
Animal Models of Autistic-like Behavior in Rodents: A Scoping Review and Call for a Comprehensive Scoring System



Abstract
:1. Introduction
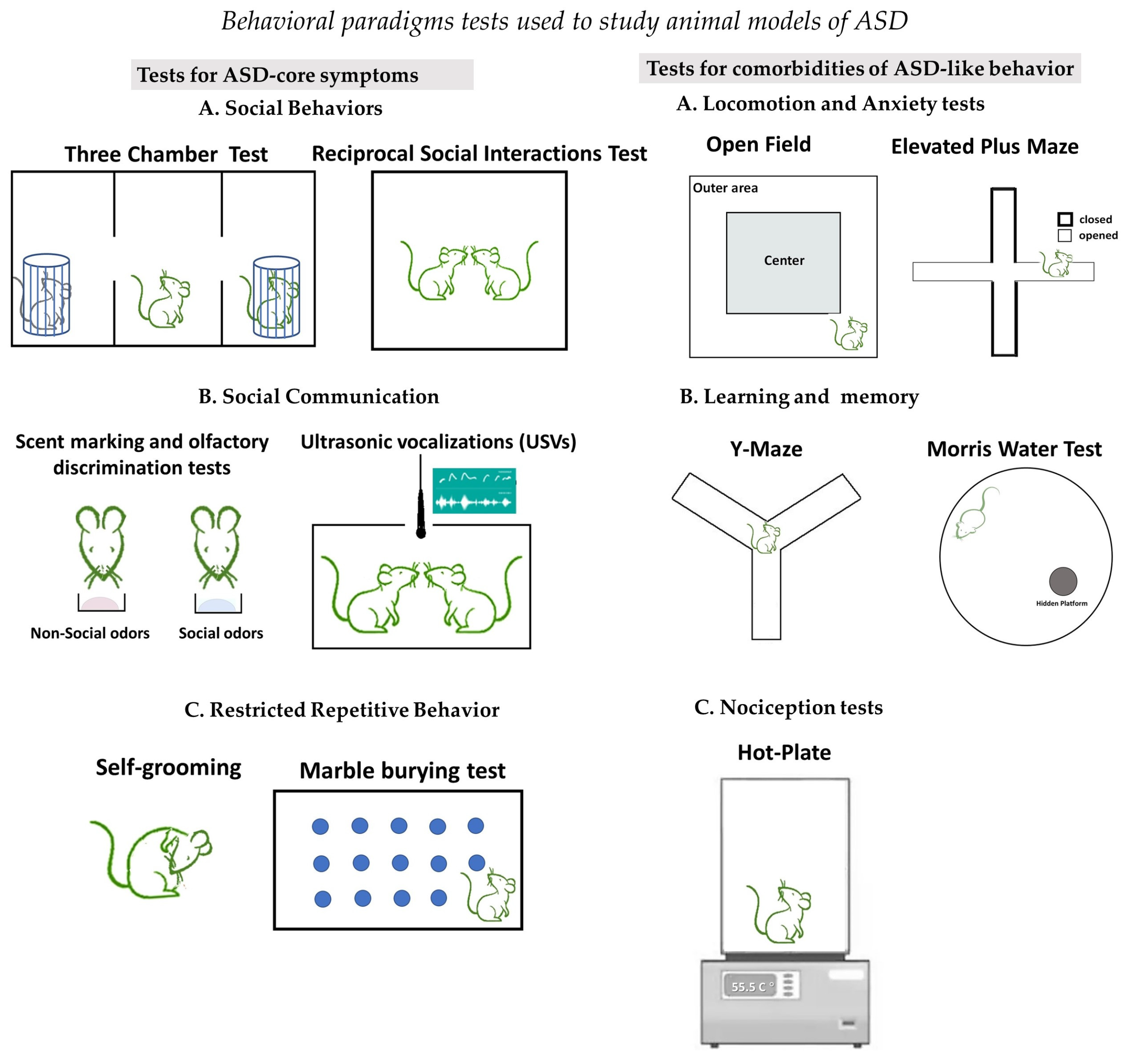
2. Brief Description of Behavioral Assays That Define ASD-like Behaviors in Rodents
2.1. Tests to Investigate Social Behaviors
2.2. Tests to Investigate Social Communication
2.3. Tests to Investigate Restricted Repetitive Behaviors
2.4. Tests to Investigate for Comorbidities of ASD-like Behaviors
3. Non-Genetic Models of ASD-like Behaviors
3.1. Biological Models of ASD-like Behaviors
3.2. Chemical Models of ASD-like Behaviors
4. Genetic Models in Rodents for ASD-like Behaviors

{kind=link}
| Gene | Conditional Animal Models | ASD-like Phenotype | Autor (Ref.) |
|---|---|---|---|
| X-linked Methyl CpG Binding Protein 2, MeCP2 | |||
Mecp2tm1.1Bird Mice Mecp2tm1.1Jae | The targeted deletion that removes exons 3 and 4 of the Mecp2 gene, resulting in a complete lack of MECP2 protein product. Mecp2tm1.1Jae mice created by condition disruption of exon 3 of the Mecp2 gene, resulting in the lack of a functional MECP2 protein. | Male hemizygous Mecp2-null mice develop a Rett-like phenotype from the 4 weeks of age with raid regression, and die between 6 and 12 weeks of age. Hind limb clasping, tremors, breathing irregularities, loss of muscle tone, reduced locomotion, reduced brain weight and body weight, are the experience of a rapid phenotypic. Female heterogeneous Mecp2-null mice develop the same features at 4–6 months of age and typically live a normal lifespan. Two weeks of treatment with Mirtazapine rescue dendritic arborization and spine density of pyramidal neurons and improve phenotypic score. | Guy et al. [99] Bittalo et al. [100] Flores Gutiérrez et al. [97] Chen et al. [101] |
| Mice Mecp2-308/y | Stop codon at amino acid position 308, leading to the production of a truncated MeCP2 protein that lacks the C-terminal domain. | Displaying a milder RTT phenotype, a delayed onset of symptoms, and an extended lifespan, due to the presence of partially functional truncated protein. | Shahbazian et al. [102] |
| Rats Mecp2308 | Expressing a truncated allele of Mecp2. | Displayed RTT phenotype: growth retardation, reduced locomotion, impaired social behavior, breathing abnormalities, excessive spontaneous firing activity of neurons in the locus coeruleus. | Wu et al. [103] |
| Viaat-Mecp2−/y | Male Viaat-Mecp2−/y mice are nearly absent MeCP2 protein from >90% of GABAergic neurons. | Male Viaat-Mecp2−/y mice developed RTT and ASD-like phenotype from 5 weeks of age: Motor dysfunction. Repetitive behaviors. Impaired working memory. Reduced levels of Gad1 and Gad2. Decreased GABA immunostaining in the cortex and striatum. | Chao et al. [104] |
| Mecp2lox-Stop/Y | Male Mecp2 null mice, with genetically restored Mecp2 expression in targeted GABAergic neurons, | Rescue MECP2 functions. Ablation of RTT phenotype. | Ure et al. [105] |
| X-Linked Mental Retardation FMR1 gene, FMR1 | |||
| Fmr1 KO mice and rats | Loss-of-function models; disruption knockout (KO) of the FMR1 gene homolog. | Displayed FXS phenotype: Altered social interaction and social play. Social anxiety. Defects in visual attention. Auditory dysfunctions. Cognitive deficits. Repetitive behaviors,. Hyperactivity. Differences in dendritic spines. | Baker et al. [106] Ding et al. [107] Albert et al. [108] Hamilton et al. [109] Barić et al. [110] Curnow et al. [111] |
| SH and multiple ankyrin repeat domains proteins (SHANK) | |||
| Shank3+/- mice | Deletion of the ankyrin repeat region of the Shank3 gene resulted in a lack of full-length SHANK3 protein. | Heterozygous (Shank3+/-) and homozygous (Shank3-/-) showed normal brain anatomic structure and displayed normal developmental trajectory, normal social interaction, normal spatial learning, and repetitive self-grooming in males. Reduced number of USVs. Decreased GLUR1 and AMPA receptor immunoreactivity. Altered LTP in hippocampal CA1 neurons. | Bozdagi et al. [112] Yang et al. [113] |
| Shank3e4–9 mice | Deletion of the Shank3 gene on exons 4–9 produced transcripts of truncated SHANK3 proteins | Homozygous Shank3e4–9 mice showed abnormal social communication, decreased novel object preference, impaired spatial learning and memory, increased stereotypic self-grooming, increased number of USV, and affected fine motor coordination. Reduction in brain levels of Shank3-interacting protein Homer1b/c, GKAP, and GluA1. | Wang et al. [114] |
| Shank3A−/− null mice Shank3B−/− null mice | Targeting the ankyrin repeat domain, resulting in the loss of the longest Shank3α isoform. Targeting the fragment-encoding exons 13 to 16 of the PDZ domain, which led to the complete deletion of both Shank3α and Shank3β isoforms, as well as a reduction in the Shank3γ isoform. | Shank3B−/− mice exhibited a more pronounced ASD-like phenotype, than Shank3A-/- mice: anxiety-like behavior, repetitive self-injurious grooming. Shank3B−/− mice demonstrated impaired social interaction preference for social novelty. Shank3A−/− mice preserved normal social communication, deficit for social novelty recognition, striatal hypertrophy, increased neuronal complexity, and dendritic arbors. Reduced frequency mEPSCs in striatal medium spiny neurons. Reduced protein levels of glutamate receptor subunits GluR2, NR2A, and NR2B. | Peça et al. [115] |
| Shank3+/ΔC mice | Conditional deletion of exon 21 in the C-terminal of the Shank3 gene, which leads to the expression of a truncated SHANK3 protein. | Only male Shank3+/ΔC mice developed ASD-like phenotype. Decreased level of histone acetylation. Subchronic treatment with romidepsin, class I HDAC inhibitor, transiently rescued social deficits in Shank3+/ΔC mice, elevated the transcriptional level of HDAC2 in PFC, restored β-catenin and restored NMDAR, and elevated expression of actin regulatory genes Grin2. Single I.V. injection with TAT-p-cofilin peptide rescues behavioral deficits and restores NMDAR function. Treatment with UNC0642 inhibitor of EHMT1 and EHMT2, reduced the elevated level of H3K9me2 in the PFC of Shank3+/ΔC mice and rescued autism-like social deficits, and restored NMDAR function | Qin et al. [116] Duffney et al. [117] Wang et al. [118]. |
| Neuroligin genes, NLGN | |||
| Knock-in mice Nlgn1 P89L mice | Knock-in mice with the novel missense mutation P89L in the NLGN1 gene. | Heterozygous Nlgn1 P89L mice: Affected sociability and social dominance. Impaired spatial memory Homozygous Nlgn1 P89L developed a milder ASD-like phenotype: Less impairment in sociability and spatial memory. Either homozygous or heterozygous Nlgn1 P89L mice demonstrated normal odor discrimination, object recognition, general locomotor activity, stereotypic repetitive behavior anxiety-like behavior, and altered stress-induced USVs. Decreased levels of NLGN1 protein in the brain. | Nakanishi et al. [119] |
| NL1 KO mice | NLGN1 depletion. | NL1 KO mice exhibited mild deficits in social behavior, impaired spatial memory evaluated by MWM test, and increased repetitive grooming behavior. Impaired hippocampal long-term potentiation. Decrease in the NMDA/AMPA ratio in synapses. A single administration of the NMDA receptor partial coagonist d-cycloserine abolished abnormal grooming phenotype in adult NL1 KO mice. | Blundell et al. [120] |
| R215H-Nlgn2 knock-in mice | Mice carrying the R215H mutation in the Nlgn2 gene lost NLGN protein expression. | R215H-Nlgn2 mice have growth retardation and demonstrated anxiety-like behavior, impaired spatial learning and memory, and enhanced Startle reflex. | Chen et al. [121] |
| R451C-Nlgn3 knock-in mice | Insertion of R451C mutations in an extracellular domain of the Nlgn3 gene caused partial retention of NLGN protein in the ER and further proteasomal degradation. | R451C-Nlgn3 mutant mice demonstrated controversial phenotype. Tabuchi et al. [122] reported reduced sociability facilitated spatial learning and memory increase in inhibitory synaptic transmission, elevating the inhibition-to-excitation (I/E) ratio of synaptic inputs to cerebellar Purkinje cells. Chadman et al. [123] reported normal reciprocal social interactions, learning, and memory in MWT, similar to WT controls, but demonstrated some delay in the early postnatal developmental trajectory. | Tabuchi et al. [124] Lai et al. [125] Chadman et al. [123]. |
