
CAR-T-Cell Therapy for Systemic Lupus Erythematosus: A Comprehensive Overview


Abstract
:1. Introduction
2. Implementation of B-Cell-Targeted Therapies in Lupus
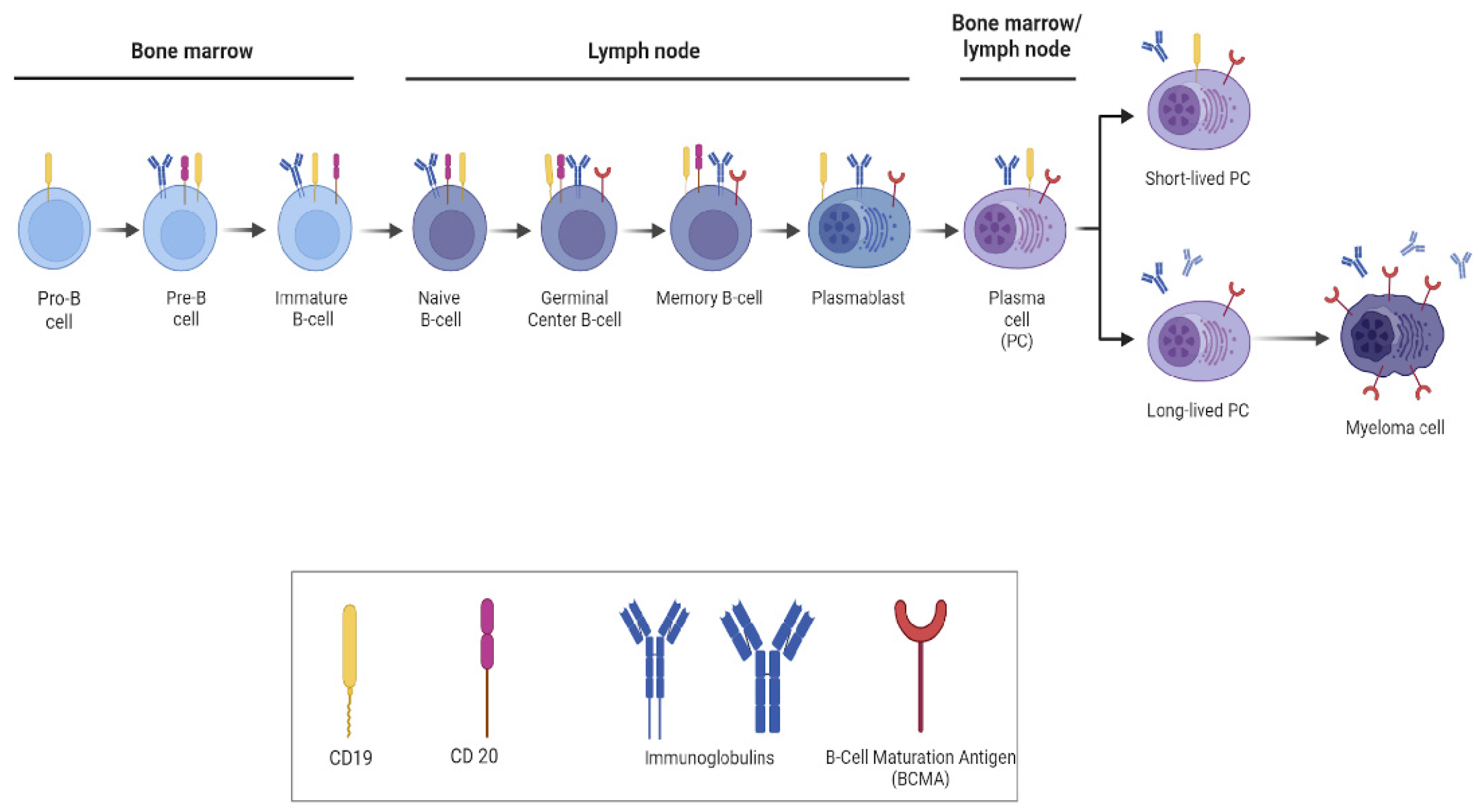
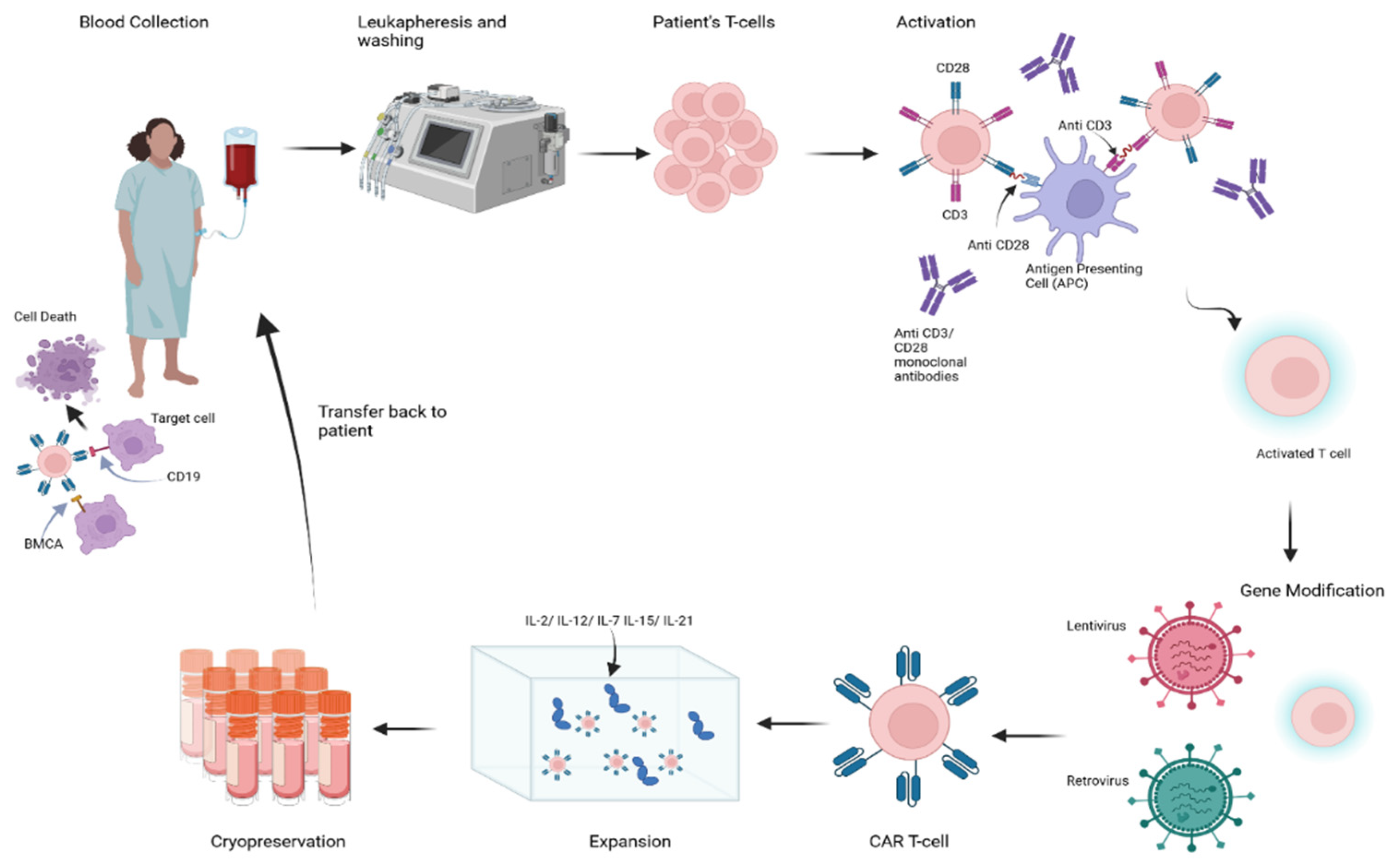
3. Principles of B-Cell Depletion via CAR-T-Cell Therapy
4. Application of CAR-T-Cell Therapy in the Management of SLE
5. CAR-T-Cell Therapy-Related Toxicity
6. Conclusions and Future Directions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fava, A.; Petri, M. Systemic lupus erythematosus: Diagnosis and clinical management. J. Autoimmun. 2019, 96, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Tsokos, G.C. Systemic lupus erythematosus. N. Engl. J. Med. 2011, 365, 2110–2121. [Google Scholar] [CrossRef] [PubMed]
- Alduraibi, F.; Fatima, H.; Hamilton, J.A.; Chatham, W.W.; Hsu, H.C.; Mountz, J.D. Lupus nephritis correlates with B cell interferon-β, anti-Smith, and anti-DNA: A retrospective study. Arthritis Res. Ther. 2022, 24, 87. [Google Scholar] [CrossRef] [PubMed]
- Bernatsky, S.; Boivin, J.F.; Joseph, L.; Manzi, S.; Ginzler, E.; Gladman, D.D.; Urowitz, M.; Fortin, P.R.; Petri, M.; Barr, S.; et al. Mortality in systemic lupus erythematosus. Arthritis Rheum. 2006, 54, 2550–2557. [Google Scholar] [CrossRef]
- Bernal, C.B.; Zamora, L.D.; Navarra, S.V. Biologic therapies in systemic lupus erythematosus. Int. J. Rheum. Dis. 2015, 18, 146–153. [Google Scholar] [CrossRef]
- Zhang, X.; Zhu, L.; Zhang, H.; Chen, S.; Xiao, Y. CAR-T cell therapy in hematological malignancies: Current opportunities and challenges. Front. Immunol. 2022, 13, 927153. [Google Scholar] [CrossRef]
- Lyu, X.; Gupta, L.; Tholouli, E.; Chinoy, H. Chimeric antigen receptor T cell therapy: A new emerging landscape in autoimmune rheumatic diseases. Rheumatology 2024, 63, 1206–1216. [Google Scholar] [CrossRef]
- Mackensen, A.; Müller, F.; Mougiakakos, D.; Böltz, S.; Wilhelm, A.; Aigner, M.; Völkl, S.; Simon, D.; Kleyer, A.; Munoz, L.; et al. Anti-CD19 CAR T cell therapy for refractory systemic lupus erythematosus. Nat. Med. 2022, 28, 2124–2132, Erratum in Nat. Med. 2023, 29, 2956. [Google Scholar] [CrossRef]
- Mougiakakos, D.; Krönke, G.; Völkl, S.; Kretschmann, S.; Aigner, M.; Kharboutli, S.; Böltz, S.; Manger, B.; Mackensen, A.; Schett, G. CD19-targeted CAR T cells in refractory systemic lupus erythematosus. N. Engl. J. Med. 2021, 385, 567–569. [Google Scholar] [CrossRef]
- Mueller, F.; Taubmann, J.; Voelkl, S.; Bucci, L.; Bergmann, C.; Aigner, M.; Wilhelm, A.; Rothe, T.; Minopoulou, I.; Knitza, J.; et al. CD19-targeted CAR-T cells in refractory systemic autoimmune diseases: A monocentric experience from the first fifteen patients. Blood 2023, 142, 220. [Google Scholar] [CrossRef]
- Taubmann, J.; Müller, F.; Boeltz, S.; Völkl, S.; Aigner, M.; Kleyer, A.; Minnopoulou, I.; Locatelli, F.; D’Agostino, M.A.; Gary, R.; et al. OP0141 long term safety and efficacy of car-t cell treatment in refractory systemic lupus erythematosus—Data from the first seven patients. Ann. Rheum. Dis. 2023, 82, 93–94. [Google Scholar] [CrossRef]
- Taubmann, J.; Müller, F.; Mutlu, M.Y.; Völkl, S.; Aigner, M.; Bozec, A.; Mackensen, A.; Grieshaber-Bouyer, R.; Schett, G. CD19 chimeric antigen receptor T cell treatment: Unraveling the role of b cells in systemic lupus erythematosus. Arthritis Rheumatol. 2024, 76, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Payne, A.S. Engineering cell therapies for autoimmune diseases: From preclinical to clinical proof of concept. Immune Netw. 2022, 22, e37. [Google Scholar] [CrossRef] [PubMed]
- Nemazee, D. Mechanisms of central tolerance for B cells. Nat. Rev. Immunol. 2017, 17, 281–294. [Google Scholar] [CrossRef]
- Kaminski, D.A.; Wei, C.; Qian, Y.; Rosenberg, A.F.; Sanz, I. Advances in human B cell phenotypic profiling. Front. Immunol. 2012, 3, 302. [Google Scholar] [CrossRef]
- Nashi, E.; Wang, Y.; Diamond, B. The role of B cells in lupus pathogenesis. Int. J. Biochem. Cell Biol. 2010, 42, 543–550. [Google Scholar] [CrossRef]
- Shlomchik, M.J.; Madaio, M.P.; Ni, D.; Trounstein, M.; Huszar, D. The role of B cells in lpr/lpr-induced autoimmunity. J. Exp. Med. 1994, 180, 1295–1306. [Google Scholar] [CrossRef]
