
Methodological Approaches to Experimental Evaluation of Neuroprotective Action of Potential Drugs
 , ,
, ,  and
and 
Abstract
:1. Introduction
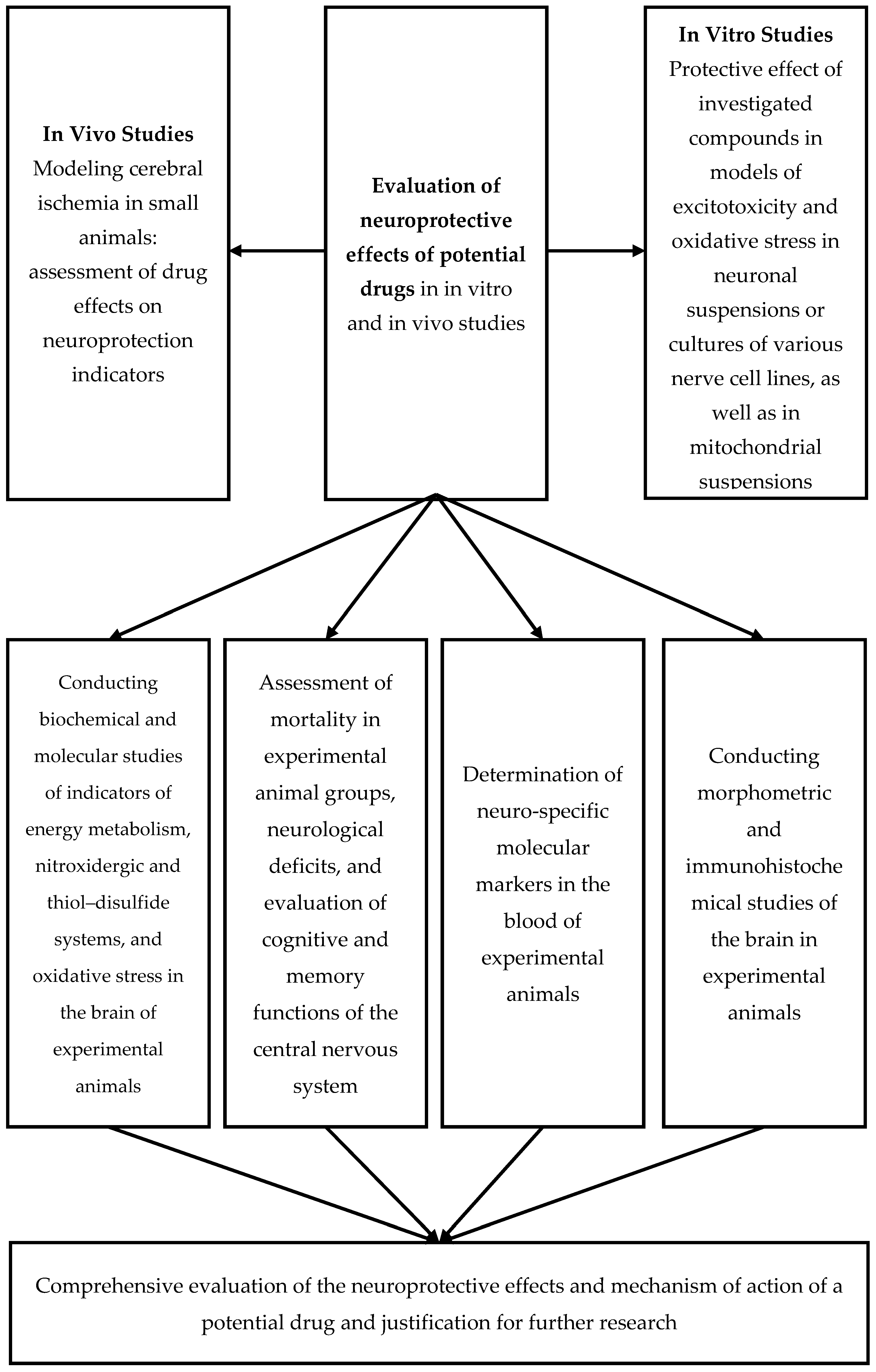
2. Preliminary Evaluation of the Neuroprotective Effects of Potential Drugs In Vitro
- -
- Intact: A suspension of neurons without the addition of initiating agents or potential neuroprotectors under investigation;
- -
- Control: A suspension of neurons to which agents that induce oxidative and nitrosative stress, glutamatergic excitotoxicity, and glutathione thiol–disulfide system deprivation are added at concentrations capable of causing the death of 50% of neurons (0.1–5 µM). To initiate oxidative stress in the neuron suspension, 0.25–1.0 mM H2O2 is added to the incubation medium. Glutamatergic “excitotoxicity” is induced by adding kainate (200–400 µM), glutamate (0.1–10 mM), or N-methyl-D-aspartate (100–150 µM) to the incubation medium. Glutathione thiol–disulfide system deprivation is achieved by introducing chloro-2,4-dinitrobenzene (CDNB) (50–500 µM), a selective inhibitor of glutathione S-transferase that forms conjugates with glutathione in cytosolic and mitochondrial fractions, into the incubation medium [16,17,18,19,20,21]. Alongside the intact and control samples, samples with the addition of initiating agents and pharmacological agents at various concentrations are prepared, followed by determining their effective concentration. The neuroprotective activity of potential neuroprotectors is assessed by counting neurons exhibiting signs of apoptosis using flow cytometry or histochemical methods. Our studies have shown that the addition of the aforementioned neurotoxins to the incubation medium led to a pronounced disruption of cellular, molecular–biochemical processes. These disruptions were consistent in nature but varied in degree of severity—accompanied by a sharp shift in the thiol–disulfide balance toward oxidized thiols (a significant decrease in reduced glutathione concentration and an increase in oxidized glutathione). An increase in the marker of oxidative protein damage—nitrotyrosine—was observed, along with a decrease in the activity of mitochondrial superoxide dismutase (Mn-SOD). We also recorded dynamic changes in the synthesis of endogenous cytoprotective factors—HSP and HIF proteins.
- MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine)-induced opening of mitochondrial pores: add 40–60 µM MPTP to the incubation medium and after 5 min, add 50 µM CaCl2;
- Ca2+-induced opening of mitochondrial pores: add 200 µM CaCl2 to the incubation medium;
- NO-induced opening of mitochondrial pores: add 20–100 µM sodium nitroprusside to the incubation medium and, after 2 min, add 50 µM CaCl2;
- H2O2-induced opening of mitochondrial pores: add 50 mM hydrogen peroxide to the incubation medium and, after 2 min, add 50 µM CaCl2.
3. Preliminary Assessment of the Neuroprotective Effects of Potential Pharmaceutical Agents Using Various Models of Cerebral Ischemia
4. Assessment of Neurological Deficit in Animals with Experimental Cerebral Ischemia Serves as an Integrative Measure of the Neuroprotective Efficacy of Potential Pharmacological Agents
- Mild: 0 to 3 points;
- Moderate: 3 to 7 points;
- Severe: 7 points and above.