| Nlgn3-KO mouse line | Nlgn3-KO knockout mouse line with completely depleted NLGN3. | Nlgn3-KO mice demonstrated increased decreased social recognition and social novelty preference. Impaired olfaction. Reduced number of USVs in males. | Radyushkin et al. [122] |
| Nlgn4-KO mice | Nlgn4-knockout mouse line with chimeric nonfunctional NLGN4 protein. | Nlgn4-KO mouse developed abnormality in reciprocal social interactions and communication, decreased USVs, and reduced total brain volume. | Jamain et al. [126] |
| Inbred model of idiopathic ASD: BTBR mice | |||
| BTBR T+ Itpr3tf/J | Carries mutations in genes including (nonagouti; Black and Tan), Itpr3tf (inositol 1,4,5-triphosphate receptor 3; tufted), and T (brachyury). | BTBR mice demonstrated natural traits of the core autism symptom: decreased social interaction, increased USVs and abnormal patterns of sonograms, repetitive grooming, lack of corpus callosum and hippocampal commissure, decreased cortical thickness, and thalamic gray matter volume. | Scattoni et al. [127] McFarlane et al. [128] Scattoni et al. [129] Wöhr et al. [130] Meyza et al. [131] Dodero et al. [132] |
| Genetic models in nonhuman primates (NHP) | |||
| Mecp2 transgenic MF | Mutant MF expressed human Mecp2 via lentiviral infection of monkey oocytes mitigating MECP2 duplication syndrome. | Mecp2 transgenic MF exhibited repetitive circular locomotion, increased stress response, reduced social interaction, mildly impaired cognition, significant enrichment in gaba-related signaling pathways, reduced β-synchronization in fronto-parieto-occipital networks EEG studies, and hyperconnectivity in prefrontal and cingulate networks. | Liu et al. [133] Cai et al. [134] |
| Mecp2 transgenic Rhesus and cynomolgus monkeys | Mecp2 mutagenesis was induced by microinjection of Mecp2- exon 3-targeted TALEN plasmids into rhesus and cynomolgus zygotes, leading to MECP2 altered expression or function. | Male mutant monkeys were embryonic lethal. Female Mecp2 mutant monkeys demonstrated stereotypical behaviors, impaired active social interaction, reduced exploration, and affected sleep patterns. | Liu et al. [135] Chen et al. [136] |
| Shank3-deficient MF | CRISPR-Cas9-targeting exon 21 of SHANK3 in Macaca fascicularis resulting in expression of non-functional SHANK3 protein | SHANK3-deficient MF capitulated most symptoms of Phelan–McDermid syndrome: Impaired sleep and motor functions. Increased repetitive behaviors. MRI: Abnormal brain global connectivity. | Zhou et al. [137] |
4.1. Animals with Single Gene Mutations
4.1.1. X-Linked Methyl CpG Binding Protein 2 (MECP2) Gene
4.1.2. X-Linked Mental Retardation FMR1 Gene (Fragile X Syndrome, FMR1)
4.1.3. SH and Multiple Ankyrin Repeat Domains Proteins (SHANK)
4.1.4. Neuroligin Genes (NLGNs)
4.1.5. Inbred Model of Idiopathic ASD: BTBR Mice
4.2. Genetic Models in Nonhuman Primates (NHP)
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADI-R | Autism Diagnostic Interview-Revised |
| ADOS | Autism Diagnostic Observation Schedule |
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| ASD | Autism spectrum disorder |
| BM | Barnes maze |
| BTBR | Black and Tan Brachyury |
| CARS | Child Autism Rating Scale |
| Cas9 | CRISPR-associated protein 9 |
| CNV | Copy number variation |
| CPF | Chlorpyrifos |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| DNM | De novo mutation |
| DSM-5 | Diagnostic and Statistical Manual |
| DTI | Diffusion tensor imaging |
| EDC | Endocrine disruptor |
| EHMT | Euchromatic histone-lysine N-methyltransferase |
| EPM | Elevated plus maze |
| FMR1 | Fragile X-linked Mental Retardation gene |
| FMRP | Fragile X Mental Retardation Protein |
| FXS | Fragile X Syndrome |
| GABA | γ-aminobutyric acid |
| Gad | Glutamic acid decarboxylase 1 |
| Gad2 | Glutamic acid decarboxylase 2 |
| GD | Gestational day |
| GKAP | Guanylate kinase-associated protein |
| GluR | Glutamate receptor subunit |
| HDAC | Histone deacetylase |
| IgG | Immunoglobulin G |
| IL-6 | interleukin 6 |
| IV | Intravenous |
| KO | Knockout |
| LPS | Lipopolysaccharide |
| MECP2 | Methyl CpG Binding Protein 2 |
| mEPSC | Miniature excitatory postsynaptic current |
| MIA | Maternal immune activation |
| MRI | Magnetic resonance imaging |
| MWM | Morris Water Maze |
| NLGN | Neuroligin |
| NMDA | N-methyl-D-aspertate |
| NOR | Novel Object Recognition |
| NR2 | NMDA receptor subunit |
| OF | Open Fields |
| P2Y | Purinergic receptor |
| PCB | Polychlorinated biphenyls |
| PFC | Prefrontal cortex |
| PMDS | Phelan–McDermid syndrome |
| PND | Post-natal day |
| PolyIC | Polyinosinic-polycytidylic acid |
| RNA | Ribonucleic acid |
| RTT | Rett Syndrome |
| SFARI | Simons Foundation Autism Research Initiative |
| SHANK | SH3 And Multiple Ankyrin Repeat Domain |
| TALEN | Transcription Activator-Like Effector Nuclease |
| USV | Ultrasonic vocalization |
| VPA | Valproic acid |
| WT | Wild type |
| WTM | Water T-maze |
References
- Lange, S.; Inal, J.M. Animal Models of Human Disease. Int. J. Mol. Sci. 2023, 24, 15821. [Google Scholar] [CrossRef] [PubMed]
- Becker, M.; Pinhasov, A.; Ornoy, A. Animal Models of Depression: What Can They Teach Us about the Human Disease? Diagnostics 2021, 11, 123. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, P.; Roy, S.; Ghosh, D.; Nandi, S.K. Role of animal models in biomedical research: A review. Lab. Anim. Res. 2022, 38, 18. [Google Scholar] [CrossRef] [PubMed]
- Dougnon, G.; Matsui, H. Modelling Autism Spectrum Disorder (ASD) and Attention-Deficit/Hyperactivity Disorder (ADHD) Using Mice and Zebrafish. Int. J. Mol. Sci. 2022, 23, 7550. [Google Scholar] [CrossRef] [PubMed]
- Ergaz, Z.; Weinstein-Fudim, L.; Ornoy, A. Genetic and non-genetic animal models for autism spectrum disorders (ASD). Reprod. Toxicol. 2016, 64, 116–140. [Google Scholar] [CrossRef]
- Li, Z.; Zhu, Y.X.; Gu, L.J.; Cheng, Y. Understanding autism spectrum disorders with animal models: Applications, insights, and perspectives. Zool. Res. 2021, 42, 800–824. [Google Scholar] [CrossRef]
- de Abreu, M.S.; Genario, R.; Giacomini, A.; Demin, K.A.; Lakstygal, A.M.; Amstislavskaya, T.G.; Fontana, B.D.; Parker, M.O.; Kalueff, A.V. Zebrafish as a Model of Neurodevelopmental Disorders. Neuroscience 2020, 445, 3–11. [Google Scholar] [CrossRef]
- Nestler, E.J.; Hyman, S.E. Animal models of neuropsychiatric disorders. Nat. Neurosci. 2010, 13, 1161–1169. [Google Scholar] [CrossRef]
- American Psychiatric Association (APA). Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Press: Washington, DC, USA, 2013. [Google Scholar]
- Sarovic, D. A Unifying Theory for Autism: The Pathogenetic Triad as a Theoretical Framework. Front. Psychiatry 2021, 12, 767075. [Google Scholar] [CrossRef]
- Frye, R.E.; Vassall, S.; Kaur, G.; Lewis, C.; Karim, M.; Rossignol, D. Emerging biomarkers in autism spectrum disorder: A systematic review. Ann. Transl. Med. 2019, 7, 792. [Google Scholar] [CrossRef]
- Crawley, J.N. Mouse Behavioral Assays Relevant to the Symptoms of Autism. Brain Pathol. 2007, 17, 448–459. [Google Scholar] [CrossRef] [PubMed]
- Berg, E.L.; Silverman, J.L. Chapter 8—Animal models of autism. In The Neuroscience of Autism; Kana, R.K., Ed.; Academic Press: Cambridge, MA, USA, 2022; pp. 157–196. [Google Scholar]
- Ornoy, A.; Gorobets, D.; Weinstein-Fudim, L.; Becker, M. Sex-Related Changes in the Clinical, Genetic, Electrophysiological, Connectivity, and Molecular Presentations of ASD: A Comparison between Human and Animal Models of ASD with Reference to Our Data. Int. J. Mol. Sci. 2023, 24, 3287. [Google Scholar] [CrossRef]
- Kaidanovich-Beilin, O.; Lipina, T.; Vukobradovic, I.; Roder, J.; Woodgett, J.R. Assessment of Social Interaction Behaviors. J. Vis. Exp. 2011, 48, e2473. [Google Scholar]
