C.; Fahn, S.; Ford, B.; Kuo, S.H.; Vanegas, N.; Shah, H.; et al. Alpha galactosidase A activity in Parkinson’s disease. Neurobiol. Dis. 2018, 112, 85–90. [Google Scholar] [CrossRef]
- Hopkin, R.J.; Feldt-Rasmussen, U.; Germain, D.P.; Jovanovic, A.; Martins, A.M.; Nicholls, K.; Ortiz, A.; Politei, J.; Ponce, E.; Varas, C.; et al. Improvement of gastrointestinal symptoms in a significant proportion of male patients with classic Fabry disease treated with agalsidase beta: A Fabry Registry analysis stratified by phenotype. Mol. Genet. Metab. Rep. 2020, 25, 100670. [Google Scholar] [CrossRef]
- Körver, S.; Vergouwe, M.; Hollak, C.E.M.; van Schaik, I.N.; Langeveld, M. Development and clinical consequences of white matter lesions in Fabry disease: A systematic review. Mol. Genet. Metab. 2018, 125, 205–216. [Google Scholar] [CrossRef]
- Rombach, S.M.; van den Bogaard, B.; de Groot, E.; Groener, J.E.M.; Poorthuis, B.J.; Linthorst, G.E.; van den Born, B.-J.H.; Hollak, C.E.M.; Aerts, J.M.F.G. Vascular aspects of Fabry disease in relation to clinical manifestations and elevations in plasma globotriaosylsphingosine. Hypertension 2012, 60, 998–1005. [Google Scholar] [CrossRef]
- Sims, K.; Politei, J.; Banikazemi, M.; Lee, P. Stroke in Fabry disease frequently occurs before diagnosis and in the absence of other clinical events: Natural history data from the Fabry Registry. Stroke 2009, 40, 788–794. [Google Scholar] [CrossRef]
- Tuttolomondo, A.; Pecoraro, R.; Simonetta, I.; Miceli, S.; Arnao, V.; Licata, G.; Pinto, A. Neurological complications of Anderson-Fabry disease. Curr. Pharm. Des. 2013, 19, 6014–6030. [Google Scholar] [CrossRef]
- Palaiodimou, L.; Kokotis, P.; Zompola, C.; Papagiannopoulou, G.; Bakola, E.; Papadopoulou, M.; Zouvelou, V.; Petras, D.; Vlachopoulos, C.; Tsivgoulis, G. Fabry Disease: Current & novel therapeutic strategies. A narrative review. Curr. Neuropharmacol. 2022, 21, 440. [Google Scholar] [CrossRef]
- Aerts, J.M.; Groener, J.E.; Kuiper, S.; Donker-Koopman, W.E.; Strijland, A.; Ottenhoff, R.; van Roomen, C.; Mirzaian, M.; Wijburg, F.A.; Linthorst, G.E.; et al. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc. Natl. Acad. Sci. USA 2008, 105, 2812–2817. [Google Scholar] [CrossRef] [PubMed]
- Kok, K.; Zwiers, K.C.; Boot, R.G.; Overkleeft, H.S.; Aerts, J.M.F.G.; Artola, M. Fabry Disease: Molecular Basis, Pathophysiology, Diagnostics and Potential Therapeutic Directions. Biomolecules 2021, 11, 271. [Google Scholar] [CrossRef] [PubMed]
- Sawai, S. Fabry Disease: Pathogenesis, Clinical Symptoms, and Treatment with Enzyme Replacement Therapy. Brain Nerve 2015, 67, 1099–1108. [Google Scholar] [CrossRef]
- Rombach, S.M.; Twickler, T.B.; Aerts, J.M.F.G.; Linthorst, G.E.; Wijburg, F.A.; Hollak, C.E.M. Vasculopathy in patients with Fabry disease: Current controversies and research directions. Mol. Genet. Metab. 2010, 99, 99–108. [Google Scholar] [CrossRef]
- Mehta, A.; Ginsberg, L. Natural history of the cerebrovascular complications of Fabry disease. Acta Paediatr. Suppl. 2005, 94, 10–24. [Google Scholar] [CrossRef]
- Bolsover, F.E.; Murphy, E.; Cipolotti, L.; Werring, D.J.; Lachmann, R.H. Cognitive dysfunction and depression in Fabry disease: A systematic review. J. Inherit. Metab. Dis. 2014, 37, 177–187. [Google Scholar] [CrossRef]
- Cole, A.L.; Lee, P.J.; Hughes, D.A.; Deegan, P.B.; Waldek, S.; Lachmann, R.H. Depression in adults with Fabry disease: A common and under-diagnosed problem. J. Inherit. Metab. Dis. 2007, 30, 943–951. [Google Scholar] [CrossRef]
- Talbot, A.; Hammerschlag, G.; Goldin, J.; Nicholls, K. Sleep Disturbance, Obstructive Sleep Apnoea and Abnormal Periodic Leg Movements: Very Common Problems in Fabry Disease. JIMD Rep. 2017, 31, 37–44. [Google Scholar] [CrossRef]