- Unilateral partial ptosis: 0.5 points;
- Unilateral ptosis: 1 point;
- Tremor: 0.5 points;
- Circling movements: 0.5 points;
- Paresis of limbs (per limb): 1 point;
- Paralysis of limbs (per limb): 2 points;
- Lateral positioning: 3 points;
- Inability to remain on the rotating rod (3 RPM) for 4 min: 3 points.
5. Determination of Oxidative Stress Markers and Antioxidant System Status
- Pentafluorophenylhydrazine (PFPH);
- Methylhydrazine (MH);
- 4-(2-phthalimidyl)benzohydrazine (FBH);
- 2,4-Dinitrophenylhydrazine (DNPH);
- o-(2,3,4,5,6-Pentafluorobenzyl)hydroxylamine hydrochloride (PFBH);
- tert-Butyldimethylchlorosilane (BDMCS);
- N,O-Di-(trimethylsilyl)-trifluoroacetamide (DTSFA);
- 2-Hydrazinobenzothiazole (HBT).
6. Determination of Antioxidant Enzyme Activity
7. α-Tocopherol
8. Determination of the Most Informative Indicators of the Thiol–Disulfide System
9. Indicators of the Nitric Oxide System in the Brain
10. Assessment of Brain Energy Metabolism Indicators
11. Morphometry of Various Brain Structures
- Density of Neurons, Glial Cells, Apoptotic, and Destructively Altered Neurons: Measured as the number of cells per 1 mm2 of tissue section;
- Cellular Composition: Determined as the percentage of neurons, glial cells, apoptotic, and destructively altered neurons in the IV–V layers of the cortex and the CA1 region of the hippocampus;
- Area of Cell Bodies: Measured in µm2 for neurons, glial cells, apoptotic, and destructively altered neurons;
- RNA Concentration: In neurons, glial cells and apoptotic and destructively altered neurons are expressed in optical density units (ODU). This is calculated as the logarithm of the ratio of the optical density of the cell body to the optical density of the extracellular matrix;
- RNA Content: In neurons, glial cells and apoptotic and destructively altered neurons are expressed in ODU. This is calculated as the product of RNA concentration and cell area;
- Neuron Survival Index: Assessed as the ratio of the number of neurons in experimental animals to the number of neurons in intact control animals.
11.1. Markers with Informational Value in CNS Pathology
11.1.1. Gold Dot (NR2 Antibody Detection)
- Independent Serum Marker: The level of antibodies to NR2 serves as an independent serum marker for cerebral ischemic events;
- Neurotoxicity Marker: NR2 antibodies are indicative of neurotoxicity associated with ischemic damage;
- Monitoring Tool: Tracking NR2 antibody levels enables monitoring the efficacy of pharmacological interventions for ischemic brain injury.
11.1.2. Neuron-Specific Enolase (NSE) (Enzyme-Linked Immunosorbent Assay, Western Blot)
11.1.3. Myelin Basic Protein (MBP) (Enzyme-Linked Immunosorbent Assay, Western Blot)
11.1.4. S-100 Protein (Enzyme-Linked Immunosorbent Assay)
11.1.5. Galanin (Immunohistochemistry Enzyme-Linked Immunosorbent Assay, Western Blot)
11.1.6. Phosphorylated Neurofilament H (pNF-H) (Enzyme-Linked Immunosorbent Assay, Western Blot)
11.1.7. Glial Fibrillary Acidic Protein (GFAP) (Enzyme-Linked Immunosorbent Assay)
11.2. Markers of Neuroplasticity
11.2.1. Neurotrophin-3 (NT3) and Neurotrophin-4/5 (NT4/5) (Immunohistochemistry, Enzyme-Linked Immunosorbent Assay, Western Blot)
11.2.2. Expression of c-fos and c-jun Proteins in the Brain (Immunohistochemistry; Western Blot)
11.2.3. Expression/Concentration of Heat Shock Proteins in the Brain (Immunohistochemistry, Western Blot, Enzyme-Linked Immunosorbent Assay)
11.2.4. Hypoxia-Inducible Factor 1α (HIF-1α) (Immunohistochemistry, Immunoblotting, Enzyme-Linked Immunosorbent Assay)
11.2.5. Brain-Derived Neurotrophic Factor (BDNF) (Immunohistochemistry, Enzyme-Linked Immunosorbent Assay, Western Blot)
11.2.6. Ciliary Neurotrophic Factor (CNTF) (Immunohistochemistry, Enzyme-Linked Immunosorbent Assay, Western Blot)
11.2.7. Pigment Epithelium-Derived Factor (PEDF) (Immunohistochemistry, Western Blot)
11.3. Markers of Apoptosis
11.3.1. Annexin V (Immunohistochemistry, Western Blot)
11.3.2. Caspase-3 (Immunohistochemistry, Western Blot, Enzyme-Linked Immunosorbent Assay)
11.3.3. Cathepsins (Immunohistochemistry, Western Blot, Enzyme-Linked Immunosorbent Assay)
11.3.4. Procathepsin B (Immunohistochemistry, Western Blot, Enzyme-Linked Immunosorbent Assay)
11.3.5. DR5 (Death Receptor) (Immunohistochemistry, Western Blot)
11.3.6. Bcl-2 Family (Immunohistochemistry, Western Blot)
- Anti-apoptotic Subfamily: includes close homologs such as Bcl-2, Bcl-XL, and Bcl-w, which inhibit apoptosis;
- Pro-apoptotic Subfamilies: include proteins such as Bax and members of the BH3-only group, which promote apoptosis.