- Chao, O.Y.; Yunger, R.; Yang, Y.M. Behavioral assessments of BTBR T+Itpr3tf/J mice by tests of object attention and elevated open platform: Implications for an animal model of psychiatric comorbidity in autism. Behav. Brain Res. 2018, 347, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Hrabovska, S.V.; Salyha, Y.T. Animal Models of Autism Spectrum Disorders and Behavioral Techniques of their Examination. Neurophysiology 2016, 48, 380–388. [Google Scholar] [CrossRef]
- Ellegood, J.; Crawley, J.N. Behavioral and Neuroanatomical Phenotypes in Mouse Models of Autism. Neurotherapeutics 2015, 12, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Jabarin, R.; Netser, S.; Wagner, S. Beyond the three-chamber test: Toward a multimodal and objective assessment of social behavior in rodents. Mol. Autism 2022, 13, 41. [Google Scholar] [CrossRef]
- Luhach, K.; Kulkarni, G.T.; Singh, V.P.; Sharma, B. Attenuation of neurobehavioural abnormalities by papaverine in prenatal valproic acid rat model of ASD. Eur. J. Pharmacol. 2021, 890, 173663. [Google Scholar] [CrossRef]
- Moy, S.S.; Nadler, J.J.; Perez, A.; Barbaro, R.P.; Johns, J.M.; Magnuson, T.R.; Piven, J.; Crawley, J.N. Sociability and preference for social novelty in five inbred strains: An approach to assess autistic-like behavior in mice. Genes Brain Behav. 2004, 3, 287–302. [Google Scholar] [CrossRef]
- Kudryavtseva, N.N. Use of the “partition” test in behavioral and pharmacological experiments. Neurosci. Behav. Physiol. 2003, 33, 461–471. [Google Scholar] [CrossRef]
- Hirsch, M.M.; Deckmann, I.; Fontes-Dutra, M.; Bauer-Negrini, G.; Nunes, G.D.-F.; Nunes, W.; Rabelo, B.; Riesgo, R.; Margis, R.; Bambini-Junior, V. Behavioral alterations in autism model induced by valproic acid and translational analysis of circulating microRNA. Food Chem. Toxicol. 2018, 115, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Brunner, D.; Kabitzke, P.; He, D.; Cox, K.; Thiede, L.; Hanania, T.; Sabath, E.; Alexandrov, V.; Saxe, M.; Peles, E.; et al. Comprehensive Analysis of the 16p11.2 Deletion and Null Cntnap2 Mouse Models of Autism Spectrum Disorder. PLoS ONE 2015, 10, e0134572. [Google Scholar] [CrossRef] [PubMed]
- Wöhr, M.; Roullet, F.I.; Crawley, J.N. Reduced scent marking and ultrasonic vocalizations in the BTBR T+tf/J mouse model of autism. Genes Brain Behav. 2011, 10, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Wöhr, M.; Roullet, F.I.; Hung, A.Y.; Sheng, M.; Crawley, J.N. Communication impairments in mice lacking Shank1: Reduced levels of ultrasonic vocalizations and scent marking behavior. PLoS ONE 2011, 6, e20631. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-C.; Cole, T.B.; Costa, L.G. Behavioral Phenotyping for Autism Spectrum Disorders in Mice. Curr. Protoc. Toxicol. 2017, 72, 11.22.11–11.22.21. [Google Scholar] [CrossRef]
- Fischer, J.; Hammerschmidt, K. Ultrasonic vocalizations in mouse models for speech and socio-cognitive disorders: Insights into the evolution of vocal communication. Genes Brain Behav. 2011, 10, 17–27. [Google Scholar] [CrossRef]
- Premoli, M.; Pietropaolo, S.; Wöhr, M.; Simola, N.; Bonini, S.A. Mouse and rat ultrasonic vocalizations in neuroscience and neuropharmacology: State of the art and future applications. Eur. J. Neurosci. 2023, 57, 2062–2096. [Google Scholar] [CrossRef]
- Simola, N.; Granon, S. Ultrasonic vocalizations as a tool in studying emotional states in rodent models of social behavior and brain disease. Neuropharmacol. Soc. Behav. Bench Bedside 2019, 159, 107420. [Google Scholar] [CrossRef]
- Becker, M.; Gorobets, D.; Shmerkin, E.; Weinstein-Fudim, L.; Pinhasov, A.; Ornoy, A. Prenatal SAMe Treatment Changes via Epigenetic Mechanism/s USVs in Young Mice and Hippocampal Monoamines Turnover at Adulthood in a Mouse Model of Social Hierarchy and Depression. Int. J. Mol. Sci. 2023, 24, 10721. [Google Scholar] [CrossRef]
- Gzielo, K.; Potasiewicz, A.; Hołuj, M.; Litwa, E.; Popik, P.; Nikiforuk, A. Valproic acid exposure impairs ultrasonic communication in infant, adolescent and adult rats. Eur. Neuropsychopharmacol. 2020, 41, 52–62. [Google Scholar] [CrossRef]
- Shekel, I.; Giladi, S.; Raykin, E.; Weiner, M.; Chalifa-Caspi, V.; Lederman, D.; Kofman, O.; Golan, H.M. Isolation-Induced Ultrasonic Vocalization in Environmental and Genetic Mice Models of Autism. Front. Neurosci. 2021, 15, 769670. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Gao, X.; Yang, L. Repetitive Restricted Behaviors in Autism Spectrum Disorder: From Mechanism to Development of Therapeutics. Front. Neurosci. 2022, 16, 780407. [Google Scholar] [CrossRef] [PubMed]
- Eshraghi, A.A.; Memis, I.; Wang, F.; White, I.; Furar, E.; Mittal, J.; Moosa, M.; Atkins, C.M.; Mittal, R. Genetic ablation of metabotropic glutamate receptor 5 in rats results in an autism-like behavioral phenotype. PLoS ONE 2022, 17, e0275937. [Google Scholar] [CrossRef] [PubMed]
- Avraham, Y.; Berry, E.M.; Donskoy, M.; Ahmad, W.A.; Vorobiev, L.; Albeck, A.; Mankuta, D. Beta-carotene as a novel therapy for the treatment of “Autistic like behavior” in animal models of Autism. Behav. Brain Res. 2019, 364, 469–479. [Google Scholar] [CrossRef]
- Hirsch, M.M.; Deckmann, I.; Santos-Terra, J.; Staevie, G.Z.; Fontes-Dutra, M.; Carello-Collar, G.; Körbes-Rockenbach, M.; Brum Schwingel, G.; Bauer-Negrini, G.; Rabelo, B.; et al. Effects of single-dose antipurinergic therapy on behavioral and molecular alterations in the valproic acid-induced animal model of autism. Neuropharmacology 2020, 167, 107930. [Google Scholar] [CrossRef] [PubMed]
- Takumi, T.; Tamada, K.; Hatanaka, F.; Nakai, N.; Bolton, P.F. Behavioral neuroscience of autism. Neurosci. Biobehav. Rev. 2020, 110, 60–76. [Google Scholar] [CrossRef]
- Ornoy, A.; Weinstein-Fudim, L.; Ergaz, Z. Prevention or Amelioration of Autism-Like Symptoms in Animal Models: Will it Bring Us Closer to Treating Human ASD? Int. J. Mol. Sci. 2019, 20, 1074. [Google Scholar] [CrossRef]
- Ornoy, A.; Weinstein-Fudim, L.; Tfilin, M.; Ergaz, Z.; Yanai, J.; Szyf, M.; Turgeman, G. S-adenosyl methionine prevents ASD like behaviors triggered by early postnatal valproic acid exposure in very young mice. Neurotoxicology Teratol. 2019, 71, 64–74. [Google Scholar] [CrossRef]
- Barnhart, C.D.; Yang, D.; Lein, P.J. Using the Morris Water Maze to Assess Spatial Learning and Memory in Weanling Mice. PLoS ONE 2015, 10, e0124521. [Google Scholar] [CrossRef]
- Markram, K.; Rinaldi, T.; La Mendola, D.; Sandi, C.; Markram, H. Abnormal fear conditioning and amygdala processing in an animal model of autism. Neuropsychopharmacology 2008, 33, 901–912. [Google Scholar] [CrossRef]
- Cording, K.R.; Bateup, H.S. Altered motor learning and coordination in mouse models of autism spectrum disorder. Front. Cell. Neurosci. 2023, 17, 1270489. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.L.; Pride, M.C.; Edmiston, E.; Yang, M.; Silverman, J.L.; Crawley, J.N.; Van de Water, J. Autism-specific maternal autoantibodies produce behavioral abnormalities in an endogenous antigen-driven mouse model of autism. Mol. Psychiatry 2020, 25, 2994–3009. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Poulson, S.J.; Mannan, E.; Sivaselvachandran, S.; Cho, M.; Setak, F.; Chan, C. Altered nociceptive behavior and emotional contagion of pain in mouse models of autism. Genes Brain Behav. 2022, 21, e12778. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, S.; Becker, M.; Herman, I.; Shamir, D.; Rigai, T.; Bar-Hamburger, R.; Pick, C. Mianserin and trazodone significantly attenuate the intensity of opioid withdrawal symptoms in mice. Addict. Biol. 2003, 8, 107–114. [Google Scholar] [PubMed]
- Guo, Q.; Yin, X.; Qiao, M.; Jia, Y.; Chen, D.; Shao, J.; Lebaron, T.W.; Gao, Y.; Shi, H.; Jia, B. Hydrogen-Rich Water Ameliorates Autistic-Like Behavioral Abnormalities in Valproic Acid-Treated Adolescent Mice Offspring. Front. Behav. Neurosci. 2018, 12, 170. [Google Scholar] [CrossRef]
- Fereshetyan, K.; Chavushyan, V.; Danielyan, M.; Yenkoyan, K. Assessment of behavioral, morphological and electrophysiological changes in prenatal and postnatal valproate induced rat models of autism spectrum disorder. Sci. Rep. 2021, 11, 23471. [Google Scholar] [CrossRef]
- Ornoy, A.; Echefu, B.; Becker, M. Valproic Acid in Pregnancy Revisited: Neurobehavioral, Biochemical and Molecular Changes Affecting the Embryo and Fetus in Humans and in Animals: A Narrative Review. Int. J. Mol. Sci. 2023, 25, 390. [Google Scholar] [CrossRef]