- Parini, R.; Pintos-Morell, G.; Hennermann, J.B.; Hsu, T.-R.; Karabul, N.; Kalampoki, V.; Gurevich, A.; Ramaswami, U. Analysis of Renal and Cardiac Outcomes in Male Participants in the Fabry Outcome Survey Starting Agalsidase Alfa Enzyme Replacement Therapy Before and After 18 Years of Age. Drug Des. Devel. Ther. 2020, 14, 2149–2158. [Google Scholar] [CrossRef]
- Hughes, D.; Linhart, A.; Gurevich, A.; Kalampoki, V.; Jazukeviciene, D.; Feriozzi, S. Prompt Agalsidase Alfa Therapy Initiation is Associated with Improved Renal and Cardiovascular Outcomes in a Fabry Outcome Survey Analysis. Drug Des. Devel. Ther. 2021, 15, 3561–3572. [Google Scholar] [CrossRef] [PubMed]
- Gragnaniello, V.; Burlina, A.P.; Commone, A.; Gueraldi, D.; Puma, A.; Porcù, E.; Stornaiuolo, M.; Cazzorla, C.; Burlina, A.B. Newborn Screening for Fabry Disease: Current Status of Knowledge. Int. J. Neonatal Screen. 2023, 9, 31. [Google Scholar] [CrossRef] [PubMed]
- Monda, E.; Diana, G.; Graziani, F.; Rubino, M.; Bakalakos, A.; Linhart, A.; Germain, D.P.; Scarpa, M.; Biagini, E.; Pieroni, M.; et al. Impact of GLA Variant Classification on the Estimated Prevalence of Fabry Disease: A Systematic Review and Meta-Analysis of Screening Studies. Circ. Genom. Precis. Med. 2023, 16, e004252. [Google Scholar] [CrossRef] [PubMed]
- Hopkin, R.J.; Cabrera, G.; Charrow, J.; Lemay, R.; Martins, A.M.; Mauer, M.; Ortiz, A.; Patel, M.R.; Sims, K.; Waldek, S.; et al. Risk factors for severe clinical events in male and female patients with Fabry disease treated with agalsidase beta enzyme replacement therapy: Data from the Fabry Registry. Mol. Genet. Metab. 2016, 119, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Talbot, A.; Nicholls, K.; Fletcher, J.M.; Fuller, M. A simple method for quantification of plasma globotriaosylsphingosine: Utility for Fabry disease. Mol. Genet. Metab. 2017, 122, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Nowak, A.; Mechtler, T.P.; Hornemann, T.; Gawinecka, J.; Theswet, E.; Hilz, M.J.; Kasper, D.C. Genotype, phenotype and disease severity reflected by serum LysoGb3 levels in patients with Fabry disease. Mol. Genet. Metab. 2018, 123, 148–153. [Google Scholar] [CrossRef]
- Burlina, A.; Brand, E.; Hughes, D.; Kantola, I.; Krämer, J.; Nowak, A.; Tøndel, C.; Wanner, C.; Spada, M. An expert consensus on the recommendations for the use of biomarkers in Fabry disease. Mol. Genet. Metab. 2023, 139, 107585. [Google Scholar] [CrossRef]
- Wang, Y.; Zhu, H.; Madabushi, R.; Liu, Q.; Huang, S.-M.; Zineh, I. Model-Informed Drug Development: Current US Regulatory Practice and Future Considerations. Clin. Pharmacol. Ther. 2019, 105, 899–911. [Google Scholar] [CrossRef]
- Van Nieuwenhove, E.; Lagou, V.; Van Eyck, L.; Dooley, J.; Bodenhofer, U.; Roca, C.; Vandebergh, M.; Goris, A.; Humblet-Baron, S.; Wouters, C.; et al. Machine learning identifies an immunological pattern associated with multiple juvenile idiopathic arthritis subtypes. Ann. Rheum. Dis. 2019, 78, 617–628. [Google Scholar] [CrossRef]
- Zhang, P.; Itan, Y. Biological Network Approaches and Applications in Rare Disease Studies. Genes 2019, 10, 797. [Google Scholar] [CrossRef]
- Segú-Vergés, C.; Coma, M.; Kessel, C.; Smeets, S.; Foell, D.; Aldea, A. Application of systems biology-based in silico tools to optimize treatment strategy identification in Still’s disease. Arthritis Res. Ther. 2021, 23, 126. [Google Scholar] [CrossRef] [PubMed]
- Jorba, G.; Aguirre-Plans, J.; Junet, V.; Segú-Vergés, C.; Ruiz, J.L.; Pujol, A.; Fernández-Fuentes, N.; Mas, J.M.; Oliva, B. In-silico simulated prototype-patients using TPMS technology to study a potential adverse effect of sacubitril and valsartan. PLoS ONE 2020, 15, e0228926. [Google Scholar] [CrossRef] [PubMed]