- Furie, R.; Petri, M.; Zamani, O.; Cervera, R.; Wallace, D.J.; Tegzová, D.; Sanchez-Guerrero, J.; Schwarting, A.; Merrill, J.T.; Chatham, W.W.; et al. A phase III, randomized, placebo-controlled study of belimumab, a monoclonal antibody that inhibits B lymphocyte stimulator, in patients with systemic lupus erythematosus. Arthritis Rheum. 2011, 63, 3918–3930. [Google Scholar] [CrossRef]
- Furie, R.A.; Aroca, G.; Cascino, M.D.; Garg, J.P.; Rovin, B.H.; Alvarez, A.; Fragoso-Loyo, H.; Zuta-Santillan, E.; Schindler, T.; Brunetta, P.; et al. B-cell depletion with obinutuzumab for the treatment of proliferative lupus nephritis: A randomised, double-blind, placebo-controlled trial. Ann. Rheum. Dis. 2022, 81, 100–107. [Google Scholar] [CrossRef]
- Weiner, G.J. Rituximab: Mechanism of action. Semin. Hematol. 2010, 47, 115–123. [Google Scholar] [CrossRef]
- Forsthuber, T.G.; Cimbora, D.M.; Ratchford, J.N.; Katz, E.; Stüve, O. B cell-based therapies in CNS autoimmunity: Differentiating CD19 and CD20 as therapeutic targets. Ther. Adv. Neurol. Disord. 2018, 11, 1756286418761697. [Google Scholar] [CrossRef] [PubMed]
- Thurlings, R.M.; Teng, O.; Vos, K.; Gerlag, D.M.; Aarden, L.; Stapel, S.O.; Van Laar, J.M.; Tak, P.P.; Wolbink, G.J. Clinical response, pharmacokinetics, development of human anti-chimaeric antibodies, and synovial tissue response to rituximab treatment in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2010, 69, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Anolik, J.H.; Barnard, J.; Owen, T.; Zheng, B.; Kemshetti, S.; Looney, R.J.; Sanz, I. Delayed memory B cell recovery in peripheral blood and lymphoid tissue in systemic lupus erythematosus after B cell depletion therapy. Arthritis Rheum. 2007, 56, 3044–3056. [Google Scholar] [CrossRef] [PubMed]
- Kamburova, E.G.; Koenen, H.J.; Borgman, K.J.; Ten Berge, I.J.; Joosten, I.; Hilbrands, L.B. A single dose of rituximab does not deplete B cells in secondary lymphoid organs but alters phenotype and function. Am. J. Transplant. 2013, 13, 1503–1511. [Google Scholar] [CrossRef]
- Merrill, J.T.; Neuwelt, C.M.; Wallace, D.J.; Shanahan, J.C.; Latinis, K.M.; Oates, J.C.; Utset, T.O.; Gordon, C.; Isenberg, D.A.; Hsieh, H.J.; et al. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: The randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum. 2010, 62, 222–233. [Google Scholar] [CrossRef]
- Rovin, B.H.; Furie, R.; Latinis, K.; Looney, R.J.; Fervenza, F.C.; Sanchez-Guerrero, J.; Maciuca, R.; Zhang, D.; Garg, J.P.; Brunetta, P.; et al. Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: The lupus nephritis assessment with rituximab study. Arthritis Rheum. 2012, 64, 1215–1226. [Google Scholar] [CrossRef]
- Mysler, E.F.; Spindler, A.J.; Guzman, R.; Bijl, M.; Jayne, D.; Furie, R.A.; Houssiau, F.A.; Drappa, J.; Close, D.; Maciuca, R.; et al. Efficacy and safety of ocrelizumab in active proliferative lupus nephritis: Results from a randomized, double-blind, phase III study. Arthritis Rheum. 2013, 65, 2368–2379. [Google Scholar] [CrossRef]
- Arnold, J.; Dass, S.; Twigg, S.; Jones, C.H.; Rhodes, B.; Hewins, P.; Chakravorty, M.; Courtney, P.; Ehrenstein, M.; Yusof, M.Y.M.; et al. Efficacy and safety of obinutuzumab in systemic lupus erythematosus patients with secondary non-response to rituximab. Rheumatology 2022, 61, 4905–4909. [Google Scholar] [CrossRef]
- Dörner, T.; Kaufmann, J.; Wegener, W.A.; Teoh, N.; Goldenberg, D.M.; Burmester, G.R. Initial clinical trial of epratuzumab (humanized anti-CD22 antibody) for immunotherapy of systemic lupus erythematosus. Arthritis Res. Ther. 2006, 8, R74, Erratum in Arthritis Res. Ther. 2008, 10, 406. [Google Scholar] [CrossRef]
- Clowse, M.E.; Wallace, D.J.; Furie, R.A.; Petri, M.A.; Pike, M.C.; Leszczyński, P.; Neuwelt, C.M.; Hobbs, K.; Keiserman, M.; Duca, L.; et al. Efficacy and safety of epratuzumab in moderately to severely active systemic lupus erythematosus: Results from two phase III randomized, double-blind, placebo-controlled trials. Arthritis Rheumatol. 2017, 69, 362–375. [Google Scholar] [CrossRef]
- Dubey, A.K.; Handu, S.S.; Dubey, S.; Sharma, P.; Sharma, K.K.; Ahmed, Q.M. Belimumab: First targeted biological treatment for systemic lupus erythematosus. J. Pharmacol. Pharmacother. 2011, 2, 317–319. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.J.; Stohl, W.; Furie, R.A.; Lisse, J.R.; McKay, J.D.; Merrill, J.T.; Petri, M.A.; Ginzler, E.M.; Chatham, W.W.; McCune, W.J.; et al. A phase II, randomized, double-blind, placebo-controlled, dose-ranging study of belimumab in patients with active systemic lupus erythematosus. Arthritis Rheum. 2009, 61, 1168–1178. [Google Scholar] [CrossRef] [PubMed]