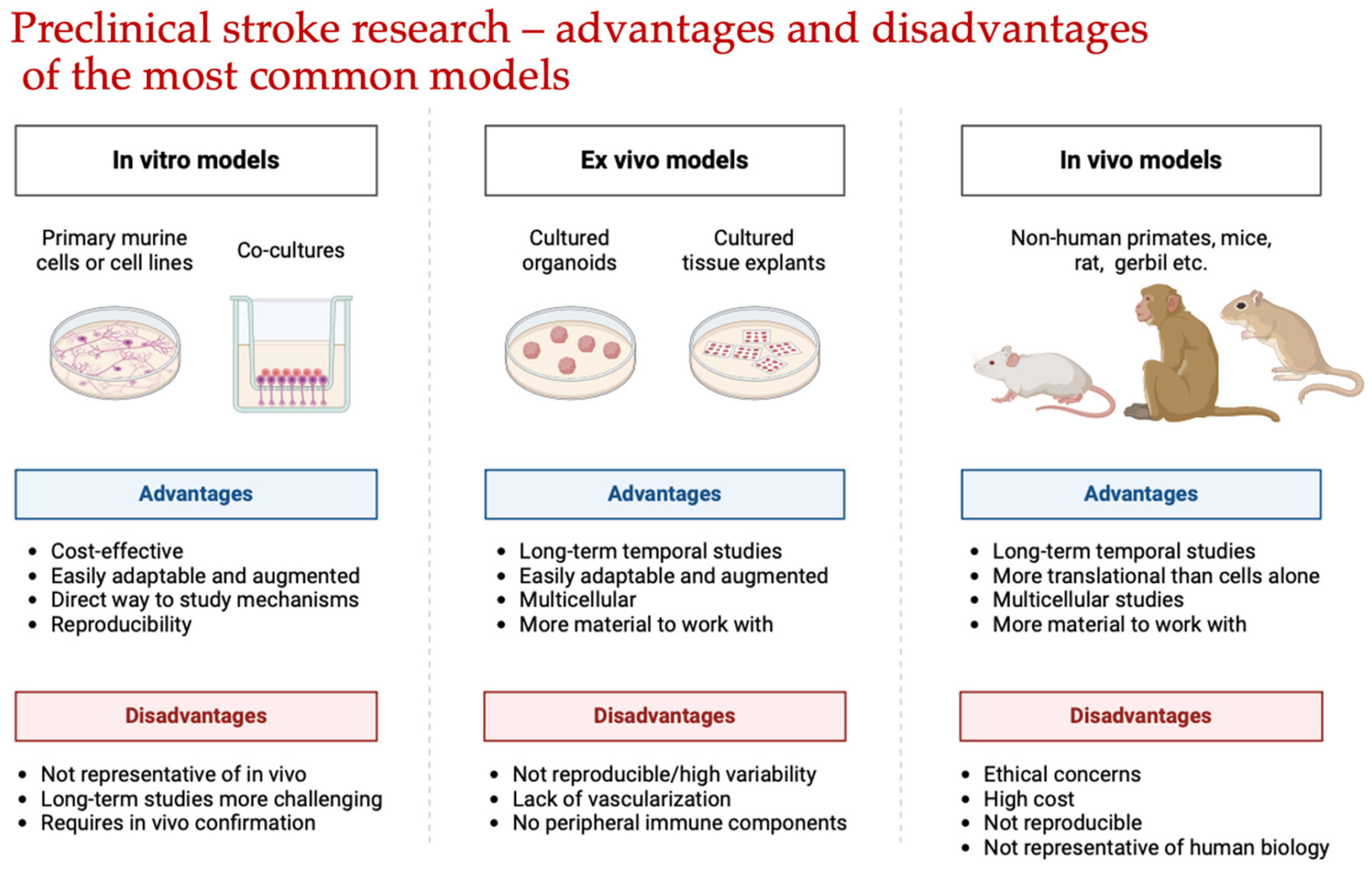
- Perform Preliminary Screening**: Evaluate a large number of molecules while preserving animal lives;
- Investigate Mechanisms**: Examine the impact of potential neuroprotectors on specific aspects of the ischemic brain damage pathogenesis, such as oxidative nitrosative stress, glutamate excitotoxicity, and thiol–disulfide balance shifts.
- Diffuse Decrease in Cerebral Blood Flow;
- Energy Deficit;
- Glutamate–Calcium Excitotoxicity;
- Oxidative Stress;
- Expression of Early Response Genes.
Author Contributions
Funding
Conflicts of Interest
References
- Donkor, E.S. Stroke in the 21st Century: A Snapshot of the Burden, Epidemiology, and Quality of Life. Stroke Res. Treat. 2018, 2018, 3238165. [Google Scholar] [CrossRef] [PubMed]
- Salaudeen, M.A.; Bello, N.; Danraka, R.N.; Ammani, M.L. Understanding the Pathophysiology of Ischemic Stroke: The Basis of Current Therapies and Opportunity for New Ones. Biomolecules 2024, 14, 305. [Google Scholar] [CrossRef] [PubMed]
- Belov Kirdajova, D.; Kriska, J.; Tureckova, J.; Anderova, M. Ischemia-Triggered Glutamate Excitotoxicity from the Perspective of Glial Cells. Front. Cell. Neurosci. 2020, 14, 51. [Google Scholar] [CrossRef] [PubMed]
- Xing, C.; Arai, K.; Lo, E.H.; Hommel, M. Pathophysiologic Cascades in Ischemic Stroke. Int. J. Stroke 2012, 7, 378–385. [Google Scholar] [CrossRef]
- Xie, X.; Liu, J. New Role of Astrocytes in Neuroprotective Mechanisms After Ischemic Stroke. Arq. Neuro-Psiquiatr. 2023, 81, 748–755. [Google Scholar] [CrossRef]
- Zhu, G.; Wang, X.; Chen, L.; Lenahan, C.; Fu, Z.; Fang, Y.; Yu, W. Crosstalk Between the Oxidative Stress and Glia Cells After Stroke: From Mechanism to Therapies. Front. Immunol. 2022, 13, 852416. [Google Scholar] [CrossRef]
- Paul, S.; Candelario-Jalil, E. Emerging Neuroprotective Strategies for the Treatment of Ischemic Stroke: An Overview of Clinical and Preclinical Studies. Exp. Neurol. 2021, 335, 113518. [Google Scholar] [CrossRef]
- Belenichev, I.F.; Cherniy, V.I.; Nagornaya, E.A.; Pavlov, S.V.; Cherniy, T.V. Neuroprotection and Neuroplasticity; Logos: Kyiv, Ukraine, 2015; p. 512. [Google Scholar]
- Ben-Shushan, S.; Miller, Y. Neuropeptides: Roles and Activities as Metal Chelators in Neurodegenerative Diseases. J. Phys. Chem. B 2021, 125, 2796–2811. [Google Scholar] [CrossRef]
- Belenichev, I.; Ryzhenko, V.; Popazova, O.; Bukhtiyarova, N.; Gorchakova, N.; Oksenych, V.; Kamyshnyi, O. Optimization of the Search for Neuroprotectors among Bioflavonoids. Pharmaceuticals 2024, 17, 877. [Google Scholar] [CrossRef]
- Jiang, X.; Wu, K.; Ye, X.Y.; Xie, T.; Zhang, P.; Blass, B.E.; Bai, R. Novel Druggable Mechanism of Parkinson’s Disease: Potential Therapeutics and Underlying Pathogenesis Based on Ferroptosis. Med. Res. Rev. 2023, 43, 872–896. [Google Scholar] [CrossRef]
- Kim, A.N.; Ngamnithiporn, A.; Du, E.; Stoltz, B.M. Recent Advances in the Total Synthesis of the Tetrahydroisoquinoline Alkaloids (2002–2020). Chem. Rev. 2023, 123, 9447–9496. [Google Scholar] [CrossRef] [PubMed]
- Amado, B.; Melo, L.; Pinto, R.; Lobo, A.; Barros, P.; Gomes, J.R. Ischemic Stroke, Lessons from the Past towards Effective Preclinical Models. Biomedicines 2022, 10, 2561. [Google Scholar] [CrossRef] [PubMed]
- Trotman-Lucas, M.; Gibson, C.L. A Review of Experimental Models of Focal Cerebral Ischemia Focusing on the Middle Cerebral Artery Occlusion Model. F1000Research 2021, 10, 242. [Google Scholar] [CrossRef] [PubMed]
- Belenichev, I.F.; Odnokoz, O.V.; Pavlov, S.V.; Belenicheva, O.I.; Polyakova, E.N. The Neuroprotective Activity of Tamoxifen and Tibolone during Glutathione Depletion in Vitro. Neurochem. J. 2012, 6, 202–212. [Google Scholar] [CrossRef]
- Kritis, A.A.; Stamoula, E.G.; Paniskaki, K.A.; Vavilis, T.D. Researching Glutamate-Induced Cytotoxicity in Different Cell Lines: A Comparative/Collective Analysis/Study. Front. Cell. Neurosci. 2015, 9, 91. [Google Scholar] [CrossRef]
- Xin, X.; Gong, T.; Hong, Y. Hydrogen Peroxide Initiates Oxidative Stress and Proteomic Alterations in Meningothelial Cells. Sci. Rep. 2022, 12, 14519. [Google Scholar] [CrossRef]
- Ransy, C.; Vaz, C.; Lombès, A.; Bouillaud, F. Use of H2O2 to Cause Oxidative Stress, the Catalase Issue. Int. J. Mol. Sci. 2020, 21, 9149. [Google Scholar] [CrossRef]
- Kirchgessner, A.L.; Liu, M.T.; Alcantara, F. Excitotoxicity in the Enteric Nervous System. J. Neurosci. 1997, 17, 8804–8816. [Google Scholar] [CrossRef]
- Hu, B.; Huang, H.; Wei, Q.; Ren, M.; Mburu, D.K.; Tian, X.; Su, J. Transcription Factors CncC/Maf and AhR/ARNT Coordinately Regulate the Expression of Multiple GSTs Conferring Resistance to Chlorpyrifos and Cypermethrin in Spodoptera exigua. Pest Manag. Sci. 2019, 75, 2009–2019. [Google Scholar] [CrossRef]
- Lin, H.; Wu, H.; Li, H.; Song, A.; Yin, W. The essential role of GSTP1 I105V polymorphism in the prediction of CDNB metabolism and toxicity: In silico and in vitro insights. Toxicol. Vitr. 2023, 90, 105601. [Google Scholar] [CrossRef]
- Bajpai, P.; Sangar, M.C.; Singh, S.; Tang, W.; Bansal, S.; Chowdhury, G.; Cheng, Q.; Fang, J.K.; Martin, M.V.; Guengerich, F.P.; et al. Metabolism of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine by mitochondrion-targeted cytochrome P450 2D6: Implications in Parkinson disease. J. Biol. Chem. 2013, 288, 4436–4451. [Google Scholar] [CrossRef] [PubMed]