- Artigas, L.; Coma, M.; Matos-Filipe, P.; Aguirre-Plans, J.; Farrés, J.; Valls, R.; Fernandez-Fuentes, N.; de la Haba-Rodriguez, J.; Olvera, A.; Barbera, J.; et al. In-silico drug repurposing study predicts the combination of pirfenidone and melatonin as a promising candidate therapy to reduce SARS-CoV-2 infection progression and respiratory distress caused by cytokine storm. PLoS ONE 2020, 15, e0240149. [Google Scholar] [CrossRef] [PubMed]
- Romeo-Guitart, D.; Forés, J.; Herrando-Grabulosa, M.; Valls, R.; Leiva-Rodríguez, T.; Galea, E.; González-Pérez, F.; Navarro, X.; Petegnief, V.; Bosch, A.; et al. Neuroprotective Drug for Nerve Trauma Revealed Using Artificial Intelligence. Sci. Rep. 2018, 8, 1879. [Google Scholar] [CrossRef]
- Gimenez, N.; Tripathi, R.; Giró, A.; Rosich, L.; López-Guerra, M.; López-Oreja, I.; Playa-Albinyana, H.; Arenas, F.; Mas, J.M.; Pérez-Galán, P.; et al. Systems biology drug screening identifies statins as enhancers of current therapies in chronic lymphocytic leukemia. Sci. Rep. 2020, 10, 22153. [Google Scholar] [CrossRef]
- Bayes-Genis, A.; Iborra-Egea, O.; Spitaleri, G.; Domingo, M.; Revuelta-López, E.; Codina, P.; Cediel, G.; Santiago-Vacas, E.; Cserkóová, A.; Pascual-Figal, D.; et al. Decoding empagliflozin’s molecular mechanism of action in heart failure with preserved ejection fraction using artificial intelligence. Sci. Rep. 2021, 11, 12025. [Google Scholar] [CrossRef]
- Lorén, V.; Garcia-Jaraquemada, A.; Naves, J.E.; Carmona, X.; Mañosa, M.; Aransay, A.M.; Lavin, J.L.; Sánchez, I.; Cabré, E.; Manyé, J.; et al. ANP32E, a Protein Involved in Steroid-Refractoriness in Ulcerative Colitis, Identified by a Systems Biology Approach. J. Crohns. Colitis 2019, 13, 351–361. [Google Scholar] [CrossRef]
- Moncunill, G.; Scholzen, A.; Mpina, M.; Nhabomba, A.; Hounkpatin, A.B.; Osaba, L.; Valls, R.; Campo, J.J.; Sanz, H.; Jairoce, C.; et al. Antigen-stimulated PBMC transcriptional protective signatures for malaria immunization. Sci. Transl. Med. 2020, 12, eaay8924. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Clough, E.; Barrett, T. The Gene Expression Omnibus Database. Methods Mol. Biol. 2016, 1418, 93–110. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef] [PubMed]
- Nanjappa, V.; Thomas, J.K.; Marimuthu, A.; Muthusamy, B.; Radhakrishnan, A.; Sharma, R.; Ahmad Khan, A.; Balakrishnan, L.; Sahasrabuddhe, N.A.; Kumar, S.; et al. Plasma Proteome Database as a resource for proteomics research: 2014 update. Nucleic Acids Res. 2014, 42, D959–D965. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Oksvold, P.; Fagerberg, L.; Lundberg, E.; Jonasson, K.; Forsberg, M.; Zwahlen, M.; Kampf, C.; Wester, K.; Hober, S.; et al. Towards a knowledge-based Human Protein Atlas. Nat. Biotechnol. 2010, 28, 1248–1250. [Google Scholar] [CrossRef]
- Nowak, A.; Mechtler, T.P.; Desnick, R.J.; Kasper, D.C. Plasma LysoGb3: A useful biomarker for the diagnosis and treatment of Fabry disease heterozygotes. Mol. Genet. Metab. 2017, 120, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Auray-Blais, C.; Lavoie, P.; Abaoui, M.; Côté, A.-M.; Boutin, M.; Akbari, A.; Levin, A.; Mac-Way, F.; Tr Clarke, J. High-risk screening for Fabry disease in a Canadian cohort of chronic kidney disease patients. Clin. Chim. Acta. 2020, 501, 234–240. [Google Scholar] [CrossRef]
- Carnicer-Cáceres, C.; Arranz-Amo, J.A.; Cea-Arestin, C.; Camprodon-Gomez, M.; Moreno-Martinez, D.; Lucas-Del-Pozo, S.; Moltó-Abad, M.; Tigri-Santiña, A.; Agraz-Pamplona, I.; Rodriguez-Palomares, J.F.; et al. Biomarkers in Fabry Disease. Implications for Clinical Diagnosis and Follow-up. J. Clin. Med. 2021, 10, 1664. [Google Scholar] [CrossRef]
- Vojtová, L.; Zima, T.; Tesař, V.; Michalová, J.; Přikryl, P.; Dostálová, G.; Linhart, A. Study of urinary proteomes in Anderson-Fabry disease. Ren. Fail. 2010, 32, 1202–1209. [Google Scholar] [CrossRef]
- Heo, S.H.; Kang, E.; Kim, Y.-M.; Go, H.; Kim, K.Y.; Jung, J.Y.; Kang, M.; Kim, G.-H.; Kim, J.-M.; Choi, I.-H.; et al. Fabry disease: Characterisation of the plasma proteome pre- and post-enzyme replacement therapy. J. Med. Genet. 2017, 54, 771–780. [Google Scholar] [CrossRef]