- Manzi, S.; Sánchez-Guerrero, J.; Merrill, J.T.; Furie, R.; Gladman, D.; Navarra, S.V.; Ginzler, E.M.; D’Cruz, D.P.; Doria, A.; Cooper, S.; et al. Effects of belimumab, a B lymphocyte stimulator-specific inhibitor, on disease activity across multiple organ domains in patients with systemic lupus erythematosus: Combined results from two phase III trials. Ann. Rheum. Dis. 2012, 71, 1833–1838. [Google Scholar] [CrossRef] [PubMed]
- Furie, R.; Rovin, B.H.; Houssiau, F.; Malvar, A.; Teng, Y.K.O.; Contreras, G.; Amoura, Z.; Yu, X.; Mok, C.C.; Santiago, M.B.; et al. Two-year, randomized, controlled trial of belimumab in lupus nephritis. N. Engl. J. Med. 2020, 383, 1117–1128. [Google Scholar] [CrossRef]
- Merrill, J.T.; Van Vollenhoven, R.F.; Buyon, J.P.; Furie, R.A.; Stohl, W.; Morgan-Cox, M.; Dickson, C.; Anderson, P.W.; Lee, C.; Berclaz, P.Y.; et al. Efficacy and safety of subcutaneous tabalumab, a monoclonal antibody to B-cell activating factor, in patients with systemic lupus erythematosus: Results from ILLUMINATE-2, a 52-week, phase III, multicentre, randomised, double-blind, placebo-controlled study. Ann. Rheum. Dis. 2016, 75, 332–340. [Google Scholar] [CrossRef]
- Furie, R.A.; Leon, G.; Thomas, M.; Petri, M.A.; Chu, A.D.; Hislop, C.; Martin, R.S.; Scheinberg, M.A. A phase 2, randomised, placebo-controlled clinical trial of blisibimod, an inhibitor of B cell activating factor, in patients with moderate-to-severe systemic lupus erythematosus, the PEARL-SC study. Ann. Rheum. Dis. 2015, 74, 1667–1675. [Google Scholar] [CrossRef]
- Merrill, J.T.; Shanahan, W.R.; Scheinberg, M.; Kalunian, K.C.; Wofsy, D.; Martin, R.S. Phase III trial results with blisibimod, a selective inhibitor of B-cell activating factor, in subjects with systemic lupus erythematosus (SLE): Results from a randomised, double-blind, placebo-controlled trial. Ann. Rheum. Dis. 2018, 77, 883–889. [Google Scholar] [CrossRef]
- Petri, M.A.; Martin, R.S.; Scheinberg, M.A.; Furie, R.A. Assessments of fatigue and disease activity in patients with systemic lupus erythematosus enrolled in the phase 2 clinical trial with blisibimod. Lupus 2017, 26, 27–37. [Google Scholar] [CrossRef]
- Isenberg, D.; Gordon, C.; Licu, D.; Copt, S.; Rossi, C.P.; Wofsy, D. Efficacy and safety of atacicept for prevention of flares in patients with moderate-to-severe systemic lupus erythematosus (SLE): 52-week data (APRIL-SLE randomised trial). Ann. Rheum. Dis. 2015, 74, 2006–2015, Erratum in Ann. Rheum. Dis. 2016, 75, 946. [Google Scholar] [CrossRef]
- Merrill, J.T.; Wallace, D.J.; Wax, S.; Kao, A.; Fraser, P.A.; Chang, P.; Isenberg, D. Efficacy and safety of atacicept in patients with systemic lupus erythematosus: Results of a twenty-four-week, multicenter, randomized, double-blind, placebo-controlled, parallel-arm, phase IIb study. Arthritis Rheumatol. 2018, 70, 266–276, Erratum in Arthritis Rheumatol. 2018, 70, 467; Erratum in Arthritis Rheumatol. 2021, 73, 2043. [Google Scholar] [CrossRef]
- Wallace, D.J.; Isenberg, D.A.; Morand, E.F.; Vazquez-Mateo, C.; Kao, A.H.; Aydemir, A.; Pudota, K.; Ona, V.; Aranow, C.; Merrill, J.T. Safety and clinical activity of atacicept in the long-term extension of the phase 2b ADDRESS II study in systemic lupus erythematosus. Rheumatology 2021, 60, 5379–5389. [Google Scholar] [CrossRef] [PubMed]
- Radic, M.; Neeli, I.; Marion, T. Prospects for CAR T cell immunotherapy in autoimmune diseases: Clues from Lupus. Expert Opin. Biol. Ther. 2022, 22, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Jayne, D.; Passweg, J.; Marmont, A.; Farge, D.; Zhao, X.; Arnold, R.; Hiepe, F.; Lisukov, I.; Musso, M.; Ou-Yang, J.; et al. Autologous stem cell transplantation for systemic lupus erythematosus. Lupus 2004, 13, 168–176. [Google Scholar] [CrossRef] [PubMed]
- De Buys, P.; Khanna, D.; Furst, D.E. Hemopoietic stem cell transplantation in rheumatic diseases—An update. Autoimmun. Rev. 2005, 4, 442–449. [Google Scholar] [CrossRef]
- Schett, G.; Mackensen, A.; Mougiakakos, D. CAR T-cell therapy in autoimmune diseases. Lancet 2023, 402, 2034–2044. [Google Scholar] [CrossRef]
- Jayaraman, J.; Mellody, M.P.; Hou, A.J.; Desai, R.P.; Fung, A.W.; Pham, A.H.T.; Chen, Y.Y.; Zhao, W. CAR-T design: Elements and their synergistic function. EBioMedicine 2020, 58, 102931. [Google Scholar] [CrossRef]