- Stepanova, A.; Galkin, A. Measurement of mitochondrial H2O2 production under varying O2 tensions. Methods Cell Biol. 2020, 155, 273–293. [Google Scholar] [PubMed]
- Saviani, E.E.; Orsi, C.H.; Oliveira, J.F.; Pinto-Maglio, C.A.; Salgado, I. Participation of the mitochondrial permeability transition pore in nitric oxide-induced plant cell death. FEBS Lett. 2002, 510, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Shin, T.; Hirokata, Y.; Shigematsu, A. Inhibition of mitochondrial respiration by sodium nitroprusside and the mechanism of cyanide liberation. Br. J. Anaesth. 1977, 49, 1239–1244. [Google Scholar] [CrossRef]
- Blomeyer, C.A.; Bazil, J.N.; Stowe, D.F.; Pradhan, R.K.; Dash, R.K.; Camara, A.K. Dynamic buffering of mitochondrial Ca2+ during Ca2+ uptake and Na+-induced Ca2+ release. J. Bioenerg. Biomembr. 2013, 45, 189–202. [Google Scholar] [CrossRef]
- Bernardi, P.; Di Lisa, F. The mitochondrial permeability transition pore: Molecular nature and role as a target in cardioprotection. J. Mol. Cell. Cardiol. 2015, 78, 100–106. [Google Scholar] [CrossRef]
- Sommer, C.J. Ischemic stroke: Experimental models and reality. Acta Neuropathol. 2017, 133, 245–261. [Google Scholar] [CrossRef]
- Bacigaluppi, M.; Comi, G.; Hermann, D.M. Animal models of ischemic stroke. Part two: Modeling cerebral ischemia. Open Neurol. J. 2010, 4, 34–38. [Google Scholar] [CrossRef]
- Fluri, F.; Schuhmann, M.K.; Kleinschnitz, C. Animal models of ischemic stroke and their application in clinical research. Drug Des. Dev. Ther. 2015, 9, 3445–3454. [Google Scholar]
- Traystman, R.J. Animal Models of Focal and Global Cerebral Ischemia. ILAR J. 2003, 44, 85–95. [Google Scholar] [CrossRef]
- Netzley, A.H.; Pelled, G. The Pig as a Translational Animal Model for Biobehavioral and Neurotrauma Research. Biomedicines 2023, 11, 2165. [Google Scholar] [CrossRef] [PubMed]
- Lovasova, V.; Bem, R.; Chlupac, J.; Dubsky, M.; Husakova, J.; Nemcova, A.; Fronek, J. Animal experimental models of ischemic limbs—A systematic review. Vasc. Pharmacol. 2023, 153, 107237. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, P.; Kushwaha, V.; Shukla, V.; Kumar, A. Animal research ethics: An overview. Eur. J. Biomed. Pharm. Sci. 2023, 10, 78–87. [Google Scholar]
- Martin, A.K. Animal Research. In The Moral Implications of Human and Animal Vulnerability; Palgrave Macmillan: Cham, Switzerland, 2023. [Google Scholar] [CrossRef]
- Jones, D. Genetic engineering of a mouse: Dr. Frank Ruddle and somatic cell genetics. Yale J. Biol. Med. 2011, 84, 117–124. [Google Scholar] [PubMed]
- Ruan, J.; Yao, Y. Behavioral tests in rodent models of stroke. Brain Hemorrhages 2020, 1, 171–184. [Google Scholar] [CrossRef]
- Topol, I.; Kamyshny, A. Study of expression of TLR2, TLR4 and transckription factor NF-kB structures of galt of rats in the conditions of the chronic social stress and modulation of structure of intestinal microflora. Georgian Med. News 2013, 225, 115–122. [Google Scholar] [PubMed]
- Krafft, P.R.; Bailey, E.L.; Lekic, T.; Rolland, W.B.; Altay, O.; Tang, J.; Wardlaw, J.M.; Zhang, J.H.; Sudlow, C.L. Etiology of stroke and choice of models. Int. J. Stroke 2012, 7, 398–406. [Google Scholar] [CrossRef]
- Koukalova, L.; Chmelova, M.; Amlerova, Z.; Vargova, L. Out of the core: The impact of focal ischemia in regions beyond the penumbra. Front. Cell. Neurosci. 2024, 18, 1336886. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, J. Animal models of stroke. Anim. Model. Exp. Med. 2021, 4, 204–219. [Google Scholar] [CrossRef]
- Sicard, K.M.; Fisher, M. Animal models of focal brain ischemia. Exp. Transl. Stroke Med. 2009, 1, 7. [Google Scholar] [CrossRef]
- Zeng, L.; Hu, S.; Zeng, L.; Chen, R.; Li, H.; Yu, J.; Yang, H. Animal Models of Ischemic Stroke with Different Forms of Middle Cerebral Artery Occlusion. Brain Sci. 2023, 13, 1007. [Google Scholar] [CrossRef] [PubMed]
- Nowak, B.; Rogujski, P.; Guzman, R.; Walczak, P.; Andrzejewska, A.; Janowski, M. Animal models of focal ischemic stroke: Brain size matters. Front. Stroke 2023, 2, 1165231. [Google Scholar] [CrossRef]
- Tamura, A.; Graham, D.I.; McCulloch, J.; Teasdale, G.M. Focal cerebral ischaemia in the rat: 1. Description of technique and early neuropathological consequences following middle cerebral artery occlusion. J. Cereb. Blood Flow. Metab. 1981, 1, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.F.; Chuang, T.Y.; Hung, Y.W.; Lan, M.Y.; Tsai, C.H.; Huang, H.X.; Lin, Y.Y. Development of a Modified Surgical Technique for Simulating Ischemic Cerebral Cortex Injury in Rats. In Vivo 2019, 33, 1175–1181. [Google Scholar] [CrossRef] [PubMed]
- Kunz, A.; Iadecola, C. Cerebral vascular dysregulation in the ischemic brain. Handb. Clin. Neurol. 2009, 92, 283–305. [Google Scholar] [CrossRef]
- Windle, V.; Szymanska, A.; Granter-Button, S.; White, C.; Buist, R.; Peeling, J.; Corbett, D. An analysis of four different methods of producing focal cerebral ischemia with endothelin-1 in the rat. Exp. Neurol. 2006, 201, 324–334. [Google Scholar] [CrossRef]
- Ma, J.; Peng, C.; Guo, W.; Dong, Y.F.; Dong, X.H.; Sun, X.; Xie, H.H. A modified model of middle cerebral artery electrocoagulation in mice. CNS Neurosci. Ther. 2012, 18, 796–798. [Google Scholar] [CrossRef]
- Wang, C.X.; Yang, Y.; Yang, T.; Shuaib, A. A focal embolic model of cerebral ischemia in rats: Introduction and evaluation. Brain Res. Brain Res. Protoc. 2001, 7, 115–120. [Google Scholar] [CrossRef]