- Brimberg, L.; Mader, S.; Jeganathan, V.; Berlin, R.; Coleman, T.R.; Gregersen, P.K.; Huerta, P.T.; Volpe, B.T.; Diamond, B. Caspr2-reactive antibody cloned from a mother of an ASD child mediates an ASD-like phenotype in mice. Mol. Psychiatry 2016, 21, 1663–1671. [Google Scholar] [CrossRef]
- Jeon, S.J.; Gonzales, E.L.; Mabunga, D.F.N.; Valencia, S.T.; Kim, D.G.; Kim, Y.; Adil, K.J.L.; Shin, D.; Park, D.; Shin, C.Y. Sex-specific Behavioral Features of Rodent Models of Autism Spectrum Disorder. Exp. Neurobiol. 2018, 27, 321–343. [Google Scholar] [CrossRef]
- Patel, S.; Dale, R.C.; Rose, D.; Heath, B.; Nordahl, C.W.; Rogers, S.; Guastella, A.J.; Ashwood, P. Maternal immune conditions are increased in males with autism spectrum disorders and are associated with behavioural and emotional but not cognitive co-morbidity. Transl. Psychiatry 2020, 10, 286. [Google Scholar] [CrossRef]
- Bruce, M.R.; Couch, A.C.M.; Grant, S.; McLellan, J.; Ku, K.; Chang, C.; Bachman, A.; Matson, M.; Berman, R.F.; Maddock, R.J.; et al. Altered behavior, brain structure, and neurometabolites in a rat model of autism-specific maternal autoantibody exposure. Mol. Psychiatry 2023, 28, 2136–2147. [Google Scholar] [CrossRef] [PubMed]
- Careaga, M.; Murai, T.; Bauman, M.D. Maternal Immune Activation and Autism Spectrum Disorder: From Rodents to Nonhuman and Human Primates. Biol. Psychiatry 2017, 81, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Bauman, M.D.; Iosif, A.M.; Ashwood, P.; Braunschweig, D.; Lee, A.; Schumann, C.M.; Van de Water, J.; Amaral, D.G. Maternal antibodies from mothers of children with autism alter brain growth and social behavior development in the rhesus monkey. Transl. Psychiatry 2013, 3, e278. [Google Scholar] [CrossRef] [PubMed]
- Haida, O.; Al Sagheer, T.; Balbous, A.; Francheteau, M.; Matas, E.; Soria, F.; Fernagut, P.O.; Jaber, M. Sex-dependent behavioral deficits and neuropathology in a maternal immune activation model of autism. Transl. Psychiatry 2019, 9, 124. [Google Scholar] [CrossRef] [PubMed]
- Tartaglione, A.M.; Villani, A.; Ajmone-Cat, M.A.; Minghetti, L.; Ricceri, L.; Pazienza, V.; De Simone, R.; Calamandrei, G. Maternal immune activation induces autism-like changes in behavior, neuroinflammatory profile and gut microbiota in mouse offspring of both sexes. Transl. Psychiatry 2022, 12, 384. [Google Scholar] [CrossRef]
- Zhang, H.L.; Hu, S.; Qu, S.T.; Lv, M.D.; Wang, J.J.; Liu, X.T.; Yao, J.H.; Ding, Y.Y.; Xu, G.Y. Inhibition of NKCC1 Ameliorates Anxiety and Autistic Behaviors Induced by Maternal Immune Activation in Mice. Curr. Issues Mol. Biol. 2024, 46, 1851–1864. [Google Scholar] [CrossRef]
- Carlezon, W.A., Jr.; Kim, W.; Missig, G.; Finger, B.C.; Landino, S.M.; Alexander, A.J.; Mokler, E.L.; Robbins, J.O.; Li, Y.; Bolshakov, V.Y.; et al. Maternal and early postnatal immune activation produce sex-specific effects on autism-like behaviors and neuroimmune function in mice. Sci. Rep. 2019, 9, 16928. [Google Scholar] [CrossRef]
- Dutra, M.L.; Dias, P.; Freiberger, V.; Ventura, L.; Comim, C.M.; Martins, D.F.; Bobinski, F. Maternal immune activation induces autism-like behavior and reduces brain-derived neurotrophic factor levels in the hippocampus and offspring cortex of C57BL/6 mice. Neurosci. Lett. 2023, 793, 136974. [Google Scholar] [CrossRef]
- Wu, J.; Lin, X.; Wu, D.; Yan, B.; Bao, M.; Zheng, P.; Wang, J.; Yang, C.; Li, Z.; Jin, X.; et al. Poly(I:C)-exposed zebrafish shows autism-like behaviors which are ameliorated by fabp2 gene knockout. Front. Mol. Neurosci. 2022, 15, 1068019. [Google Scholar] [CrossRef]
- Zeng, X.; Fan, L.; Li, M.; Qin, Q.; Pang, X.; Shi, S.; Zheng, D.; Jiang, Y.; Wang, H.; Wu, L.; et al. Resveratrol regulates Thoc5 to improve maternal immune activation-induced autism-like behaviors in adult mouse offspring. J. Nutr. Biochem. 2024, 129, 109638. [Google Scholar] [CrossRef]
- Manjeese, W.; Mvubu, N.E.; Steyn, A.J.C.; Mpofana, T. Mycobacterium tuberculosis-Induced Maternal Immune Activation Promotes Autism-Like Phenotype in Infected Mice Offspring. Int. J. Environ. Res. Public Health 2021, 18, 4513. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Qi, F.; Song, D.; He, Z.; Zuo, Z.; Yang, Y.; Liu, Q.; Hu, S.; Wang, X.; Zheng, X.; et al. Prenatal influenza vaccination rescues impairments of social behavior and lamination in a mouse model of autism. J. Neuroinflammation 2018, 15, 228. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, V.L.; Michita, R.T.; Ellwanger, J.H.; Veit, T.D.; Schuch, J.B.; Riesgo, R.D.S.; Roman, T.; Chies, J.A.B. Exploring potential impacts of pregnancy-related maternal immune activation and extracellular vesicles on immune alterations observed in autism spectrum disorder. Heliyon 2023, 9, e15593. [Google Scholar] [CrossRef] [PubMed]
- Ornoy, A.; Weinstein-Fudim, L.; Becker, M. SAMe, Choline, and Valproic Acid as Possible Epigenetic Drugs: Their Effects in Pregnancy with a Special Emphasis on Animal Studies. Pharmaceuticals 2022, 15, 192. [Google Scholar] [CrossRef] [PubMed]
- Ornoy, A.; Becker, M.; Weinstein-Fudim, L.; Ergaz, Z. S-Adenosine Methionine (SAMe) and Valproic Acid (VPA) as Epigenetic Modulators: Special Emphasis on their Interactions Affecting Nervous Tissue during Pregnancy. Int. J. Mol. Sci. 2020, 21, 3721. [Google Scholar] [CrossRef]
- Ornoy, A.; Weinstein-Fudim, L.; Becker, M.; Gorobets, D.; Szyf, M. Gender specific neurobehavioral and gene expression changes in a valproic acid (VPA)–induced mouse model of autistic like behavior and correction by S-adenosylmethionine (SAMe). In Sex, Gender, and Epigenetics: From Molecule to Bedside; Legato, J.M., Feldberg, D., Glezerman, M., Eds.; Academic Press: Cambridge, MA, USA; Elsevier: Amsterdam, The Netherlands, 2023; pp. 180–197. ISBN 9780128239384. [Google Scholar]
- Chen, J.; Lei, L.; Tian, L.; Hou, F.; Roper, C.; Ge, X.; Zhao, Y.; Chen, Y.; Dong, Q.; Tanguay, R.L.; et al. Developmental and behavioral alterations in zebrafish embryonically exposed to valproic acid (VPA): An aquatic model for autism. Neurotoxicology Teratol. 2018, 66, 8–16. [Google Scholar] [CrossRef]
- Jolous-Jamshidi, B.; Cromwell, H.C.; McFarland, A.M.; Meserve, L.A. Perinatal exposure to polychlorinated biphenyls alters social behaviors in rats. Toxicol. Lett. 2010, 199, 136–143. [Google Scholar] [CrossRef]
- Watanabe, S.; Kurotani, T.; Oga, T.; Noguchi, J.; Isoda, R.; Nakagami, A.; Sakai, K.; Nakagaki, K.; Sumida, K.; Hoshino, K.; et al. Functional and molecular characterization of a non-human primate model of autism spectrum disorder shows similarity with the human disease. Nat. Commun. 2021, 12, 5388. [Google Scholar] [CrossRef]
- Lan, A.; Kalimian, M.; Amram, B.; Kofman, O. Prenatal chlorpyrifos leads to autism-like deficits in C57Bl6/J mice. Environ. Health 2017, 16, 43. [Google Scholar] [CrossRef]
- Messina, A.; Sovrano, V.A.; Baratti, G.; Musa, A.; Gobbo, A.; Adiletta, A.; Sgadò, P. Valproic acid exposure affects social visual lateralization and asymmetric gene expression in zebrafish larvae. Sci. Rep. 2024, 14, 4474. [Google Scholar] [CrossRef]
- Koide, T.; Goto, T.; Takano-Shimizu, T. Genomic mixing to elucidate the genetic system of complex traits. Exp. Anim. 2012, 61, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Yalcin, B.; Nicod, J.; Bhomra, A.; Davidson, S.; Cleak, J.; Farinelli, L.; Østerås, M.; Whitley, A.; Yuan, W.; Gan, X.; et al. Commercially available outbred mice for genome-wide association studies. PLoS Genet. 2010, 6, e1001085. [Google Scholar] [CrossRef] [PubMed]
- Weinstein-Fudim, L.; Ergaz, Z.; Turgeman, G.; Yanai, J.; Szyf, M.; Ornoy, A. Gender Related Changes in Gene Expression Induced by Valproic Acid in A Mouse Model of Autism and the Correction by S-adenosyl Methionine. Does It Explain the Gender Differences in Autistic Like Behavior? Int. J. Mol. Sci. 2019, 20, 5278. [Google Scholar] [CrossRef] [PubMed]
- Panesar, H.K.; Kennedy, C.L.; Keil Stietz, K.P.; Lein, P.J. Polychlorinated Biphenyls (PCBs): Risk Factors for Autism Spectrum Disorder? Toxics 2020, 8, 70. [Google Scholar] [CrossRef] [PubMed]