- Chmielewski, M.; Hombach, A.; Heuser, C.; Adams, G.P.; Abken, H. T cell activation by antibody-like immunoreceptors: Increase in affinity of the single-chain fragment domain above threshold does not increase T cell activation against antigen-positive target cells but decreases selectivity. J. Immunol. 2004, 173, 7647–7653. [Google Scholar] [CrossRef]
- Liu, X.; Jiang, S.; Fang, C.; Yang, S.; Olalere, D.; Pequignot, E.C.; Cogdill, A.P.; Li, N.; Ramones, M.; Granda, B.; et al. Affinity-tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Res. 2015, 75, 3596–3607. [Google Scholar] [CrossRef]
- Dejenie, T.A.; Tiruneh, G.M.M.; Terefe, G.D.; Admasu, F.T.; Tesega, W.W.; Abebe, E.C. Current updates on generations, approvals, and clinical trials of CAR T-cell therapy. Hum. Vaccin. Immunother. 2022, 18, 2114254. [Google Scholar] [CrossRef]
- Brocker, T. Chimeric Fv-zeta or Fv-epsilon receptors are not sufficient to induce activation or cytokine production in peripheral T cells. Blood 2000, 96, 1999–2001. [Google Scholar] [CrossRef]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Acuto, O.; Michel, F. CD28-mediated co-stimulation: A quantitative support for TCR signalling. Nat. Rev. Immunol. 2003, 3, 939–951. [Google Scholar] [CrossRef] [PubMed]
- Hombach, A.; Wieczarkowiecz, A.; Marquardt, T.; Heuser, C.; Usai, L.; Pohl, C.; Seliger, B.; Abken, H. Tumor-specific T cell activation by recombinant immunoreceptors: CD3 zeta signaling and CD28 costimulation are simultaneously required for efficient IL-2 secretion and can be integrated into one combined CD28/CD3 zeta signaling receptor molecule. J. Immunol. 2001, 167, 6123–6131, Erratum in J. Immunol. 2004, 173, 695. [Google Scholar] [CrossRef] [PubMed]
- Imai, C.; Mihara, K.; Andreansky, M.; Nicholson, I.C.; Pui, C.H.; Geiger, T.L.; Campana, D. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 2004, 18, 676–684. [Google Scholar] [CrossRef]
- Jenkins, M.K.; Burrell, E.; Ashwell, J.D. Antigen presentation by resting B cells. Effectiveness at inducing T cell proliferation is determined by costimulatory signals, not T cell receptor occupancy. J. Immunol. 1990, 144, 1585–1590. [Google Scholar] [CrossRef]
- Maher, J.; Brentjens, R.J.; Gunset, G.; Rivière, I.; Sadelain, M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta /CD28 receptor. Nat. Biotechnol. 2002, 20, 70–75. [Google Scholar] [CrossRef]
- Ramos, C.A.; Rouce, R.; Robertson, C.S.; Reyna, A.; Narala, N.; Vyas, G.; Mehta, B.; Zhang, H.; Dakhova, O.; Carrum, G.; et al. In vivo fate and activity of second- versus third-generation CD19-specific CAR-T cells in B cell non-Hodgkin’s lymphomas. Mol. Ther. 2018, 26, 2727–2737. [Google Scholar] [CrossRef]
- Sadelain, M.; Brentjens, R.; Rivière, I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef]
- Schubert, M.L.; Schmitt, A.; Neuber, B.; Hückelhoven-Krauss, A.; Kunz, A.; Wang, L.; Gern, U.; Michels, B.; Sellner, L.; Hofmann, S.; et al. Third-generation CAR T cells targeting CD19 are associated with an excellent safety profile and might improve persistence of CAR T cells in treated patients. Blood 2019, 134, 51. [Google Scholar] [CrossRef]
- Chmielewski, M.; Abken, H. TRUCKs: The fourth generation of CARs. Expert Opin. Biol. Ther. 2015, 15, 1145–1154. [Google Scholar] [CrossRef]
- Vormittag, P.; Gunn, R.; Ghorashian, S.; Veraitch, F.S. A guide to manufacturing CAR T cell therapies. Curr. Opin. Biotechnol. 2018, 53, 164–181. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; An, L.; Huang, R.; Xiong, J.; Yang, H.; Wang, X.; Zhang, X. Strategies to enhance CAR-T persistence. Biomark. Res. 2022, 10, 86. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, R.; Graham, C.; Yallop, D.; Jozwik, A.; Mirci-Danicar, O.C.; Lucchini, G.; Pinner, D.; Jain, N.; Kantarjian, H.; Boissel, N.; et al. Genome-edited, donor-derived allogeneic anti-CD19 chimeric antigen receptor T cells in paediatric and adult B-cell acute lymphoblastic leukaemia: Results of two phase 1 studies. Lancet 2020, 396, 1885–1894. [Google Scholar] [CrossRef] [PubMed]
- Chong, E.A.; Ruella, M.; Schuster, S.J. Five-year outcomes for refractory B-cell lymphomas with CAR T-cell therapy. N. Engl. J. Med. 2021, 384, 673–674. [Google Scholar] [CrossRef]