- Justić, H.; Barić, A.; Šimunić, I.; Radmilović, M.; Ister, R.; Škokić, S.; Dobrivojević Radmilović, M. Redefining the Koizumi model of mouse cerebral ischemia: A comparative longitudinal study of cerebral and retinal ischemia in the Koizumi and Longa middle cerebral artery occlusion models. Cereb. Blood Flow. Metab. 2022, 42, 2080–2094. [Google Scholar] [CrossRef]
- Shahjouei, S.; Cai, P.Y.; Ansari, S.; Sharififar, S.; Azari, H.; Ganji, S.; Zand, R. Middle cerebral artery occlusion model of stroke in rodents: A step-by-step approach. J. Vasc. Interv. Neurol. 2016, 8, 1–8. [Google Scholar]
- Fukuda, S.; del Zoppo, G.J. Models of focal cerebral ischemia in the nonhuman primate. ILAR J. 2003, 44, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Wang, H.; Chen, J.; Zhao, P.; Wen, M.; Bingwa, L.A.; Jin, K.; Zhuge, Q.; Yang, S. Nonhuman primate models of ischemic stroke and neurological evaluation after stroke. J. Neurosci. Methods 2022, 376, 109611. [Google Scholar] [CrossRef] [PubMed]
- Themistoklis, K.M.; Papasilekas, T.I.; Melanis, K.S.; Boviatsis, K.A.; Korfias, S.I.; Vekrellis, K.; Sakas, D.E. Transient intraluminal filament middle cerebral artery occlusion stroke model in rats: A step-by-step guide and technical considerations. World Neurosurg. 2022, 168, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, B.A.; Neuhaus, A.A.; Couch, Y.; Balami, J.S.; DeLuca, G.C.; Hadley, G.; Harris, S.L.; Grey, A.N.; Buchan, A.M. The transient intraluminal filament middle cerebral artery occlusion model as a model of endovascular thrombectomy in stroke. J. Cereb. Blood Flow. Metab. 2016, 36, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Ozdemir, Y.G.; Bolay, H.; Erdem, E.; Dalkara, T. Occlusion of the MCA by an intraluminal filament may cause disturbances in the hippocampal blood flow due to anomalies of circle of Willis and filament thickness. Brain Res. 1999, 822, 260–264. [Google Scholar] [CrossRef]
- Watson, B.D.; Dietrich, W.D.; Busto, R.; Wachtel, M.S.; Ginsberg, M.D. Induction of reproducible brain infarction by photochemically initiated thrombosis. Ann. Neurol. 1985, 17, 497–504. [Google Scholar] [CrossRef]
- Montalbetti, L.; Rozza, A.; Rizzo, V.; Favalli, L.; Scavini, C.; Lanza, E.; Savoldi, F.; Racagni, G.; Scelsi, R. Aminoacid recovery via microdialysis and photoinduced focal cerebral ischemia in brain cortex of rats. Neurosci. Lett. 1995, 192, 153–156. [Google Scholar] [CrossRef]
- Ito, H.; Hashimoto, A.; Matsumoto, Y.; Yao, H.; Miyakoda, G. Cilostazol, a phosphodiesterase inhibitor, attenuates photothrombotic focal ischemic brain injury in hypertensive rats. J. Cereb. Blood Flow. Metab. 2010, 30, 343–351. [Google Scholar] [CrossRef]
- Atochin, D.N.; Chernysheva, G.A.; Aliev, O.I.; Smolyakova, V.I.; Osipenko, A.N.; Logvinov, S.V.; Zhdankina, A.A.; Plotnikova, T.M.; Plotnikov, M.B. An improved three-vessel occlusion model of global cerebral ischemia in rats. Brain Res. Bull. 2017, 132, 213–221. [Google Scholar] [CrossRef]
- Neumann, J.T.; Cohan, C.H.; Dave, K.R.; Wright, C.B.; Perez-Pinzon, M.A. Global cerebral ischemia: Synaptic and cognitive dysfunction. Curr. Drug Targets 2013, 14, 20–35. [Google Scholar] [CrossRef]
- Mansoorali, K.P.; Prakash, T.; Kotresha, D.; Prabhu, K.; Rama Rao, N. Cerebroprotective effect of Eclipta alba against global model of cerebral ischemia induced oxidative stress in rats. Phytomedicine 2012, 19, 1108–1116. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, E.M.; Snyder, J.V.; Carroll, R.G.; Morita, H. Global ischemia in dogs: Cerebrovascular CO2 reactivity and autoregulation. Stroke 1975, 6, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, E.M.; Bleyaert, A.L.; Stezoski, S.W.; Moossy, J.; Rao, G.R.; Safar, P. Global brain ischemia: A reproducible monkey model. Stroke 1977, 8, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Bleyaert, A.; Safar PNemoto, E.; Moossy, J.; Sassano, J. Effect of postcirculatory-arrest life-support on neurological recovery in monkeys. Crit. Care Med. 1980, 8, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Dave, K.R.; Della-Morte, D.; Saul, I.; Prado, R.; Perez-Pinzon, M.A. Ventricular fibrillation-induced cardiac arrest in the rat as a model of global cerebral ischemia. Transl. Stroke Res. 2013, 4, 571–578. [Google Scholar] [CrossRef]
- Deng, G.; Yonchek, J.C.; Quillinan, N.; Strnad, F.A.; Exo, J.; Herson, P.S.; Traystman, R.J. A novel mouse model of pediatric cardiac arrest and cardiopulmonary resuscitation reveals age-dependent neuronal sensitivities to ischemic injury. J. Neurosci. Methods 2014, 222, 34–41. [Google Scholar] [CrossRef]
- Kofler, J.; Hattori, K.; Sawada, M.; DeVries, A.C.; Martin, L.J.; Hurn, P.D.; Traystman, R.J. Histopathological and behavioral characterization of a novel model of cardiac arrest and cardiopulmonary resuscitation in mice. J. Neurosci. Methods 2004, 136, 33–44. [Google Scholar] [CrossRef]
- Kaya, A.H.; Erdogan, H.; Tasdemiroglu, E. Searching evidences of stroke in animal models: A review of discrepancies. Turk. Neurosurg. 2017, 27, 167–173. [Google Scholar] [CrossRef]
- Woodruff, T.M.; Thundyil, J.; Tang, S.C.; Sobey, C.G.; Taylor, S.M.; Arumugam, T.V. Pathophysiology, treatment, and animal and cellular models of human ischemic stroke. Mol. Neurodegener. 2011, 6, 11. [Google Scholar] [CrossRef]