- Xiao, K.; Lu, D.; Hoepfner, J.; Santer, L.; Gupta, S.; Pfanne, A.; Thum, S.; Lenders, M.; Brand, E.; Nordbeck, P.; et al. Circulating microRNAs in Fabry Disease. Sci. Rep. 2019, 9, 15277. [Google Scholar] [CrossRef]
- Salamon, I.; Biagini, E.; Kunderfranco, P.; Roncarati, R.; Ferracin, M.; Taglieri, N.; Nardi, E.; Laprovitera, N.; Tomasi, L.; Santostefano, M.; et al. Circulating miR-184 is a potential predictive biomarker of cardiac damage in Anderson-Fabry disease. Cell Death Dis. 2021, 12, 1150. [Google Scholar] [CrossRef]
- Nowak, A.; Haddad, G.; Kistler, A.D.; Nlandu-Khodo, S.; Beuschlein, F.; Wüthrich, R.P.; Lorenzen, J.M.; Kölling, M. Circular RNA-based biomarkers in blood of patients with Fabry disease and related phenotypes. J. Med. Genet. 2022, 59, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Schiffmann, R.; Waldek, S.; Auray-Blais, C. Biomarkers of Fabry disease nephropathy. Clin. J. Am. Soc. Nephrol. 2010, 5, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Riccio, E.; Sabbatini, M.; Capuano, I.; Pisani, A. Early Biomarkers of Fabry Nephropathy: A Review of the Literature. Nephron 2019, 143, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Heinecke, J.W. Oxidized amino acids: Culprits in human atherosclerosis and indicators of oxidative stress. Free Radic. Biol. Med. 2002, 32, 1090–1101. [Google Scholar] [CrossRef] [PubMed]
- Shu, L.; Vivekanandan-Giri, A.; Pennathur, S.; Smid, B.E.; Aerts, J.M.F.G.; Hollak, C.E.M.; Shayman, J.A. Establishing 3-nitrotyrosine as a biomarker for the vasculopathy of Fabry disease. Kidney Int. 2014, 86, 58–66. [Google Scholar] [CrossRef]
- Shishehbor, M.H.; Aviles, R.J.; Brennan, M.L.; Fu, X.; Goormastic, M.; Pearce, G.L.; Gokce, N.; Keaney, J.F.; Penn, M.S.; Sprecher, D.L.; et al. Association of Nitrotyrosine Levels with Cardiovascular Disease and Modulation by Statin Therapy. JAMA 2003, 289, 1675–1680. [Google Scholar] [CrossRef]
- Rauchhaus, M.; Doehner, W.; Francis, D.P.; Davos, C.; Kemp, M.; Liebenthal, C.; Niebauer, J.; Hooper, J.; Volk, H.D.; Coats, A.J.S.; et al. Plasma cytokine parameters and mortality in patients with chronic heart failure. Circulation 2000, 102, 3060–3067. [Google Scholar] [CrossRef]
- Maeda, K.; Tsutamoto, T.; Wada, A.; Mabuchi, N.; Hayashi, M.; Tsutsui, T.; Ohnishi, M.; Sawaki, M.; Fujii, M.; Matsumoto, T.; et al. High levels of plasma brain natriuretic peptide and interleukin-6 after optimized treatment for heart failure are independent risk factors for morbidity and mortality in patients with congestive heart failure. J. Am. Coll. Cardiol. 2000, 36, 1587–1593. [Google Scholar] [CrossRef]
- González-Nicolás, M.Á.; González-Guerrero, C.; Goicoechea, M.; Boscá, L.; Valiño-Rivas, L.; Lázaro, A. Biomarkers in Contrast-Induced Acute Kidney Injury: Towards A New Perspective. Int. J. Mol. Sci. 2024, 25, 3438. [Google Scholar] [CrossRef]
- Braga, M.C.; Fonseca, F.L.A.; Marins, M.M.; Gomes, C.P.; Bacci, M.R.; Martins, A.M.; D’Almeida, V. Evaluation of Beta 2-Microglobulin, Cystatin C, and Lipocalin-2 as Renal Biomarkers for Patients with Fabry Disease. Nephron 2019, 143, 217–227. [Google Scholar] [CrossRef]
- Chien, Y.; Chien, C.-S.; Chiang, H.-C.; Huang, W.-L.; Chou, S.-J.; Chang, W.-C.; Chang, Y.-L.; Leu, H.-B.; Chen, K.-H.; Wang, K.-L.; et al. Interleukin-18 deteriorates Fabry cardiomyopathy and contributes to the development of left ventricular hypertrophy in Fabry patients with GLA IVS4+919 G>A mutation. Oncotarget 2016, 7, 87161–87179. [Google Scholar] [CrossRef] [PubMed]
- Plow, E.F.; Simon, D.I. Implicating ILK in inflammation. Blood 2020, 136, 2097–2099. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.N.; Sbaizero, O.; Taylor, M.R.G.; Mestroni, L. Lamin A/C Cardiomyopathy: Implications for Treatment. Curr. Cardiol. Rep. 2019, 21, 160. [Google Scholar] [CrossRef] [PubMed]