- DiNofia, A.M.; Grupp, S.A. Will allogeneic CAR T cells for CD19+ malignancies take autologous CAR T cells ‘off the shelf’? Nat. Rev. Clin. Oncol. 2021, 18, 195–196. [Google Scholar] [CrossRef]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, Ö.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef]
- Levine, B.L.; Miskin, J.; Wonnacott, K.; Keir, C. Global manufacturing of CAR T cell therapy. Mol. Ther. Methods Clin. Dev. 2017, 4, 92–101. [Google Scholar] [CrossRef]
- Lee, D.W.; Gardner, R.; Porter, D.L.; Louis, C.U.; Ahmed, N.; Jensen, M.; Grupp, S.A.; Mackall, C.L. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014, 124, 188–195, Erratum in Blood 2015, 126, 1048; Erratum in Blood 2016, 128, 1533. [Google Scholar] [CrossRef]
- Wang, X.; Rivière, I. Clinical manufacturing of CAR T cells: Foundation of a promising therapy. Mol. Ther. Oncolytics 2016, 3, 16015. [Google Scholar] [CrossRef]
- Poorebrahim, M.; Sadeghi, S.; Fakhr, E.; Abazari, M.F.; Poortahmasebi, V.; Kheirollahi, A.; Askari, H.; Rajabzadeh, A.; Rastegarpanah, M.; Linē, A.; et al. Production of CAR T-cells by GMP-grade lentiviral vectors: Latest advances and future prospects. Crit. Rev. Clin. Lab. Sci. 2019, 56, 393–419. [Google Scholar] [CrossRef]
- Akhavan, D.; Alizadeh, D.; Wang, D.; Weist, M.R.; Shepphird, J.K.; Brown, C.E. CAR T cells for brain tumors: Lessons learned and road ahead. Immunol. Rev. 2019, 290, 60–84. [Google Scholar] [CrossRef] [PubMed]
- Kambayana, G.; Rini, S.S. Autologous CD19-targeted chimeric antigen receptor (CAR)T-cells as the future of systemic lupus erythematosus treatment. Curr. Rheumatol. Rev. 2023, 19, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571, Erratum in Nat. Med. 2021, 27, 561. [Google Scholar] [CrossRef] [PubMed]
- Ghassemi, S.; Nunez-Cruz, S.; O’Connor, R.S.; Fraietta, J.A.; Patel, P.R.; Scholler, J.; Barrett, D.M.; Lundh, S.M.; Davis, M.M.; Bedoya, F.; et al. Reducing ex vivo culture improves the antileukemic activity of chimeric antigen receptor (CAR) T cells. Cancer Immunol. Res. 2018, 6, 1100–1109. [Google Scholar] [CrossRef]
- Melenhorst, J.J.; Chen, G.M.; Wang, M.; Porter, D.L.; Chen, C.; Collins, M.A.; Gao, P.; Bandyopadhyay, S.; Sun, H.; Zhao, Z.; et al. Decade-long leukaemia remissions with persistence of CD4+ CAR T cells. Nature 2022, 602, 503–509, Erratum in Nature 2022, 612, E22. [Google Scholar] [CrossRef]
- Müller, F.; Taubmann, J.; Bucci, L.; Wilhelm, A.; Bergmann, C.; Völkl, S.; Aigner, M.; Rothe, T.; Minopoulou, I.; Tur, C.; et al. CD19 CAR T-cell therapy in autoimmune disease—A case series with follow-up. N. Engl. J. Med. 2024, 390, 687–700. [Google Scholar] [CrossRef]
- Wang, W.; He, S.; Zhang, W.; Zhang, H.; DeStefano, V.M.; Wada, M.; Pinz, K.; Deener, G.; Shah, D.; Hagag, N.; et al. BCMA-CD19 compound CAR T cells for systemic lupus erythematosus: A phase 1 open-label clinical trial. Ann. Rheum. Dis. 2024. [CrossRef]
- Hernández, J.C.; Barba, P.; Alberich, M.L.; Fischer, O.; Kovacs, B.; Calzascia, T.; Pearson, D.; Garrotte, A.L.J.; Kirsilae, T.; Siegel, R. An open-label, multi-center, phase 1/2 study to assess safety, efficacy and cellular kinetics of YTB323, a rapid manufacturing CAR-T cell therapy targeting CD19 on B cells, for severe refractory systemic lupus erythematosus: Preliminary results. Arthritis Rheumatol. 2023, 75. [Google Scholar] [CrossRef]
- Krickau, T.; Naumann-Bartsch, N.; Aigner, M.; Kharboutli, S.; Kretschmann, S.; Spoerl, S.; Vasova, I.; Völkl, S.; Woelfle, J.; Mackensen, A.; et al. CAR T-cell therapy rescues adolescent with rapidly progressive lupus nephritis from haemodialysis. Lancet 2024, 403, 1627–1630. [Google Scholar] [CrossRef]
- Podoll, A.; Furie, R.; Kim, F.; Chou, J.; Sengupta, R.; Bayer, R.; Gutman, J.; Chung, J. First two US patients with lupus nephritis (LN) treated with anti-CD19 chimeric antigen receptor (CAR) T-cell therapy: Preliminary results from the KYSA-1 phase 1, multicenter study of KYV-101. Lupus Sci. Med. 2024, 11, A109. [Google Scholar] [CrossRef]
- Marasco, E.; Bracaglia, C.; Merli, P.; Alvarez, P.; Nicolai, R.; Algeri, M.; Cefalo, M.; Becilli, M.; Benedetti, F.; Locatelli, F. Anti-CD19 CAR-T cell therapy for refractory childhood-onset systemic lupus erythematosus. Lupus Sci. Med. 2024, 11, A113. [Google Scholar] [CrossRef]
- European Alliance of Associations for Rheumatology. Welcome to EULAR’s Abstract Archives. Available online: https://scientific.sparx-ip.net/archiveeular/?c=s&view=1&searchfor (accessed on 24 June 2024).