- Bell, M.R.; Thompson, L.M.; Rodriguez, K.; Gore, A.C. Two-hit exposure to polychlorinated biphenyls at gestational and juvenile life stages: 1. Sexually dimorphic effects on social and anxiety-like behaviors. Horm. Behav. 2016, 78, 168–177. [Google Scholar] [CrossRef]
- Berg, E.L.; Ching, T.M.; Bruun, D.A.; Rivera, J.K.; Careaga, M.; Ellegood, J.; Lerch, J.P.; Wöhr, M.; Lein, P.J.; Silverman, J.L. Translational outcomes relevant to neurodevelopmental disorders following early life exposure of rats to chlorpyrifos. J. Neurodev. Disord. 2020, 12, 40. [Google Scholar] [CrossRef]
- De Felice, A.; Scattoni, M.L.; Ricceri, L.; Calamandrei, G. Prenatal exposure to a common organophosphate insecticide delays motor development in a mouse model of idiopathic autism. PLoS ONE 2015, 10, e0121663. [Google Scholar] [CrossRef]
- Venerosi, A.; Ricceri, L.; Scattoni, M.L.; Calamandrei, G. Prenatal chlorpyrifos exposure alters motor behavior and ultrasonic vocalization in CD-1 mouse pups. Environ. Health 2009, 8, 12. [Google Scholar] [CrossRef]
- Morales-Navas, M.; Castaño-Castaño, S.; Pérez-Fernández, C.; Sánchez-Gil, A.; Teresa Colomina, M.; Leinekugel, X.; Sánchez-Santed, F. Similarities between the Effects of Prenatal Chlorpyrifos and Valproic Acid on Ultrasonic Vocalization in Infant Wistar Rats. Int. J. Environ. Res. Public Health 2020, 17, 6376. [Google Scholar] [CrossRef]
- Werling, D.M.; Geschwind, D.H. Sex differences in autism spectrum disorders. Curr. Opin. Neurol. 2013, 26, 146–153. [Google Scholar] [CrossRef]
- Bailey, A.; Le Couteur, A.; Gottesman, I.; Bolton, P.; Simonoff, E.; Yuzda, E.; Rutter, M. Autism as a strongly genetic disorder: Evidence from a British twin study. Psychol. Med. 1995, 25, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Zuko, A.; Kleijer, K.T.E.; Oguro-Ando, A.; Kas, M.J.H.; van Daalen, E.; van der Zwaag, B.; Burbach, J.P.H. Contactins in the neurobiology of autism. Eur. J. Pharmacol. 2013, 719, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, B.S.; Geschwind, D.H. Advances in autism genetics: On the threshold of a new neurobiology. Nat. Rev. Genet. 2008, 9, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Bourgeron, T. From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat. Rev. Neurosci. 2015, 16, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Weiner, D.J.; Wigdor, E.M.; Ripke, S.; Walters, R.K.; Kosmicki, J.A.; Grove, J.; Samocha, K.E.; Goldstein, J.I.; Okbay, A.; Bybjerg-Grauholm, J.; et al. Polygenic transmission disequilibrium confirms that common and rare variation act additively to create risk for autism spectrum disorders. Nat. Genet. 2017, 49, 978–985. [Google Scholar] [CrossRef]
- Zhao, X.; Leotta, A.; Kustanovich, V.; Lajonchere, C.; Geschwind, D.H.; Law, K.; Law, P.; Qiu, S.; Lord, C.; Sebat, J.; et al. A unified genetic theory for sporadic and inherited autism. Proc. Natl. Acad. Sci. USA 2007, 104, 12831–12836. [Google Scholar] [CrossRef]
- Parikshak, N.N.; Luo, R.; Zhang, A.; Won, H.; Lowe, J.K.; Chandran, V.; Horvath, S.; Geschwind, D.H. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 2013, 155, 1008–1021. [Google Scholar] [CrossRef]
- Basu, S.N.; Kollu, R.; Banerjee-Basu, S. AutDB: A gene reference resource for autism research. Nucleic Acids Res. 2009, 37, D832–D836. [Google Scholar] [CrossRef] [PubMed]
- Möhrle, D.; Fernández, M.; Peñagarikano, O.; Frick, A.; Allman, B.; Schmid, S. What we can learn from a genetic rodent model about autism. Neurosci. Biobehav. Rev. 2020, 109, 29–53. [Google Scholar] [CrossRef]
- Pasciuto, E.; Borrie, S.C.; Kanellopoulos, A.K.; Santos, A.R.; Cappuyns, E.; D’Andrea, L.; Pacini, L.; Bagni, C. Autism Spectrum Disorders: Translating human deficits into mouse behavior. Neurobiol. Learn. Mem. 2015, 124, 71–87. [Google Scholar] [CrossRef]
- Provenzano, G.; Chelini, G.; Bozzi, Y. Genetic control of social behavior: Lessons from mutant mice. Behav. Brain Res. 2017, 325, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Wadhawan, R.; Swanwick, C.C.; Kollu, R.; Basu, S.N.; Banerjee-Basu, S. Animal model integration to AutDB, a genetic database for autism. BMC Med. Genom. 2011, 4, 15. [Google Scholar] [CrossRef] [PubMed]
- Vorstman, J.; Staal, W.; Hochstenbach, P.; Franke, L.; Van Daalen, E.; Van Engeland, H. Overview of cytogenetic regions of interest (CROIs) associated with the autism phenotype across the human genome. Mol. Psychiatry 2006, 11, 1. [Google Scholar] [CrossRef] [PubMed]
- Flores Gutiérrez, J.; De Felice, C.; Natali, G.; Leoncini, S.; Signorini, C.; Hayek, J.; Tongiorgi, E. Protective role of mirtazapine in adult female Mecp2(+/-) mice and patients with Rett syndrome. J. Neurodev. Disord. 2020, 12, 26. [Google Scholar] [CrossRef] [PubMed]
- Vermudez, S.A.D.; Gogliotti, R.G.; Arthur, B.; Buch, A.; Morales, C.; Moxley, Y.; Rajpal, H.; Conn, P.J.; Niswender, C.M. Profiling beneficial and potential adverse effects of MeCP2 overexpression in a hypomorphic Rett syndrome mouse model. Genes Brain Behav. 2022, 21, e12752. [Google Scholar] [CrossRef]
- Guy, J.; Hendrich, B.; Holmes, M.; Martin, J.E.; Bird, A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 2001, 27, 322–326. [Google Scholar] [CrossRef]
- Bittolo, T.; Raminelli, C.A.; Deiana, C.; Baj, G.; Vaghi, V.; Ferrazzo, S.; Bernareggi, A.; Tongiorgi, E. Pharmacological treatment with mirtazapine rescues cortical atrophy and respiratory deficits in MeCP2 null mice. Sci. Rep. 2016, 6, 19796. [Google Scholar] [CrossRef]
- Chen, R.Z.; Akbarian, S.; Tudor, M.; Jaenisch, R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat. Genet. 2001, 27, 327–331. [Google Scholar] [CrossRef]
- Shahbazian, M.D.; Young, J.I.; Yuva-Paylor, L.A.; Spencer, C.M.; Antalffy, B.A.; Noebels, J.L.; Armstrong, D.L.; Paylor, R.; Zoghbi, H.Y. Mice with Truncated MeCP2 Recapitulate Many Rett Syndrome Features and Display Hyperacetylation of Histone H3. Neuron 2002, 35, 243–254. [Google Scholar] [CrossRef]
- Wu, Y.; Zhong, W.; Cui, N.; Johnson, C.M.; Xing, H.; Zhang, S.; Jiang, C. Characterization of Rett Syndrome-like phenotypes in Mecp2-knockout rats. J. Neurodev. Disord. 2016, 8, 23. [Google Scholar] [CrossRef]
- Chao, H.-T.; Chen, H.; Samaco, R.C.; Xue, M.; Chahrour, M.; Yoo, J.; Neul, J.L.; Gong, S.; Lu, H.-C.; Heintz, N. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature 2010, 468, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Ure, K.; Lu, H.; Wang, W.; Ito-Ishida, A.; Wu, Z.; He, L.J.; Sztainberg, Y.; Chen, W.; Tang, J.; Zoghbi, H.Y. Restoration of Mecp2 expression in GABAergic neurons is sufficient to rescue multiple disease features in a mouse model of Rett syndrome. eLife 2016, 5, e14198. [Google Scholar] [CrossRef] [PubMed]
- Baker, K.B.; Wray, S.P.; Ritter, R.; Mason, S.; Lanthorn, T.H.; Savelieva, K.V. Male and female Fmr1 knockout mice on C57 albino background exhibit spatial learning and memory impairments. Genes Brain Behav. 2010, 9, 562–574. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Sethna, F.; Wang, H. Behavioral analysis of male and female Fmr1 knockout mice on C57BL/6 background. Behav. Brain Res. 2014, 271, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Albert, P.R.; Vahid-Ansari, F.; Luckhart, C. Serotonin-prefrontal cortical circuitry in anxiety and depression phenotypes: Pivotal role of pre- and post-synaptic 5-HT1A receptor expression. Front. Behav. Neurosci. 2014, 8, 199. [Google Scholar] [CrossRef]
- Hamilton, S.M.; Green, J.R.; Veeraragavan, S.; Yuva, L.; McCoy, A.; Wu, Y.; Warren, J.; Little, L.; Ji, D.; Cui, X.; et al. Fmr1 and Nlgn3 knockout rats: Novel tools for investigating autism spectrum disorders. Behav. Neurosci. 2014, 128, 103–109. [Google Scholar] [CrossRef]
- Barić, I.; Staufner, C.; Augoustides-Savvopoulou, P.; Chien, Y.-H.; Dobbelaere, D.; Grünert, S.C.; Opladen, T.; Petković Ramadža, D.; Rakić, B.; Wedell, A.; et al. Consensus recommendations for the diagnosis, treatment and follow-up of inherited methylation disorders. J. Inherit. Metab. Dis. 2017, 40, 5–20. [Google Scholar] [CrossRef]
- Curnow, E.; Wang, Y. New Animal Models for Understanding FMRP Functions and FXS Pathology. Cells 2022, 11, 1628. [Google Scholar] [CrossRef]