- DeGraba, T.; Azhar, S.; Dignat-George, F.; Brown, E.; Boutière, B.; Altarescu, G.; McCarron, R.; Schiffmann, R. Profile of endothelial and leukocyte activation in Fabry patients. Ann. Neurol. 2000, 47, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Campillo, S.; Gutiérrez-Calabrés, E.; García-Miranda, S.; Griera, M.; Fernández Rodríguez, L.; de Frutos, S.; Rodríguez-Puyol, D.; Calleros, L. Integrin-linked kinase mRNA expression in circulating mononuclear cells as a biomarker of kidney and vascular damage in experimental chronic kidney disease. Cell Commun. Signal. 2024, 22, 264. [Google Scholar] [CrossRef]
- Camici, P.G.; Crea, F. Coronary microvascular dysfunction. N. Engl. J. Med. 2007, 356, 830–840. [Google Scholar] [CrossRef]
- He, Q.; Li, Z. The dysregulated expression and functional effect of CaMK2 in cancer. Cancer Cell Int. 2021, 21, 326. [Google Scholar] [CrossRef]
- Yasuda, R.; Hayashi, Y.; Hell, J.W. CaMKII: A central molecular organizer of synaptic plasticity, learning and memory. Nat. Rev. Neurosci. 2022, 23, 666–682. [Google Scholar] [CrossRef]
- Briata, P.; Bordo, D.; Puppo, M.; Gorlero, F.; Rossi, M.; Perrone-Bizzozero, N.; Gherzi, R. Diverse roles of the nucleic acid-binding protein KHSRP in cell differentiation and disease. Wiley Interdiscip. Rev. RNA 2016, 7, 227–240. [Google Scholar] [CrossRef]
- Körver, S.; Geurtsen, G.J.; Hollak, C.E.M.; van Schaik, I.N.; Longo, M.G.F.; Lima, M.R.; Dijkgraaf, M.G.W.; Langeveld, M. Cognitive functioning and depressive symptoms in Fabry disease: A follow-up study. J. Inherit. Metab. Dis. 2020, 43, 1070–1081. [Google Scholar] [CrossRef]
- Cortés-Saladelafont, E.; Fernández-Martín, J.; Ortolano, S. Fabry Disease and Central Nervous System Involvement: From Big to Small, from Brain to Synapse. Int. J. Mol. Sci. 2023, 24, 5246. [Google Scholar] [CrossRef] [PubMed]
- Ryckman, A.E.; Brockhausen, I.; Walia, J.S. Metabolism of Glycosphingolipids and Their Role in the Pathophysiology of Lysosomal Storage Disorders. Int. J. Mol. Sci. 2020, 21, 6881. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, M.M.; Changsila, E.; Iaonou, C.; Goker-Alpan, O. Impaired autophagic and mitochondrial functions are partially restored by ERT in Gaucher and Fabry diseases. PLoS ONE 2019, 14, e0210617. [Google Scholar] [CrossRef] [PubMed]
- McKenna, M.C.; Schuck, P.F.; Ferreira, G.C. Fundamentals of CNS energy metabolism and alterations in lysosomal storage diseases. J. Neurochem. 2019, 148, 590–599. [Google Scholar] [CrossRef]
- Pourhamzeh, M.; Moravej, F.G.; Arabi, M.; Shahriari, E.; Mehrabi, S.; Ward, R.; Ahadi, R.; Joghataei, M.T. The Roles of Serotonin in Neuropsychiatric Disorders. Cell. Mol. Neurobiol. 2022, 42, 1671–1692. [Google Scholar] [CrossRef]
- Ring, H.Z.; Vameghi-Meyers, V.; Nikolic, J.M.; Min, H.; Black, D.L.; Francke, U. Mapping of the KHSRP gene to a region of conserved synteny on human chromosome 19p13.3 and mouse chromosome 17. Genomics 1999, 56, 350–352. [Google Scholar] [CrossRef]
- Palzer, K.A.; Bolduan, V.; Käfer, R.; Kleinert, H.; Bros, M.; Pautz, A. The Role of KH-Type Splicing Regulatory Protein (KSRP) for Immune Functions and Tumorigenesis. Cells 2022, 11, 1482. [Google Scholar] [CrossRef]
- Olguin, S.L.; Patel, P.; Buchanan, C.N.; Dell’Orco, M.; Gardiner, A.S.; Cole, R.; Vaughn, L.S.; Sundararajan, A.; Mudge, J.; Allan, A.M.; et al. KHSRP loss increases neuronal growth and synaptic transmission and alters memory consolidation through RNA stabilization. Commun. Biol. 2022, 5, 672. [Google Scholar] [CrossRef]
- Califf, R.M. Biomarker definitions and their applications. Exp. Biol. Med. 2018, 243, 213–221. [Google Scholar] [CrossRef]