- Ahuja, A.; Shupe, J.; Dunn, R.; Kashgarian, M.; Kehry, M.R.; Shlomchik, M.J. Depletion of B cells in murine lupus: Efficacy and resistance. J. Immunol. 2007, 179, 3351–3361. [Google Scholar] [CrossRef] [PubMed]
- Bekar, K.W.; Owen, T.; Dunn, R.; Ichikawa, T.; Wang, W.; Wang, R.; Barnard, J.; Brady, S.; Nevarez, S.; Goldman, B.I.; et al. Prolonged effects of short-term anti-CD20 B cell depletion therapy in murine systemic lupus erythematosus. Arthritis Rheum. 2010, 62, 2443–2457. [Google Scholar] [CrossRef]
- Kansal, R.; Richardson, N.; Neeli, I.; Khawaja, S.; Chamberlain, D.; Ghani, M.; Ghani, Q.U.; Balazs, L.; Beranova-Giorgianni, S.; Giorgianni, F.; et al. Sustained B cell depletion by CD19-targeted CAR T cells is a highly effective treatment for murine lupus. Sci. Transl. Med. 2019, 11, eaav1648. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.L.; Hwang, W.T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7, 303ra139. [Google Scholar] [CrossRef]
- Gardner, R.A.; Finney, O.; Annesley, C.; Brakke, H.; Summers, C.; Leger, K.; Bleakley, M.; Brown, C.; Mgebroff, S.; Kelly-Spratt, K.S.; et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood 2017, 129, 3322–3331. [Google Scholar] [CrossRef]
- Dickinson, M.J.; Barba, P.; Jäger, U.; Shah, N.N.; Blaise, D.; Briones, J.; Shune, L.; Boissel, N.; Bondanza, A.; Mariconti, L.; et al. A novel autologous CAR-T therapy, YTB323, with preserved T-cell stemness shows enhanced CAR T-cell efficacy in preclinical and early clinical development. Cancer Discov. 2023, 13, 1982–1997. [Google Scholar] [CrossRef]
- Maude, S.L.; Barrett, D.; Teachey, D.T.; Grupp, S.A. Managing cytokine release syndrome associated with novel T cell-engaging therapies. Cancer J. 2014, 20, 119–122. [Google Scholar] [CrossRef]
- Hay, K.A.; Hanafi, L.A.; Li, D.; Gust, J.; Liles, W.C.; Wurfel, M.M.; López, J.A.; Chen, J.; Chung, D.; Harju-Baker, S.; et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T-cell therapy. Blood 2017, 130, 2295–2306. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517, Erratum in N. Engl. J. Med. 2016, 374, 998. [Google Scholar] [CrossRef]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518, Erratum in N. Engl. J. Med. 2016, 374, 998. [Google Scholar] [CrossRef] [PubMed]
- Davila, M.L.; Riviere, I.; Wang, X.; Bartido, S.; Park, J.; Curran, K.; Chung, S.S.; Stefanski, J.; Borquez-Ojeda, O.; Olszewska, M.; et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med. 2014, 6, 224ra225. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.L.; Ma, C.; O’Connell, R.M.; Mehta, A.; DiLoreto, R.; Heath, J.R.; Baltimore, D. Conversion of danger signals into cytokine signals by hematopoietic stem and progenitor cells for regulation of stress-induced hematopoiesis. Cell Stem Cell 2014, 14, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Scheinberg, P.; Wu, C.O.; Samsel, L.; Nunez, O.; Prince, C.; Ganetzky, R.D.; McCoy, J.P.; Maciejewski, J.P.; Young, N.S. Cytokine signature profiles in acquired aplastic anemia and myelodysplastic syndromes. Haematologica 2011, 96, 602–606. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez Mdel, C.; Bernad, A.; Aracil, M. Interleukin-6 deficiency affects bone marrow stromal precursors, resulting in defective hematopoietic support. Blood 2004, 103, 3349–3354. [Google Scholar] [CrossRef]
- Tie, R.; Li, H.; Cai, S.; Liang, Z.; Shan, W.; Wang, B.; Tan, Y.; Zheng, W.; Huang, H. Interleukin-6 signaling regulates hematopoietic stem cell emergence. Exp. Mol. Med. 2019, 51, 1–12. [Google Scholar] [CrossRef]
- Damoulis, P.D.; Hauschka, P.V. Nitric oxide acts in conjunction with proinflammatory cytokines to promote cell death in osteoblasts. J. Bone Miner. Res. 1997, 12, 412–422. [Google Scholar] [CrossRef]
- Saini, N.K.; Sinha, R.; Singh, P.; Sharma, M.; Pathak, R.; Rathor, N.; Varma-Basil, M.; Bose, M. Mce4A protein of Mycobacterium tuberculosis induces pro inflammatory cytokine response leading to macrophage apoptosis in a TNF-α dependent manner. Microb. Pathog. 2016, 100, 43–50. [Google Scholar] [CrossRef]
- Diorio, C.; Shraim, R.; Myers, R.; Behrens, E.M.; Canna, S.; Bassiri, H.; Aplenc, R.; Burudpakdee, C.; Chen, F.; DiNofia, A.M.; et al. Comprehensive serum proteome profiling of cytokine release syndrome and immune effector cell-associated neurotoxicity syndrome patients with B-cell all receiving CAR T19. Clin. Cancer Res. 2022, 28, 3804–3813. [Google Scholar] [CrossRef]
- Buechner, J.; Grupp, S.A.; Hiramatsu, H.; Teachey, D.T.; Rives, S.; Laetsch, T.W.; Yanik, G.A.; Wood, P.; Awasthi, R.; Yi, L.; et al. Practical guidelines for monitoring and management of coagulopathy following tisagenlecleucel CAR T-cell therapy. Blood Adv. 2021, 5, 593–601. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, Y.; Shan, M.; Zong, X.; Geng, H.; Li, J.; Chen, G.; Yu, L.; Xu, Y.; Li, C.; et al. Cytopenia after chimeric antigen receptor T cell immunotherapy in relapsed or refractory lymphoma. Front. Immunol. 2022, 13, 997589. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Hong, R.; Zhou, L.; Wang, Y.; Lv, Y.; Ni, F.; Zhang, M.; Zhao, H.; Ding, S.; Chang, A.H.; et al. Cytokine profiles are associated with prolonged hematologic toxicities after B-cell maturation antigen targeted chimeric antigen receptor-T-cell therapy. Cytotherapy 2023, 25, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Qi, K.; Yan, Z.; Cheng, H.; Chen, W.; Wang, Y.; Wang, X.; Cao, J.; Zhang, H.; Sang, W.; Zhu, F.; et al. An analysis of cardiac disorders associated with chimeric antigen receptor T cell therapy in 126 patients: A single-centre retrospective study. Front. Oncol. 2021, 11, 691064. [Google Scholar] [CrossRef] [PubMed]