- Bozdagi, O.; Sakurai, T.; Papapetrou, D.; Wang, X.; Dickstein, D.L.; Takahashi, N.; Kajiwara, Y.; Yang, M.; Katz, A.M.; Scattoni, M.L.; et al. Haploinsufficiency of the autism-associated Shank3 gene leads to deficits in synaptic function, social interaction, and social communication. Mol. Autism 2010, 1, 15. [Google Scholar] [CrossRef]
- Yang, M.; Bozdagi, O.; Scattoni, M.L.; Wöhr, M.; Roullet, F.I.; Katz, A.M.; Abrams, D.N.; Kalikhman, D.; Simon, H.; Woldeyohannes, L.; et al. Reduced excitatory neurotransmission and mild autism-relevant phenotypes in adolescent Shank3 null mutant mice. J. Neurosci. 2012, 32, 6525–6541. [Google Scholar] [CrossRef]
- Wang, X.; McCoy, P.A.; Rodriguiz, R.M.; Pan, Y.; Je, H.S.; Roberts, A.C.; Kim, C.J.; Berrios, J.; Colvin, J.S.; Bousquet-Moore, D.; et al. Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3. Hum. Mol. Genet. 2011, 20, 3093–3108. [Google Scholar] [CrossRef] [PubMed]
- Peça, J.; Feliciano, C.; Ting, J.T.; Wang, W.; Wells, M.F.; Venkatraman, T.N.; Lascola, C.D.; Fu, Z.; Feng, G. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 2011, 472, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Ma, K.; Wang, Z.J.; Hu, Z.; Matas, E.; Wei, J.; Yan, Z. Social deficits in Shank3-deficient mouse models of autism are rescued by histone deacetylase (HDAC) inhibition. Nat. Neurosci. 2018, 21, 564–575. [Google Scholar] [CrossRef] [PubMed]
- Duffney, L.J.; Zhong, P.; Wei, J.; Matas, E.; Cheng, J.; Qin, L.; Ma, K.; Dietz, D.M.; Kajiwara, Y.; Buxbaum, J.D.; et al. Autism-like Deficits in Shank3-Deficient Mice Are Rescued by Targeting Actin Regulators. Cell Rep. 2015, 11, 1400–1413. [Google Scholar] [CrossRef]
- Wang, Z.J.; Zhong, P.; Ma, K.; Seo, J.S.; Yang, F.; Hu, Z.; Zhang, F.; Lin, L.; Wang, J.; Liu, T.; et al. Amelioration of autism-like social deficits by targeting histone methyltransferases EHMT1/2 in Shank3-deficient mice. Mol. Psychiatry 2020, 25, 2517–2533. [Google Scholar] [CrossRef]
- Nakanishi, M.; Nomura, J.; Ji, X.; Tamada, K.; Arai, T.; Takahashi, E.; Bućan, M.; Takumi, T. Functional significance of rare neuroligin 1 variants found in autism. PLoS Genet. 2017, 13, e1006940. [Google Scholar] [CrossRef]
- Blundell, J.; Blaiss, C.A.; Etherton, M.R.; Espinosa, F.; Tabuchi, K.; Walz, C.; Bolliger, M.F.; Südhof, T.C.; Powell, C.M. Neuroligin-1 deletion results in impaired spatial memory and increased repetitive behavior. J. Neurosci. 2010, 30, 2115–2129. [Google Scholar] [CrossRef]
- Chen, C.H.; Lee, P.W.; Liao, H.M.; Chang, P.K. Neuroligin 2 R215H Mutant Mice Manifest Anxiety, Increased Prepulse Inhibition, and Impaired Spatial Learning and Memory. Front. Psychiatry 2017, 8, 257. [Google Scholar] [CrossRef]
- Radyushkin, K.; Hammerschmidt, K.; Boretius, S.; Varoqueaux, F.; El-Kordi, A.; Ronnenberg, A.; Winter, D.; Frahm, J.; Fischer, J.; Brose, N.; et al. Neuroligin-3-deficient mice: Model of a monogenic heritable form of autism with an olfactory deficit. Genes Brain Behav. 2009, 8, 416–425. [Google Scholar] [CrossRef]
- Chadman, K.K.; Gong, S.; Scattoni, M.L.; Boltuck, S.E.; Gandhy, S.U.; Heintz, N.; Crawley, J.N. Minimal aberrant behavioral phenotypes of neuroligin-3 R451C knockin mice. Autism Res. 2008, 1, 147–158. [Google Scholar] [CrossRef]
- Tabuchi, K.; Blundell, J.; Etherton, M.R.; Hammer, R.E.; Liu, X.; Powell, C.M.; Südhof, T.C. A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science 2007, 318, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Lai, E.S.K.; Nakayama, H.; Miyazaki, T.; Nakazawa, T.; Tabuchi, K.; Hashimoto, K.; Watanabe, M.; Kano, M. An Autism-Associated Neuroligin-3 Mutation Affects Developmental Synapse Elimination in the Cerebellum. Front. Neural Circuits 2021, 15, 676891. [Google Scholar] [CrossRef] [PubMed]
- Jamain, S.; Radyushkin, K.; Hammerschmidt, K.; Granon, S.; Boretius, S.; Varoqueaux, F.; Ramanantsoa, N.; Gallego, J.; Ronnenberg, A.; Winter, D.; et al. Reduced social interaction and ultrasonic communication in a mouse model of monogenic heritable autism. Proc. Natl. Acad. Sci. USA 2008, 105, 1710–1715. [Google Scholar] [CrossRef] [PubMed]
- Scattoni, M.L.; Martire, A.; Cartocci, G.; Ferrante, A.; Ricceri, L. Reduced social interaction, behavioural flexibility and BDNF signalling in the BTBR T+ tf/J strain, a mouse model of autism. Behav. Brain Res. 2013, 251, 35–40. [Google Scholar] [CrossRef] [PubMed]
- McFarlane, H.G.; Kusek, G.K.; Yang, M.; Phoenix, J.L.; Bolivar, V.J.; Crawley, J.N. Autism-like behavioral phenotypes in BTBR T+tf/J mice. Genes Brain Behav. 2008, 7, 152–163. [Google Scholar] [CrossRef]
- Scattoni, M.L.; Gandhy, S.U.; Ricceri, L.; Crawley, J.N. Unusual Repertoire of Vocalizations in the BTBR T+tf/J Mouse Model of Autism. PLoS ONE 2008, 3, e3067. [Google Scholar] [CrossRef]
- Wöhr, M.; Silverman, J.L.; Scattoni, M.L.; Turner, S.M.; Harris, M.J.; Saxena, R.; Crawley, J.N. Developmental delays and reduced pup ultrasonic vocalizations but normal sociability in mice lacking the postsynaptic cell adhesion protein neuroligin2. Behav. Brain Res. 2013, 251, 50–64. [Google Scholar] [CrossRef]
- Meyza, K.Z.; Defensor, E.B.; Jensen, A.L.; Corley, M.J.; Pearson, B.L.; Pobbe, R.L.; Bolivar, V.J.; Blanchard, D.C.; Blanchard, R.J. The BTBR T+ tf/J mouse model for autism spectrum disorders-in search of biomarkers. Behav. Brain Res. 2013, 251, 25–34. [Google Scholar] [CrossRef]
- Dodero, L.; Damiano, M.; Galbusera, A.; Bifone, A.; Tsaftsaris, S.A.; Scattoni, M.L.; Gozzi, A. Neuroimaging evidence of major morpho-anatomical and functional abnormalities in the BTBR T+TF/J mouse model of autism. PLoS ONE 2013, 8, e76655. [Google Scholar] [CrossRef]
- Liu, Z.; Li, X.; Zhang, J.T.; Cai, Y.J.; Cheng, T.L.; Cheng, C.; Wang, Y.; Zhang, C.C.; Nie, Y.H.; Chen, Z.F.; et al. Autism-like behaviours and germline transmission in transgenic monkeys overexpressing MeCP2. Nature 2016, 530, 98–102. [Google Scholar] [CrossRef]
- Cai, D.-C.; Wang, Z.; Bo, T.; Yan, S.; Liu, Y.; Liu, Z.; Zeljic, K.; Chen, X.; Zhan, Y.; Xu, X.; et al. MECP2 Duplication Causes Aberrant GABA Pathways, Circuits and Behaviors in Transgenic Monkeys: Neural Mappings to Patients with Autism. J. Neurosci. 2020, 40, 3799. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Chen, Y.; Niu, Y.; Zhang, K.; Kang, Y.; Ge, W.; Liu, X.; Zhao, E.; Wang, C.; Lin, S.; et al. TALEN-mediated gene mutagenesis in rhesus and cynomolgus monkeys. Cell Stem Cell 2014, 14, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yu, J.; Niu, Y.; Qin, D.; Liu, H.; Li, G.; Hu, Y.; Wang, J.; Lu, Y.; Kang, Y.; et al. Modeling Rett Syndrome Using TALEN-Edited MECP2 Mutant Cynomolgus Monkeys. Cell 2017, 169, 945–955.910. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Sharma, J.; Ke, Q.; Landman, R.; Yuan, J.; Chen, H.; Hayden, D.S.; Fisher, J.W.; Jiang, M.; Menegas, W.; et al. Atypical behaviour and connectivity in SHANK3-mutant macaques. Nature 2019, 570, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, B. Clinical manifestations and stages of rett syndrome. Ment. Retard. Dev. Disabil. Res. Rev. 2002, 8, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Amir, R.E.; Van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef]
- Cuddapah, V.A.; Pillai, R.B.; Shekar, K.V.; Lane, J.B.; Motil, K.J.; Skinner, S.A.; Tarquinio, D.C.; Glaze, D.G.; McGwin, G.; Kaufmann, W.E.; et al. Methyl-CpG-binding protein 2 (MECP2) mutation type is associated with disease severity in Rett syndrome. J. Med. Genet. 2014, 51, 152–158. [Google Scholar] [CrossRef]
- Van den Veyver, I.B.; Zoghbi, H.Y. Mutations in the gene encoding methyl-CpG-binding protein 2 cause Rett syndrome. Brain Dev. 2001, 23, S147–S151. [Google Scholar] [CrossRef]
- Percy, A.K. Rett syndrome: Exploring the autism link. Arch. Neurol. 2011, 68, 985–989. [Google Scholar] [CrossRef]
- Boxer, L.D.; Renthal, W.; Greben, A.W.; Whitwam, T.; Silberfeld, A.; Stroud, H.; Li, E.; Yang, M.G.; Kinde, B.; Griffith, E.C.; et al. MeCP2 Represses the Rate of Transcriptional Initiation of Highly Methylated Long Genes. Mol. Cell 2020, 77, 294–309.e299. [Google Scholar] [CrossRef]