- Iborra-Egea, O.; Gálvez-Montón, C.; Prat-Vidal, C.; Roura, S.; Soler-Botija, C.; Revuelta-López, E.; Ferrer-Curriu, G.; Segú-Vergés, C.; Mellado-Bergillos, A.; Gomez-Puchades, P.; et al. Deep Learning Analyses to Delineate the Molecular Remodeling Process after Myocardial Infarction. Cells 2021, 10, 3268. [Google Scholar] [CrossRef]
- Aerts, J.M.F.G.; Kallemeijn, W.W.; Wegdam, W.; Joao Ferraz, M.; van Breemen, M.J.; Dekker, N.; Kramer, G.; Poorthuis, B.J.; Groener, J.E.M.; Cox-Brinkman, J.; et al. Biomarkers in the diagnosis of lysosomal storage disorders: Proteins, lipids, and inhibodies. J. Inherit. Metab. Dis. 2011, 34, 605–619. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Yao, T.; Fujimura, T.; Murayama, K.; Okumura, K.; Hagiwara, N.; Seko, Y. Oxidative stress-responsive apoptosis-inducing protein in patients with heterozygous familial hypercholesterolemia. Heart Vessels 2021, 36, 1923–1932. [Google Scholar] [CrossRef] [PubMed]
- Tseng, W.-L.; Chou, S.-J.; Chiang, H.-C.; Wang, M.-L.; Chien, C.-S.; Chen, K.-H.; Leu, H.-B.; Wang, C.-Y.; Chang, Y.-L.; Liu, Y.-Y.; et al. Imbalanced Production of Reactive Oxygen Species and Mitochondrial Antioxidant SOD2 in Fabry Disease-Specific Human Induced Pluripotent Stem Cell-Differentiated Vascular Endothelial Cells. Cell Transplant. 2017, 26, 513–527. [Google Scholar] [CrossRef] [PubMed]
- Vujkovac, A.C.; Vujkovac, B.; Novaković, S.; Števanec, M.; Šabovič, M. Characteristics of Vascular Phenotype in Fabry Patients. Angiology 2021, 72, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Yazd, H.S.; Bazargani, S.F.; Vanbeek, C.A.; King-Morris, K.; Heldermon, C.; Segal, M.S.; Clapp, W.L.; Garrett, T.J. LC-MS lipidomics of renal biopsies for the diagnosis of Fabry disease. J. Mass Spectrom. Adv. Clin. Lab 2021, 22, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Viceconti, M.; Pappalardo, F.; Rodriguez, B.; Horner, M.; Bischoff, J.; Musuamba Tshinanu, F. In silico trials: Verification, validation and uncertainty quantification of predictive models used in the regulatory evaluation of biomedical products. Methods 2021, 185, 120–127. [Google Scholar] [CrossRef]
- Whybra, C.; Kampmann, C.; Krummenauer, F.; Ries, M.; Mengel, E.; Miebach, E.; Baehner, F.; Kim, K.; Bajbouj, M.; Schwarting, A.; et al. The Mainz Severity Score Index: A new instrument for quantifying the Anderson-Fabry disease phenotype, and the response of patients to enzyme replacement therapy. Clin. Genet. 2004, 65, 299–307. [Google Scholar] [CrossRef]
- Simats, A.; Ramiro, L.; Valls, R.; de Ramón, H.; García-Rodríguez, P.; Orset, C.; Artigas, L.; Sardon, T.; Rosell, A.; Montaner, J. Ceruletide and Alpha-1 Antitrypsin as a Novel Combination Therapy for Ischemic Stroke. Neurother. J. Am. Soc. Exp. Neurother. 2022, 19, 513–527. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. Data, information, knowledge and principle: Back to metabolism in KEGG. Nucleic Acids Res. 2014, 42, D199–D205. [Google Scholar] [CrossRef]
- Croft, D.; Mundo, A.F.; Haw, R.; Milacic, M.; Weiser, J.; Wu, G.; Caudy, M.; Garapati, P.; Gillespie, M.; Kamdar, M.R.; et al. The Reactome pathway knowledgebase. Nucleic Acids Res. 2014, 42, D472–D477. [Google Scholar] [CrossRef]
- Orchard, S.; Ammari, M.; Aranda, B.; Breuza, L.; Briganti, L.; Broackes-Carter, F.; Campbell, N.H.; Chavali, G.; Chen, C.; del-Toro, N.; et al. The MIntAct project--IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014, 42, D358–D363. [Google Scholar] [CrossRef] [PubMed]
- Salwinski, L.; Licata, L.; Winter, A.; Thorneycroft, D.; Khadake, J.; Ceol, A.; Aryamontri, A.C.; Oughtred, R.; Livstone, M.; Boucher, L.; et al. Recurated protein interaction datasets. Nat. Methods 2009, 6, 860–861. [Google Scholar] [CrossRef]