- Wahul, A.B.; Joshi, P.C.; Kumar, A.; Chakravarty, S. Transient global cerebral ischemia differentially affects cortex, striatum and hippocampus in bilateral common carotid arterial occlusion (BCCAo) mouse model. J. Chem. Neuroanat. 2018, 92, 1–15. [Google Scholar] [CrossRef]
- Sun, W.; Chen, Y.; Zhang, Y.; Geng, Y.; Tang, X.; Guo, R.; Zhang, Z.; Xu, H.; Tian, X. A modified four vessel occlusion model of global cerebral ischemia in rats. J. Neurosci. Methods 2021, 352, 109090. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Wu, Y.; Qu, Y.; Shi, F.; Hu, J.; Gao, B.; Wang, B.; Gao, G.; He, S.; Zhao, T. A modified method to reduce variable outcomes in a rat model of four-vessel arterial occlusion. Neurol. Res. 2016, 38, 1102–1110. [Google Scholar] [CrossRef] [PubMed]
- Radenovic, L. In Vivo Experimental Models of Cerebral Ischemia: Analysis, Translational Potential and Clinical Relevance. Cardiol. Res. Cardiovasc. Med. 2024, 9, 232. [Google Scholar] [CrossRef]
- McGraw, C.P. Experimental cerebral infarction: Effects of pentobarbital in Mongolian gerbils. Arch. Neurol. 1977, 34, 334–336. [Google Scholar] [CrossRef] [PubMed]
- Laas, R. Common carotid artery stump pressure in the gerbil stroke model. J. Neurol. Neurosurg. Psychiatry 1984, 47, 365–371. [Google Scholar] [CrossRef]
- Zhang, T.; Yue, Y.; Li, C.; Wu, X.; Park, S. Vagus nerve suppression in ischemic stroke by carotid artery occlusion: Implications for metabolic regulation, cognitive function, and gut microbiome in a gerbil model. Int. J. Mol. Sci. 2024, 25, 7831. [Google Scholar] [CrossRef]
- Belenichev, I.F.; Kolesnik, Y.M.; Pavlov, S.V.; Sokolik, E.P.; Bukhtiyarova, N.V. Malate-aspartate shunt in neuronal adaptation to ischemic conditions: Molecular-biochemical mechanisms of activation and regulation. Neurochem. J. 2012, 6, 22–28. [Google Scholar] [CrossRef]
- Pavlov, S.V.; Belenichev, I.F. Molecular and biochemical aspects of the neuroprotective effect of the selective estrogen receptor modulator tamoxifen in a model of acute cerebral ischemia. Neurochem. J. 2014, 8, 28–32. [Google Scholar] [CrossRef]
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free radicals: Properties, sources, targets, and their implication in various diseases. Indian. J. Clin. Biochem. 2015, 30, 11–26. [Google Scholar] [CrossRef]
- Lee, K.H.; Cha, M.; Lee, B.H. Crosstalk between neuron and glial cells in oxidative injury and neuroprotection. Int. J. Mol. Sci. 2021, 22, 13315. [Google Scholar] [CrossRef] [PubMed]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef] [PubMed]
- Cordiano, R.; Di Gioacchino, M.; Mangifesta, R.; Panzera, C.; Gangemi, S.; Minciullo, P.L. Malondialdehyde as a potential oxidative stress marker for allergy-oriented diseases: An update. Molecules 2023, 28, 5979. [Google Scholar] [CrossRef] [PubMed]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative stress: Harms and benefits for human health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Raptova, R.; Alomar, S.Y.; Alwasel, S.H.; Nepovimova, E.; Kuca, K.; Valko, M. Reactive oxygen species, toxicity, oxidative stress, and antioxidants: Chronic diseases and aging. Arch. Toxicol. 2023, 97, 2499–2574. [Google Scholar] [CrossRef] [PubMed]
- Alzaid, F.; Patel, V.B.; Preedy, V.R. Biomarkers of oxidative stress in blood. Biomark. Dis. Methods Discov. Appl. 2015, 1–2, 567–594. [Google Scholar] [CrossRef]
- Frijhoff, J.; Winyard, P.G.; Zarkovic, N.; Davies, S.S.; Stocker, R.; Cheng, D.; Knight, A.R.; Taylor, E.L.; Oettrich, J.; Ruskovska, T.; et al. Clinical relevance of biomarkers of oxidative stress. Antioxid. Redox Signal. 2015, 23, 1144–1170. [Google Scholar] [CrossRef]
- Ng, S.C.W.; Furman, R.; Axelsen, P.H.; Shchepinov, M.S. Free radical chain reactions and polyunsaturated fatty acids in brain lipids. ACS Omega 2022, 7, 25337–25345. [Google Scholar] [CrossRef]
- Guéraud, F.; Atalay, M.; Bresgen, N.; Cipak, A.; Eckl, P.M.; Huc, L.; Jouanin, I.; Siems, W.; Uchida, K. Chemistry and biochemistry of lipid peroxidation products. Free Radic. Res. 2010, 44, 1098–1124. [Google Scholar] [CrossRef]
- Meagher, E.A.; FitzGerald, G.A. Indices of lipid peroxidation in vivo: Strengths and limitations. Free Radic. Biol. Med. 2000, 28, 1745–1750. [Google Scholar] [CrossRef]
- Praticò, D. Lipid peroxidation in mouse models of atherosclerosis. Trends Cardiovasc. Med. 2001, 11, 112–116. [Google Scholar] [CrossRef]
- Bilska-Wilkosz, A.; Iciek, M.; Górny, M. Chemistry and biochemistry aspects of the 4-hydroxy-2,3-trans-nonenal. Biomolecules 2022, 12, 145. [Google Scholar] [CrossRef] [PubMed]
- Abeyrathne, E.D.N.S.; Nam, K.; Ahn, D.U. Analytical methods for lipid oxidation and antioxidant capacity in food systems. Antioxidants 2021, 10, 1587. [Google Scholar] [CrossRef] [PubMed]
- Zeb, A.; Ullah, F. A simple spectrophotometric method for the determination of thiobarbituric acid reactive substances in fried fast foods. J. Anal. Methods Chem. 2016, 2016, 9412767. [Google Scholar] [CrossRef]
- Grotto, D.; Santa Maria, L.D.; Boeira, S.; Valentini, J.; Charão, M.F.; Moro, A.M.; Nascimento, P.C.; Pomblum, V.J.; Garcia, S.C. Rapid quantification of malondialdehyde in plasma by high performance liquid chromatography-visible detection. J. Pharm. Biomed. Anal. 2007, 43, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Steghens, J.P.; van Kappel, A.L.; Denis, I.; Collombel, C. Diaminonaphtalene, a new highly specific reagent for HPLC-UV measurement of total and free malondialdehyde in human plasma or serum. Free Radic. Biol. Med. 2001, 31, 242–249, Erratum in: Free Radic. Biol. Med. 2002, 32, 299. [Google Scholar] [CrossRef]