- Young, J.I.; Hong, E.P.; Castle, J.C.; Crespo-Barreto, J.; Bowman, A.B.; Rose, M.F.; Kang, D.; Richman, R.; Johnson, J.M.; Berget, S.; et al. Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proc. Natl. Acad. Sci. USA 2005, 102, 17551–17558. [Google Scholar] [CrossRef] [PubMed]
- Skene, P.J.; Illingworth, R.S.; Webb, S.; Kerr, A.R.; James, K.D.; Turner, D.J.; Andrews, R.; Bird, A.P. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol. Cell 2010, 37, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Shiota, K. Methyl-CpG-binding protein, MeCP2, is a target molecule for maintenance DNA methyltransferase, Dnmt1. J. Biol. Chem. 2003, 278, 4806–4812. [Google Scholar] [CrossRef] [PubMed]
- Olson, C.O.; Zachariah, R.M.; Ezeonwuka, C.D.; Liyanage, V.R.; Rastegar, M. Brain region-specific expression of MeCP2 isoforms correlates with DNA methylation within Mecp2 regulatory elements. PLoS ONE 2014, 9, e90645. [Google Scholar] [CrossRef] [PubMed]
- Moretti, P.; Bouwknecht, J.A.; Teague, R.; Paylor, R.; Zoghbi, H.Y. Abnormalities of social interactions and home-cage behavior in a mouse model of Rett syndrome. Hum. Mol. Genet. 2005, 14, 205–220. [Google Scholar] [CrossRef]
- Katz, D.M.; Berger-Sweeney, J.E.; Eubanks, J.H.; Justice, M.J.; Neul, J.L.; Pozzo-Miller, L.; Blue, M.E.; Christian, D.; Crawley, J.N.; Giustetto, M.; et al. Preclinical research in Rett syndrome: Setting the foundation for translational success. Dis. Models Mech. 2012, 5, 733–745. [Google Scholar] [CrossRef]
- Samaco, R.C.; McGraw, C.M.; Ward, C.S.; Sun, Y.; Neul, J.L.; Zoghbi, H.Y. Female Mecp2+/− mice display robust behavioral deficits on two different genetic backgrounds providing a framework for pre-clinical studies. Hum. Mol. Genet. 2012, 22, 96–109. [Google Scholar] [CrossRef]
- Díaz de León-Guerrero, S.; Pedraza-Alva, G.; Pérez-Martínez, L. In sickness and in health: The role of methyl-CpG binding protein 2 in the central nervous system. Eur. J. Neurosci. 2011, 33, 1563–1574. [Google Scholar] [CrossRef]
- Peyser, A.; Singer, T.; Mullin, C.; Hershlag, A. Reduction in the number of CGG repeats on the FMR1 gene in carriers of genetic disorders versus noncarriers. JBRA Assist. Reprod. 2017, 21, 327–329. [Google Scholar] [CrossRef]
- Dahlhaus, R. Of Men and Mice: Modeling the Fragile X Syndrome. Front. Mol. Neurosci. 2018, 11, 41. [Google Scholar] [CrossRef]
- Maurin, T.; Zongaro, S.; Bardoni, B. Fragile X Syndrome: From molecular pathology to therapy. Neurosci. Biobehav. Rev. 2014, 46 Pt 2, 242–255. [Google Scholar] [CrossRef]
- Christie, S.B.; Akins, M.R.; Schwob, J.E.; Fallon, J.R. The FXG: A presynaptic fragile X granule expressed in a subset of developing brain circuits. J. Neurosci. 2009, 29, 1514–1524. [Google Scholar] [CrossRef] [PubMed]
- Stefani, G.; Fraser, C.E.; Darnell, J.C.; Darnell, R.B. Fragile X mental retardation protein is associated with translating polyribosomes in neuronal cells. J. Neurosci. 2004, 24, 7272–7276. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.C.; Van Driesche, S.J.; Zhang, C.; Hung, K.Y.; Mele, A.; Fraser, C.E.; Stone, E.F.; Chen, C.; Fak, J.J.; Chi, S.W.; et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 2011, 146, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Comery, T.A.; Harris, J.B.; Willems, P.J.; Oostra, B.A.; Irwin, S.A.; Weiler, I.J.; Greenough, W.T. Abnormal dendritic spines in fragile X knockout mice: Maturation and pruning deficits. Proc. Natl. Acad. Sci. USA 1997, 94, 5401–5404. [Google Scholar] [CrossRef]
- He, C.X.; Portera-Cailliau, C. The trouble with spines in fragile X syndrome: Density, maturity and plasticity. Neuroscience 2013, 251, 120–128. [Google Scholar] [CrossRef]
- Gauducheau, M.; Lemaire-Mayo, V.; D’Amato, F.R.; Oddi, D.; Crusio, W.E.; Pietropaolo, S. Age-specific autistic-like behaviors in heterozygous Fmr1-KO female mice. Autism Res. 2017, 10, 1067–1078. [Google Scholar] [CrossRef]
- Portales-Casamar, E.; Lussier, A.A.; Jones, M.J.; MacIsaac, J.L.; Edgar, R.D.; Mah, S.M.; Barhdadi, A.; Provost, S.; Lemieux-Perreault, L.P.; Cynader, M.S.; et al. DNA methylation signature of human fetal alcohol spectrum disorder. Epigenetics Chromatin 2016, 9, 25. [Google Scholar] [CrossRef]
- Boeckers, T.M.; Bockmann, J.; Kreutz, M.R.; Gundelfinger, E.D. ProSAP/Shank proteins—A family of higher order organizing molecules of the postsynaptic density with an emerging role in human neurological disease. J. Neurochem. 2002, 81, 903–910. [Google Scholar] [CrossRef]
- Meyer, G.; Varoqueaux, F.; Neeb, A.; Oschlies, M.; Brose, N. The complexity of PDZ domain-mediated interactions at glutamatergic synapses: A case study on neuroligin. Neuropharmacology 2004, 47, 724–733. [Google Scholar] [CrossRef]
- Sato, D.; Lionel, A.C.; Leblond, C.S.; Prasad, A.; Pinto, D.; Walker, S.; O’Connor, I.; Russell, C.; Drmic, I.E.; Hamdan, F.F.; et al. SHANK1 Deletions in Males with Autism Spectrum Disorder. Am. J. Hum. Genet. 2012, 90, 879–887. [Google Scholar] [CrossRef]
- Berkel, S.; Tang, W.; Treviño, M.; Vogt, M.; Obenhaus, H.A.; Gass, P.; Scherer, S.W.; Sprengel, R.; Schratt, G.; Rappold, G.A. Inherited and de novo SHANK2 variants associated with autism spectrum disorder impair neuronal morphogenesis and physiology. Hum. Mol. Genet. 2012, 21, 344–357. [Google Scholar] [CrossRef] [PubMed]
- Bonaglia, M.C.; Giorda, R.; Mani, E.; Aceti, G.; Anderlid, B.-M.; Baroncini, A.; Pramparo, T.; Zuffardi, O. Identification of a recurrent breakpoint within the SHANK3 gene in the 22q13. 3 deletion syndrome. J. Med. Genet. 2006, 43, 822–828. [Google Scholar] [PubMed]
- Uchino, S.; Waga, C. SHANK3 as an autism spectrum disorder-associated gene. Brain Dev. 2013, 35, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Durand, C.M.; Betancur, C.; Boeckers, T.M.; Bockmann, J.; Chaste, P.; Fauchereau, F.; Nygren, G.; Rastam, M.; Gillberg, I.C.; Anckarsäter, H.; et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 2007, 39, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Waga, C.; Okamoto, N.; Ondo, Y.; Fukumura-Kato, R.; Goto, Y.-i.; Kohsaka, S.; Uchino, S. Novel variants of the SHANK3 gene in Japanese autistic patients with severe delayed speech development. Psychiatr. Genet. 2011, 21, 208–211. [Google Scholar] [CrossRef]
- Gauthier, J.; Champagne, N.; Lafrenière, R.G.; Xiong, L.; Spiegelman, D.; Brustein, E.; Lapointe, M.; Peng, H.; Côté, M.; Noreau, A.; et al. De novo mutations in the gene encoding the synaptic scaffolding protein SHANK3 in patients ascertained for schizophrenia. Proc. Natl. Acad. Sci. USA 2010, 107, 7863–7868. [Google Scholar] [CrossRef]
- Phelan, M.C.; Rogers, R.C.; Saul, R.A.; Stapleton, G.A.; Sweet, K.; McDermid, H.; Shaw, S.R.; Claytor, J.; Willis, J.; Kelly, D.P. 22q13 deletion syndrome. Am. J. Med. Genet. 2001, 101, 91–99. [Google Scholar] [CrossRef]
- Kleefstra, T.; Kramer, J.M.; Neveling, K.; Willemsen, M.H.; Koemans, T.S.; Vissers, L.E.; Wissink-Lindhout, W.; Fenckova, M.; van den Akker, W.M.; Kasri, N.N.; et al. Disruption of an EHMT1-associated chromatin-modification module causes intellectual disability. Am. J. Hum. Genet. 2012, 91, 73–82. [Google Scholar] [CrossRef]
- Laumonnier, F.; Bonnet-Brilhault, F.; Gomot, M.; Blanc, R.; David, A.; Moizard, M.P.; Raynaud, M.; Ronce, N.; Lemonnier, E.; Calvas, P.; et al. X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am. J. Hum. Genet. 2004, 74, 552–557. [Google Scholar] [CrossRef]
- Jamain, S.; Quach, H.; Betancur, C.; Råstam, M.; Colineaux, C.; Gillberg, I.C.; Soderstrom, H.; Giros, B.; Leboyer, M.; Gillberg, C.; et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 2003, 34, 27–29. [Google Scholar] [CrossRef] [PubMed]
- Thomas, N.S.; Sharp, A.J.; Browne, C.E.; Skuse, D.; Hardie, C.; Dennis, N.R. Xp deletions associated with autism in three females. Hum. Genet. 1999, 104, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.A.; Lehr, A.W.; Roche, K.W. Neuroligins and Neurodevelopmental Disorders: X-Linked Genetics. Front. Synaptic Neurosci. 2020, 12, 33. [Google Scholar] [CrossRef] [PubMed]
- Varoqueaux, F.; Aramuni, G.; Rawson, R.L.; Mohrmann, R.; Missler, M.; Gottmann, K.; Zhang, W.; Südhof, T.C.; Brose, N. Neuroligins determine synapse maturation and function. Neuron 2006, 51, 741–754. [Google Scholar] [CrossRef] [PubMed]