- Brentjens, R.; Yeh, R.; Bernal, Y.; Riviere, I.; Sadelain, M. Treatment of chronic lymphocytic leukemia with genetically targeted autologous T cells: Case report of an unforeseen adverse event in a phase I clinical trial. Mol. Ther. 2010, 18, 666–668. [Google Scholar] [CrossRef]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011, 365, 725–733, Erratum in N. Engl. J. Med. 2016, 374, 998. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Dudley, M.E.; Feldman, S.A.; Wilson, W.H.; Spaner, D.E.; Maric, I.; Stetler-Stevenson, M.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 2012, 119, 2709–2720. [Google Scholar] [CrossRef]
- Kalos, M.; Levine, B.L.; Porter, D.L.; Katz, S.; Grupp, S.A.; Bagg, A.; June, C.H. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl. Med. 2011, 3, 95ra73. [Google Scholar] [CrossRef]
- Pepys, M.B.; Hirschfield, G.M. C-reactive protein: A critical update. J. Clin. Investig. 2003, 111, 1805–1812, Erratum in J. Clin. Investig. 2003, 112, 299. [Google Scholar] [CrossRef]
- U.S. Department of Health and Human Services. Common Terminology Criteria for Adverse Events (CTCAE) Version 5.0. 2017. Available online: https://ctep.cancer.gov/protocoldevelopment/electronic_applications/docs/CTCAE_v5_Quick_Reference_5×7.pdf (accessed on 22 April 2018).
- Mitchell, C.D.; Richards, S.M.; Kinsey, S.E.; Lilleyman, J.; Vora, A.; Eden, T.O. Benefit of dexamethasone compared with prednisolone for childhood acute lymphoblastic leukaemia: Results of the UK medical research council ALL97 randomized trial. Br. J. Haematol. 2005, 129, 734–745. [Google Scholar] [CrossRef]
- Teachey, D.T.; Rheingold, S.R.; Maude, S.L.; Zugmaier, G.; Barrett, D.M.; Seif, A.E.; Nichols, K.E.; Suppa, E.K.; Kalos, M.; Berg, R.A.; et al. Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy. Blood 2013, 121, 5154–5157. [Google Scholar] [CrossRef]
- Alten, R. Tocilizumab: A novel humanized anti-interleukin 6 receptor antibody for the treatment of patients with rheumatoid arthritis. Ther. Adv. Musculoskelet. Dis. 2011, 3, 133–149. [Google Scholar] [CrossRef] [PubMed]
- Flammiger, A.; Fiedler, W.; Bacher, U.; Bokemeyer, C.; Schneider, M.; Binder, M. Critical imbalance of TNF-α and soluble TNF receptor 1 in a patient with macrophage activation syndrome: Potential implications for diagnostics and treatment. Acta Haematol. 2012, 128, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Si, S.; Teachey, D.T. Spotlight on tocilizumab in the treatment of CAR-T-cell-induced cytokine release syndrome: Clinical evidence to date. Ther. Clin. Risk Manag. 2020, 16, 705–714. [Google Scholar] [PubMed]
- Gabay, C.; Lamacchia, C.; Palmer, G. IL-1 pathways in inflammation and human diseases. Nat. Rev. Rheumatol. 2010, 6, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Prahalad, S.; Bove, K.E.; Dickens, D.; Lovell, D.J.; Grom, A.A. Etanercept in the treatment of macrophage activation syndrome. J. Rheumatol. 2001, 28, 2120–2124. [Google Scholar]
- Hayden, P.J.; Roddie, C.; Bader, P.; Basak, G.W.; Bonig, H.; Bonini, C.; Chabannon, C.; Ciceri, F.; Corbacioglu, S.; Ellard, R.; et al. Management of adults and children receiving CAR T-cell therapy: 2021 best practice recommendations of the European society for blood and marrow transplantation (EBMT) and the joint accreditation committee of ISCT and EBMT (JACIE) and the European haematology association (EHA). Ann. Oncol. 2022, 33, 259–275. [Google Scholar] [CrossRef]
- Sheth, V.S.; Gauthier, J. Taming the beast: CRS and ICANS after CAR T-cell therapy for ALL. Bone Marrow Transplant. 2021, 56, 552–566. [Google Scholar] [CrossRef]
- Nellan, A.; McCully, C.M.L.; Garcia, R.C.; Jayaprakash, N.; Widemann, B.C.; Lee, D.W.; Warren, K.E. Improved CNS exposure to tocilizumab after cerebrospinal fluid compared to intravenous administration in rhesus macaques. Blood 2018, 132, 662–666. [Google Scholar] [CrossRef]
- Yakoub-Agha, I.; Chabannon, C.; Bader, P.; Basak, G.W.; Bonig, H.; Ciceri, F.; Corbacioglu, S.; Duarte, R.F.; Einsele, H.; Hudecek, M.; et al. Management of adults and children undergoing chimeric antigen receptor T-cell therapy: Best practice recommendations of the European society for blood and marrow transplantation (EBMT) and the joint accreditation committee of ISCT and EBMT (JACIE). Haematologica 2020, 105, 297–316. [Google Scholar] [CrossRef]
- Santomasso, B.D.; Nastoupil, L.J.; Adkins, S.; Lacchetti, C.; Schneider, B.J.; Anadkat, M.; Atkins, M.B.; Brassil, K.J.; Caterino, J.M.; Chau, I.; et al. Management of immune-related adverse events in patients treated with chimeric antigen receptor T-cell therapy: ASCO guideline. J. Clin. Oncol. 2021, 39, 3978–3992, Erratum in J. Clin. Oncol. 2022, 40, 919. [Google Scholar] [CrossRef]