- Mendonça, R.; Gning, O.; Di Cesaré, C.; Lachat, L.; Bennett, N.C.; Helfenstein, F.; Glauser, G. Sensitive and selective quantification of free and total malondialdehyde in plasma using UHPLC-HRMS. J. Lipid Res. 2017, 58, 1924–1931. [Google Scholar] [CrossRef]
- Mao, J.; Zhang, H.; Luo, J.; Li, L.; Zhao, R.; Zhang, R.; Liu, G. New method for HPLC separation and fluorescence detection of malonaldehyde in normal human plasma. J. Chromatogr. B Analyt Technol. Biomed. Life Sci. 2006, 832, 103–108. [Google Scholar] [CrossRef]
- Graille, M.; Wild, P.; Sauvain, J.; Hemmendinger, M.; Guseva Canu, I.; Hopf, N.B. Urinary 8-isoprostane as a biomarker for oxidative stress. A systematic review and meta-analysis. Toxicol. Lett. 2020, 328, 19–27. [Google Scholar] [CrossRef]
- Charlier, C.; Michaux, C. Dual inhibition of cyclooxygenase-2 (COX-2) and 5-lipoxygenase (5-LOX) as a new strategy to provide safer non-steroidal anti-inflammatory drugs. Eur. J. Med. Chem. 2003, 38, 645–659. [Google Scholar] [CrossRef]
- Nakajima, H.; Unoda, K.; Ito, T.; Kitaoka, H.; Kimura, F.; Hanafusa, T. The relation of urinary 8-OHdG, a marker of oxidative stress to DNA, and clinical outcomes for ischemic stroke. Open Neurol. J. 2012, 6, 51–57. [Google Scholar] [CrossRef]
- Makropoulos, W.; Kocher, K.; Heintz, B.; Schwarz, E.R.; Mertens, P.R.; Stefanidis, I. Urinary thymidine glycol as a biomarker for oxidative stress after kidney transplantation. Ren. Fail. 2000, 22, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Barrera, G. Oxidative stress and lipid peroxidation products in cancer progression and therapy. ISRN Oncol. 2012, 2012, 137289. [Google Scholar] [CrossRef] [PubMed]
- Altomare, A.; Baron, G.; Gianazza, E.; Banfi, C.; Carini, M.; Aldini, G. Lipid peroxidation derived reactive carbonyl species in free and conjugated forms as an index of lipid peroxidation: Limits and perspectives. Redox Biol. 2021, 42, 101899. [Google Scholar] [CrossRef] [PubMed]
- Chidambaram, S.B.; Anand, N.; Varma, S.R.; Ramamurthy, S.; Vichitra, C.; Sharma, A.; Mahalakshmi, A.M.; Essa, M.M. Superoxide dismutase and neurological disorders. IBRO Neurosci. Rep. 2024, 16, 373–394. [Google Scholar] [CrossRef] [PubMed]
- Fukui, M.; Zhu, B.T. Mitochondrial superoxide dismutase SOD2, but not cytosolic SOD1, plays a critical role in protection against glutamate-induced oxidative stress and cell death in HT22 neuronal cells. Free Radic. Biol. Med. 2010, 48, 821–830. [Google Scholar] [CrossRef]
- Lauridsen, C.; Jensen, S.K. α-Tocopherol incorporation in mitochondria and microsomes upon supranutritional vitamin E supplementation. Genes. Nutr. 2012, 7, 475–482. [Google Scholar] [CrossRef]
- Mendonça, J.D.S.; Guimarães, R.C.A.; Zorgetto-Pinheiro, V.A.; Fernandes, C.D.P.; Marcelino, G.; Bogo, D.; Freitas, K.C.; Hiane, P.A.; de Pádua Melo, E.S.; Vilela, M.L.B.; et al. Natural antioxidant evaluation: A review of detection methods. Molecules 2022, 27, 3563. [Google Scholar] [CrossRef]
- Siluk, D.; Oliveira, R.V.; Esther-Rodriguez-Rosas, M.; Ling, S.; Bos, A.; Ferrucci, L.; Wainer, I.W. A validated liquid chromatography method for the simultaneous determination of vitamins A and E in human plasma. J. Pharm. Biomed. Anal. 2007, 44, 1001–1007. [Google Scholar] [CrossRef]
- Go, Y.M.; Jones, D.P. Thiol/disulfide redox states in signaling and sensing. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 173–181. [Google Scholar] [CrossRef]
- Higashi, Y.; Aratake, T.; Shimizu, T.; Shimizu, S.; Saito, M. Protective role of glutathione in the hippocampus after brain ischemia. Int. J. Mol. Sci. 2021, 22, 7765. [Google Scholar] [CrossRef]
- Tewari, D.; Sah, A.N.; Bawari, S.; Nabavi, S.F.; Dehpour, A.R.; Shirooie, S.; Braidy, N.; Fiebich, B.L.; Vacca, R.A.; Nabavi, S.M. Role of nitric oxide in neurodegeneration: Function, regulation, and inhibition. Curr. Neuropharmacol. 2021, 19, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Contestabile, A.; Monti, B.; Contestabile, A.; Ciani, E. Brain nitric oxide and its dual role in neurodegeneration/neuroprotection: Understanding molecular mechanisms to devise drug approaches. Curr. Med. Chem. 2003, 10, 2147–2174. [Google Scholar] [CrossRef] [PubMed]
- Iova, O.M.; Marin, G.E.; Lazar, I.; Stanescu, I.; Dogaru, G.; Nicula, C.A.; Bulboacă, A.E. Nitric oxide/nitric oxide synthase system in the pathogenesis of neurodegenerative disorders—An overview. Antioxidants 2023, 12, 753. [Google Scholar] [CrossRef] [PubMed]
- Belenichev, I.; Popazova, O.; Bukhtiyarova, N.; Savchenko, D.; Oksenych, V.; Kamyshnyi, O. Modulating nitric oxide: Implications for cytotoxicity and cytoprotection. Antioxidants 2024, 13, 504. [Google Scholar] [CrossRef]
- D’Apolito, E.; Sisalli, M.J.; Tufano, M.; Annunziato, L.; Scorziello, A. Oxidative metabolism in brain ischemia and preconditioning: Two sides of the same coin. Antioxidants 2024, 13, 547. [Google Scholar] [CrossRef]
- Sifat, A.E.; Nozohouri, S.; Archie, S.R.; Chowdhury, E.A.; Abbruscato, T.J. Brain energy metabolism in ischemic stroke: Effects of smoking and diabetes. Int. J. Mol. Sci. 2022, 23, 8512. [Google Scholar] [CrossRef]
- Belenichev, I.F.; Aliyeva, O.G.; Popazova, O.O.; Bukhtiyarova, N.V. Involvement of heat shock proteins HSP70 in the mechanisms of endogenous neuroprotection: The prospect of using HSP70 modulators. Front. Cell. Neurosci. 2023, 17, 1131683. [Google Scholar] [CrossRef]