- Trobiani, L.; Meringolo, M.; Diamanti, T.; Bourne, Y.; Marchot, P.; Martella, G.; Dini, L.; Pisani, A.; De Jaco, A.; Bonsi, P. The neuroligins and the synaptic pathway in Autism Spectrum Disorder. Neurosci. Biobehav. Rev. 2020, 119, 37–51. [Google Scholar] [CrossRef]
- Parente, D.J.; Garriga, C.; Baskin, B.; Douglas, G.; Cho, M.T.; Araujo, G.C.; Shinawi, M. Neuroligin 2 nonsense variant associated with anxiety, autism, intellectual disability, hyperphagia, and obesity. Am. J. Med. Genet. Part A 2017, 173, 213–216. [Google Scholar] [CrossRef]
- Kisaretova, P.; Tsybko, A.; Bondar, N.; Reshetnikov, V. Molecular Abnormalities in BTBR Mice and Their Relevance to Schizophrenia and Autism Spectrum Disorders: An Overview of Transcriptomic and Proteomic Studies. Biomedicines 2023, 11, 289. [Google Scholar] [CrossRef]
- Bolivar, V.J.; Walters, S.R.; Phoenix, J.L. Assessing autism-like behavior in mice: Variations in social interactions among inbred strains. Behav. Brain Res. 2007, 176, 21–26. [Google Scholar] [CrossRef]
- Silverman, J.L.; Tolu, S.S.; Barkan, C.L.; Crawley, J.N. Repetitive self-grooming behavior in the BTBR mouse model of autism is blocked by the mGluR5 antagonist MPEP. Neuropsychopharmacology 2010, 35, 976–989. [Google Scholar] [CrossRef]
- Alexander, A.L.; Lee, J.E.; Lazar, M.; Boudos, R.; DuBray, M.B.; Oakes, T.R.; Miller, J.N.; Lu, J.; Jeong, E.-K.; McMahon, W.M.; et al. Diffusion tensor imaging of the corpus callosum in Autism. NeuroImage 2007, 34, 61–73. [Google Scholar] [CrossRef]
- Egaas, B.; Courchesne, E.; Saitoh, O. Reduced Size of Corpus Callosum in Autism. Arch. Neurol. 1995, 52, 794–801. [Google Scholar] [CrossRef] [PubMed]
- El-Kordi, A.; Winkler, D.; Hammerschmidt, K.; Kästner, A.; Krueger, D.; Ronnenberg, A.; Ritter, C.; Jatho, J.; Radyushkin, K.; Bourgeron, T.; et al. Development of an autism severity score for mice using Nlgn4 null mutants as a construct-valid model of heritable monogenic autism. Behav. Brain Res. 2013, 251, 41–49. [Google Scholar] [CrossRef] [PubMed]
| Animals | Testing Paradigm | Phenotype Manifestations | Authors |
|---|---|---|---|
| Pregnant C57BL/6 were treated with 300 μg of IgG on GD 13.5 | OF, Y-Maze, marble burying, clock maze | Adult offspring show abnormal sociability, impaired motivation, stereotypic and/or compulsive behavior, learning inflexibility | Brimberg et al. [50] |
| C57BL/6 dams were injected with multiple synthetic antigenic epitopes before pregnancy; inducing autoimmune response | Open arena social approach, three-chambers, self-grooming, marble burying, USVs, MWM, OF, EPM, light and dark box | Adult offspring manifest reduced number in USVs, repetitive behaviors, diminished interest in social interaction, neurodevelopmental delays | Jones et al. [44] |
| Sprague Dawley Rat dams received injections of 21 custom synthetic peptides (LDH-A, LDH-B, STIP1, and CRMP1), 4 weeks before pregnancy | Neurodevelopmental test, USVs, EPM, OF, three-chamber, pre-pulse inhibition reciprocal social behavior social novelty test | Adult offspring show impaired social behavior, dampened social reciprocity | Bruce et al. [53] |
| C57BL/6J dams were injected IP with 20 mg/kg poly(I:C) on GD 12.5 | Three-chamber Self-grooming, EPM OF | Adult offspring show declined sociability, social recognition, and anxiety Excessive self-grooming Increased NKCC1 Dendritic spines Reactive microglia in PFC | Zhang et al. [58] |
| C57BL/6J dams were treated with Poly I:C on GD 12.5 Pups were treated with LPS on PND 9 | USVs, scent marking, social recognition, OF, rotarod | ASD-like phenotype more severe in males than in females Altered social behavior Repetitive behaviors Anxiety Altered USVs in both sexes | Carlezon et al. [59] |
| C57bL/6 dams were IP injected with 15 µg/kg LPS on GD 15 | Three-chamber, stereotypic behavior test | Adult offspring show altered social interaction, stereotyped self-grooming, abnormal BDNF and interleukin 17A in the hippocampus and cortex These altered behaviors were absent at age 28 | Dutra et al. [60] |
| Female zebrafish were treated with Poly(I:C) intraperitoneally at 24 h before mating | Three-chamber, shoaling, OF social preference test | Offspring zebrafish show impaired social approach/cohesion, altered villin-1 (vil1) pathway | Wu et al. [61] |
| C57BL/6J dams were IP injected with 20 mg/kg poly (I:C) on GD 12.5 | OF, EPM, grooming test, marble burying Three-chamber test | Adult offspring show reduced locomotion, increased anxiety, higher repetitive digging, higher repetitive stereotyped behavior, impaired social interaction and recognition memory | Zeng et al. [62] |
| Balb/c dams were exposed to Mycobacterium tuberculosis (Mtb) via aerosol infection on GD 12.5 | Three-chamber test, self-grooming | No deficit in social behavior Increased repetitive self-grooming. | Manjeese et al. [63] |
| Pregnant rhesus monkey was treated on GDs 30, 44, 58, 72, 86, 100 with IgG from mother of ASD patient | Reciprocal social interaction, three-chambers, MIR | IgG-ASD offspring were asocial to conspecific; showed impaired reciprocal social interaction; had abnormal frontal lobe white matter | Bauman et al. [55] |
| C57BL/6 dams were IP injected with 75 μg/kg LPS on GD 14. | Three-chamber, OF, EPM, forced swim tail suspension | Impaired social interactions and social recognition Altered locomotion anxiety Depression | Wu et al. [64] |
| Animal | Test Paradigm | Phenotype Manifestations | Authors, Ref. |
|---|---|---|---|
| ICR mice were treated with 300 mg/kg VPA on PND 4 | Three-chamber, EPM, Water T-maze, OF test | VPA-increased grooming frequency, impaired sociability in males increased anxiety-like behaviors in females. | Ornoy, et al. [40] |
| Pregnant Wistar rats were administered 600 mg/kg of VPA on GD 12.5 | Three-chamber, reciprocal social interaction, OF/self-grooming, and EPM | Decreased social interactions and recognition, increased anxiety and nociceptive threshold | Hirsch et al. [37] |
| Treating zebrafish embryos with 5, 50, and 500 µM of VPA at 8 h post fertilization | Light and dark swim speed/preference test, larval social test, mirror attack, shoaling, and social contact test | Hyperactive movement disorder and thigmotaxis, reduced social interaction, macrocephaly | Chen et al. [69] |
| Rats administered 25 mg/kg of PCB from GD 3 to parturition | Two-chamber social paradigm | Impairment of sociability and social recognition | Jolous-Jamshidi et al. [70] |
| 200 mg VPA was orally given to pregnant marmosets from GD 60 to 66 | Pulse code modulation (PCM) audio recorder | Altered infant and juvenile vocalizations | Watanabe et al. [71] |
| 5 mg of CPF to pregnant mice from GDs 12–15 | Three-chamber, social interaction, object recognition and restricted interest tests | Enhanced restricted interest. reduced social conditioned place preference, dampened social recognition | Lan et al. [72] |
| Treated zebrafish embryos for 48 h with 1 µM of VPA starting 8 h post fertilization | Mirror test, two-chamber social paradigm | Exhibited impaired social behavior and social visual laterality, with altered brain asymmetry | Messina et al. [73] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ornoy, A.; Echefu, B.; Becker, M. Animal Models of Autistic-like Behavior in Rodents: A Scoping Review and Call for a Comprehensive Scoring System. Int. J. Mol. Sci. 2024, 25, 10469. https://doi.org/10.3390/ijms251910469
Ornoy A, Echefu B, Becker M. Animal Models of Autistic-like Behavior in Rodents: A Scoping Review and Call for a Comprehensive Scoring System. International Journal of Molecular Sciences. 2024; 25(19):10469. https://doi.org/10.3390/ijms251910469
Chicago/Turabian StyleOrnoy, Asher, Boniface Echefu, and Maria Becker. 2024. "Animal Models of Autistic-like Behavior in Rodents: A Scoping Review and Call for a Comprehensive Scoring System" International Journal of Molecular Sciences 25, no. 19: 10469. https://doi.org/10.3390/ijms251910469
APA StyleOrnoy, A., Echefu, B., & Becker, M. (2024). Animal Models of Autistic-like Behavior in Rodents: A Scoping Review and Call for a Comprehensive Scoring System. International Journal of Molecular Sciences, 25(19), 10469. https://doi.org/10.3390/ijms251910469