- Keshava Prasad, T.S.; Goel, R.; Kandasamy, K.; Keerthikumar, S.; Kumar, S.; Mathivanan, S.; Telikicherla, D.; Raju, R.; Shafreen, B.; Venugopal, A.; et al. Human Protein Reference Database--2009 update. Nucleic Acids Res. 2009, 37, D767–D772. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Cho, J.-W.; Lee, S.; Yun, A.; Kim, H.; Bae, D.; Yang, S.; Kim, C.Y.; Lee, M.; Kim, E.; et al. TRRUST v2: An expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2018, 46, D380–D386. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Cheng, D.; Shrivastava, S.; Tzur, D.; Gautam, B.; Hassanali, M. DrugBank: A knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008, 36, D901–D906. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M.; Letunic, I.; Jensen, L.J.; Bork, P. The SIDER database of drugs and side effects. Nucleic Acids Res. 2016, 44, D1075–D1079. [Google Scholar] [CrossRef]
- Chow, C.; Liu, C. Approximating discrete probability distributions with dependence trees. IEEE Trans. Inf. Theory 1968, 14, 462–467. [Google Scholar] [CrossRef]
- Peng, H.; Long, F.; Ding, C. Feature selection based on mutual information: Criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans. Pattern Anal. Mach. Intell. 2005, 27, 1226–1238. [Google Scholar] [CrossRef]
- Bishop, C.M. Pattern Recoginiton and Machine Learning; Springer: New York, NY, USA, 2006; ISBN 9780387310732. [Google Scholar]
- Guyon, I.; Weston, J.; Barnhill, S.; Vapnik, V. Gene Selection for Cancer Classification using Support Vector Machines. Mach. Learn. 2002, 46, 389–422. [Google Scholar] [CrossRef]
- Ververidis, D.; Kotropoulos, C. Fast and accurate sequential floating forward feature selection with the Bayes classifier applied to speech emotion recognition. Signal Process. 2008, 88, 2956–2970. [Google Scholar] [CrossRef]
- Christin, C.; Hoefsloot, H.C.J.; Smilde, A.K.; Hoekman, B.; Suits, F.; Bischoff, R.; Horvatovich, P. A critical assessment of feature selection methods for biomarker discovery in clinical proteomics. Mol. Cell. Proteomics 2013, 12, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Madsen, H.; Thyregod, P. Introduction to General and Generalized Linear Models; Routledge: London, UK, 2011; ISBN 9781420091557. [Google Scholar]
- Russell, S. Artificial Intelligence: A Modern Approach; Recording for the Blind & Dyslexic: Princeton, NJ, USA, 2003; ISBN 9780137903955. [Google Scholar]
- Kentsis, A.; Monigatti, F.; Dorff, K.; Campagne, F.; Bachur, R.; Steen, H. Urine proteomics for profiling of human disease using high accuracy mass spectrometry. Proteomics. Clin. Appl. 2009, 3, 1052–1061. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Tzur, D.; Knox, C.; Eisner, R.; Guo, A.C.; Young, N.; Cheng, D.; Jewell, K.; Arndt, D.; Sawhney, S.; et al. HMDB: The Human Metabolome Database. Nucleic Acids Res. 2007, 35, D521–D526. [Google Scholar] [CrossRef] [PubMed]
- Griffiths-Jones, S.; Grocock, R.J.; van Dongen, S.; Bateman, A.; Enright, A.J. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006, 34, D140–D144. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Biomarker (Gene Symbol) [50] | Biomarker (UniProt Code) [51] | Cross-Validated Accuracy (%) | Cross-Validated p Value |
|---|---|---|---|
| MAPK11 | Q15759 | 76 | 7.41 × 10−20 |
| B2M | P61769 | 71 | 1.18 × 10−10 |
| CD80 | P33681 | 68 | 2.14 × 10−7 |
| CCL2 | P13500 | 66 | 4.77 × 10−6 |
| NOS3 | P29474 | 63 | 9.04 × 10−6 |
| GPX1 | P07203 | 61 | 3.54 × 10−6 |
| ACTB | P60709 | 61 | 0.0000772 |
| TGFB1 | P01137 | 61 | 0.000801 |
| AGTR1 | P30556 | 60 | 0.004 |
| MMP9 | P14780 | 59 | 0.002 |
| Biomarker Candidate * (Gene Symbol) [50] | Biomarker Candidate (UniProt Code) [51] | Plasma Protein | Urine Protein | Cross-Validated Accuracy (%) | Location ** [51,52,53] |
|---|---|---|---|---|---|
| CAMK2A | Q9UQM7 | YES | NO | 100 | NV |
| CTSS | P25774 | YES | NO | 100 | N |
| FOSL1 | P15407 | YES | NO | 100 | N |
| PPP1CC | P36873 | YES | NO | 100 | N |
| ILK | Q13418 | YES | NO | 99 | NV |
| CDK1 | P06493 | YES | NO | 96 | N |
| GNB5 | O14775 | YES | NO | 96 | N |