- Khlyntsevaa, S.V.; Bazel Ya, R.; Vishnikin, A.B.; Andruch, V. Methods for the determination of adenosine triphosphate and other adenine nucleotides. J. Anal. Chem. 2009, 64, 657–673. [Google Scholar] [CrossRef]
- Kawabori, M.; Yenari, M.A. Inflammatory responses in brain ischemia. Curr. Med. Chem. 2015, 22, 1258–1277. [Google Scholar] [CrossRef]
- Kawtharani, S.; Omeis, I. Acute ischemic stroke biomarkers: A new era with diagnostic promise? Acute Med. Surg. 2021, 8, 696. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, Z.; Huang, Z.-X.; Liu, Z. Biomarkers and the outcomes of ischemic stroke. Front. Mol. Neurosci. 2023, 16, 1171101. [Google Scholar] [CrossRef] [PubMed]
- di Biase, L.; Bonura, A.; Pecoraro, P.M.; Carbone, S.P.; Di Lazzaro, V. Unlocking the potential of stroke blood biomarkers: Early diagnosis, ischemic vs. hemorrhagic differentiation and hemorrhagic transformation risk: A comprehensive review. Int. J. Mol. Sci. 2023, 24, 11545. [Google Scholar] [CrossRef] [PubMed]
- Kamyshna, I.I.; Pavlovych, L.B.; Maslyanko, V.A.; Kamyshnyi, A.M. Analysis of the transcriptional activity of genes of neuropeptides and their receptors in the blood of patients with thyroid pathology. J. Med. Life 2021, 14, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Rawat, V.; Goux, W.; Piechaczyk, M.; Mello, S.R. c-Fos protects neurons through a noncanonical mechanism involving HDAC3 interaction: Identification of a 21-amino acid fragment with neuroprotective activity. Mol. Neurobiol. 2016, 53, 1165–1180. [Google Scholar] [CrossRef]
- Krukoff, T.L. c-Fos Expression as a Marker of Functional Activity in the Brain. In Cell Neurobiology Techniques. Neuromethods; Humana Press: Totowa, NJ, USA, 1999; Volume 33. [Google Scholar] [CrossRef]
- Kamyshna, I.I.; Pavlovych, L.B.; Sydorchuk, L.P.; Malyk, I.V.; Kamyshnyi, A.M. BDNF blood serum linkage with BDNF gene polymorphism (rs6265) in thyroid pathology patients in the West-Ukrainian population. Endocr. Regul. 2021, 55, 193–203. [Google Scholar] [CrossRef]
- Pasquin, S.; Sharma, M.; Gauchat, J.F. Ciliary neurotrophic factor (CNTF): New facets of an old molecule for treating neurodegenerative and metabolic syndrome pathologies. Cytokine Growth Factor. Rev. 2015, 26, 507–515. [Google Scholar] [CrossRef]
- Brook, N.; Brook, E.; Dharmarajan, A.; Chan, A.; Dass, C.R. Pigment epithelium-derived factor regulation of neuronal and stem cell fate. Exp. Cell Res. 2020, 389, 111891. [Google Scholar] [CrossRef]
- Creeley, C.E. From Drug-Induced Developmental Neuroapoptosis to Pediatric Anesthetic Neurotoxicity—Where Are We Now? Brain Sci. 2016, 6, 32. [Google Scholar] [CrossRef]
- Chen, D.L.; Engle, J.T.; Griffin, E.A.; Miller, J.P.; Chu, W.; Zhou, D.; Mach, R.H. Imaging caspase-3 activation as a marker of apoptosis-targeted treatment response in cancer. Mol. Imaging Biol. 2015, 17, 384–393. [Google Scholar] [CrossRef]
- Kingham, P.J.; Pocock, J.M. Microglial secreted cathepsin B induces neuronal apoptosis. J. Neurochem. 2001, 76, 1475–1484. [Google Scholar] [CrossRef]
- Shlyakhtina, Y.; Pavet, V.; Gronemeyer, H. Dual role of DR5 in death and survival signaling leads to TRAIL resistance in cancer cells. Cell Death Dis. 2017, 8, 3025. [Google Scholar] [CrossRef] [PubMed]
- Lončarević-Vasiljković, N.; Milanović, D.; Pešić, V.; Tešić, V.; Brkić, M.; Lazić, D.; Avramović, V.; Kanazir, S. Dietary restriction suppresses apoptotic cell death, promotes Bcl-2 and Bcl-xl mRNA expression and increases the Bcl-2/Bax protein ratio in the rat cortex after cortical injury. Neurochem. Int. 2016, 96, 69–76. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Group of Oxidative Stress Reaction Products | Chemical Compounds | Detection Methods |
|---|---|---|
| I. Unstable (Radical Nature) | Alkyl, alkoxyl, peroxyl, nitrite, peroxynitrite radicals | Spontaneous CL, Fe2+ or H2O2-induced CL, EPR |
| II. Stable (Non-Radical Nature): | ||
| (1) Primary | Hydroperoxides, conjugated dienes, endoperoxides, dialkyl peroxides, epoxides | Polarography, iodometry, UV, IR, NMR, HPLC |
| (2) Secondary | Aldehydes (alkanal, alkenal), hydroxyalkenals, malondialdehyde, trienones, 8-isoprostanes, 8-hydroxy-2-deoxyguanosine, o-nitrotyrosine, o-chlorotyrosine, thymidine glycol | UV, HPLC, HPLC/Fluorescence, HPLC/UV, FS, TLC, GC, GC/MS |
| (3) End-Products | Gaseous products (pentane, heptane, etc.), Schiff bases, nitrates, and nitrites | FS, GRP |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belenichev, I.; Bukhtiyarova, N.; Ryzhenko, V.; Makyeyeva, L.; Morozova, O.; Oksenych, V.; Kamyshnyi, O. Methodological Approaches to Experimental Evaluation of Neuroprotective Action of Potential Drugs. Int. J. Mol. Sci. 2024, 25, 10475. https://doi.org/10.3390/ijms251910475
Belenichev I, Bukhtiyarova N, Ryzhenko V, Makyeyeva L, Morozova O, Oksenych V, Kamyshnyi O. Methodological Approaches to Experimental Evaluation of Neuroprotective Action of Potential Drugs. International Journal of Molecular Sciences. 2024; 25(19):10475. https://doi.org/10.3390/ijms251910475
Chicago/Turabian StyleBelenichev, Igor, Nina Bukhtiyarova, Victor Ryzhenko, Lyudmyla Makyeyeva, Oksana Morozova, Valentyn Oksenych, and Oleksandr Kamyshnyi. 2024. "Methodological Approaches to Experimental Evaluation of Neuroprotective Action of Potential Drugs" International Journal of Molecular Sciences 25, no. 19: 10475. https://doi.org/10.3390/ijms251910475