| AKT3 | Q9Y243 | YES | NO | 96 | N |
| LMNA | P02545 | YES | YES | 95 | NV |
| MAPK1 | P28482 | YES | YES | 95 | N |
| KHSRP | Q92945 | YES | NO | 93 | NV |
| PTPN13 | Q12923 | NO | YES | 92 | N |
| GNB3 | P16520 | YES | NO | 89 | N |
| STK3 | Q13188 | YES | NO | 88 | N |
| TGFB2 | P61812 | NO | YES | 88 | N |
| CIITA | P33076 | YES | NO | 87 | N |
| ELAVL1 | Q15717 | YES | NO | 87 | N |
| VIM | P08670 | YES | NO | 86 | N |
| NCAM1 | P13591 | YES | YES | 84 | NV |
| IL18 | Q14116 | YES | YES | 84 | NV |
| KPNB1 | Q14974 | YES | NO | 81 | NV |
| GRIN1 | Q05586 | YES | NO | 80 | N |
| APP | P05067 | YES | YES | 78 | N |
| DNMT1 | P26358 | YES | NO | 78 | N |
| PML | P29590 | YES | NO | 76 | NV |
| BRCA1 | P38398 | YES | NO | 76 | NV |
| TYMS | P04818 | YES | NO | 75 | NV |
| SPP1 | P10451 | YES | NO | 75 | N |
| ANK3 | Q12955 | YES | NO | 74 | N |
| ETS1 | P14921 | YES | NO | 73 | N |
| PKN2 | Q16513 | YES | NO | 72 | N |
| YWHAQ | P27348 | YES | NO | 72 | N |
| HIPK2 | Q9H2X6 | YES | NO | 71 | NV |
| ANK2 | Q01484 | YES | NO | 71 | NV |
| DCC | P43146 | YES | NO | 71 | N |
| B2M | P61769 | YES | NO | 71 | NV |
| Motive | Motive Name | Proteins |
|---|---|---|
| 1 | α-Galactosidase A mutation and Gb3 accumulation | 13 |
| 2 | Oxidative stress | 16 |
| 3 | Impaired autophagy | 8 |
| 4 | Cellular death | 10 |
| 5 | Lipid raft disruption | 4 |
| 6 | Compromised energy metabolism | 17 |
| 7 | Inflammation and immune response | 27 |
| Neuronal-Related Proteins | Vascular-Related Proteins | ||
|---|---|---|---|
| Subcellular location | THPA [52,53] | - | - |
| UniProt [51] | axon, dendrite, midbody, synaptic vesicle membrane, neuronal cell body, axon initial segment, terminal bouton, dendritic spine, postsynaptic density of dendrite, neuron projection, presynaptic membrane, synapse, postsynaptic membrane, cell body, synaptic cleft, myelin sheath, synaptic vesicle, paranode region of axon, dendritic branch, excitatory synapse | - | |
| Tissue | THPA [52,53] | caudate, cerebellum, cerebral cortex, hippocampus | smooth muscle |
| UniProt [51] | cerebrospinal fluid, peripheral nerve, pituitary, substantia nigra, temporal cortex, glial cell, brain stem, hippocampus, cerebellum, nervous system, neuron, astrocyte, Schwann cell, nerve, brain | vascular smooth muscle, smooth muscle cell, smooth muscle, endothelial cell |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gervas-Arruga, J.; Barba-Romero, M.Á.; Fernández-Martín, J.J.; Gómez-Cerezo, J.F.; Segú-Vergés, C.; Ronzoni, G.; Cebolla, J.J. In Silico Modeling of Fabry Disease Pathophysiology for the Identification of Early Cellular Damage Biomarker Candidates. Int. J. Mol. Sci. 2024, 25, 10329. https://doi.org/10.3390/ijms251910329
Gervas-Arruga J, Barba-Romero MÁ, Fernández-Martín JJ, Gómez-Cerezo JF, Segú-Vergés C, Ronzoni G, Cebolla JJ. In Silico Modeling of Fabry Disease Pathophysiology for the Identification of Early Cellular Damage Biomarker Candidates. International Journal of Molecular Sciences. 2024; 25(19):10329. https://doi.org/10.3390/ijms251910329
Chicago/Turabian StyleGervas-Arruga, Javier, Miguel Ángel Barba-Romero, Jorge Julián Fernández-Martín, Jorge Francisco Gómez-Cerezo, Cristina Segú-Vergés, Giacomo Ronzoni, and Jorge J. Cebolla. 2024. "In Silico Modeling of Fabry Disease Pathophysiology for the Identification of Early Cellular Damage Biomarker Candidates" International Journal of Molecular Sciences 25, no. 19: 10329. https://doi.org/10.3390/ijms251910329
APA StyleGervas-Arruga, J., Barba-Romero, M. Á., Fernández-Martín, J. J., Gómez-Cerezo, J. F., Segú-Vergés, C., Ronzoni, G., & Cebolla, J. J. (2024). In Silico Modeling of Fabry Disease Pathophysiology for the Identification of Early Cellular Damage Biomarker Candidates. International Journal of Molecular Sciences, 25(19), 10329. https://doi.org/10.3390/ijms251910329