- Yan, Z.; Cao, J.; Cheng, H.; Qiao, J.; Zhang, H.; Wang, Y.; Shi, M.; Lan, J.; Fei, X.; Jin, L.; et al. A combination of humanised anti-CD19 and anti-BCMA CAR T cells in patients with relapsed or refractory multiple myeloma: A single-arm, phase 2 trial. Lancet Haematol. 2019, 6, e521–e529. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.D.; Smith, M.; Shah, N.N. How I treat refractory CRS and ICANS after CAR T-cell therapy. Blood 2023, 141, 2430–2442. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Hao, H.; Yang, G.; Zhang, Y.; Fu, Y. Immunotherapy with CAR-modified T cells: Toxicities and overcoming strategies. J. Immunol. Res. 2018, 2018, 2386187. [Google Scholar] [CrossRef] [PubMed]
- Zahid, U.; Shaukat, A.A.; Hassan, N.; Anwer, F. Coccidioidomycosis, immunoglobulin deficiency: Safety challenges with CAR T cells therapy for relapsed lymphoma. Immunotherapy 2017, 9, 1061–1066. [Google Scholar] [CrossRef]
- Howard, S.C.; Jones, D.P.; Pui, C.H. The tumor lysis syndrome. N. Engl. J. Med. 2011, 364, 1844–1854, Erratum in N. Engl. J. Med. 2018, 379, 1094. [Google Scholar] [CrossRef]
- Bonifant, C.L.; Jackson, H.J.; Brentjens, R.J.; Curran, K.J. Toxicity and management in CAR T-cell therapy. Mol. Ther. Oncolytics 2016, 3, 16011. [Google Scholar] [CrossRef]
- Lamers, C.H.; Sleijfer, S.; Van Steenbergen, S.; Van Elzakker, P.; Van Krimpen, B.; Groot, C.; Vulto, A.; Den Bakker, M.; Oosterwijk, E.; Debets, R.; et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: Clinical evaluation and management of on-target toxicity. Mol. Ther. 2013, 21, 904–912. [Google Scholar] [CrossRef]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Hombach, A.; Hombach, A.A.; Abken, H. Adoptive immunotherapy with genetically engineered T cells: Modification of the IgG1 Fc ‘spacer’ domain in the extracellular moiety of chimeric antigen receptors avoids ‘off-target’ activation and unintended initiation of an innate immune response. Gene Ther. 2010, 17, 1206–1213. [Google Scholar] [CrossRef]
- Cameron, B.J.; Gerry, A.B.; Dukes, J.; Harper, J.V.; Kannan, V.; Bianchi, F.C.; Grand, F.; Brewer, J.E.; Gupta, M.; Plesa, G.; et al. Identification of a titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci. Transl. Med. 2013, 5, 197ra103. [Google Scholar] [CrossRef]
- Linette, G.P.; Stadtmauer, E.A.; Maus, M.V.; Rapoport, A.P.; Levine, B.L.; Emery, L.; Litzky, L.; Bagg, A.; Carreno, B.M.; Cimino, P.J.; et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 2013, 122, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Curran, K.J.; Pegram, H.J.; Brentjens, R.J. Chimeric antigen receptors for T cell immunotherapy: Current understanding and future directions. J. Gene Med. 2012, 14, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Lamers, C.H.; Willemsen, R.; Van Elzakker, P.; Van Steenbergen-Langeveld, S.; Broertjes, M.; Oosterwijk-Wakka, J.; Oosterwijk, E.; Sleijfer, S.; Debets, R.; Gratama, J.W. Immune responses to transgene and retroviral vector in patients treated with ex vivo-engineered T cells. Blood 2011, 117, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Maus, M.V.; Haas, A.R.; Beatty, G.L.; Albelda, S.M.; Levine, B.L.; Liu, X.; Zhao, Y.; Kalos, M.; June, C.H. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol. Res. 2013, 1, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. Considerations for the Development of Chimeric Antigen Receptor (CAR) T Cell Products. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/considerations-development-chimeric-antigen-receptor-car-t-cell-products (accessed on 3 August 2024).
- Verdun, N.; Marks, P. Secondary cancers after chimeric antigen receptor T-cell therapy. N. Engl. J. Med. 2024, 390, 584–586. [Google Scholar] [CrossRef] [PubMed]
- US Food & Drug Administration. FDA Investigating Serious Risk of T-Cell Malignancy Following BCMA-Directed or CD19-Directed Autologous Chimeric Antigen Receptor (CAR) T Cell Immunotherapies. Available online: https://www.fda.gov/vaccines-blood-biologics/safety-availability-biologics/fda-investigating-serious-risk-t-cell-malignancy-following-bcma-directed-or-cd19-directed-autologous (accessed on 28 November 2023).
- Tokarew, N.; Ogonek, J.; Endres, S.; Von Bergwelt-Baildon, M.; Kobold, S. Teaching an old dog new tricks: Next-generation CAR T cells. Br. J. Cancer 2019, 120, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.L. FDA’s Peter Marks Says Some Secondary Cancer Cases after CAR-T Therapy May Be ‘Causal,’ But Benefits Still Outweigh Risks: #JPM24. Available online: https://endpts.com/jpm24-fdas-peter-marks-says-some-secondary-cancer-cases-after-car-t-therapy-may-be-causal-but-benefits-still-outweigh-risks/ (accessed on 16 June 2024).
- US Food & Drug Administration. FDA Requires Boxed Warning for T Cell Malignancies Following Treatment with BCMA-Directed or CD19-Directed Autologous Chimeric Antigen Receptor (CAR) T Cell Immunotherapies. Available online: https://www.fda.gov/vaccines-blood-biologics/safety-availability-biologics/fda-requires-boxed-warning-t-cell-malignancies-following-treatment-bcma-directed-or-cd19-directed (accessed on 14 June 2024).
- Levine, B.L.; Pasquini, M.C.; Connolly, J.E.; Porter, D.L.; Gustafson, M.P.; Boelens, J.J.; Horwitz, E.M.; Grupp, S.A.; Maus, M.V.; Locke, F.L.; et al. Unanswered questions following reports of secondary malignancies after CAR-T cell therapy. Nat. Med. 2024, 30, 338–341. [Google Scholar] [CrossRef]
- Harrison, S.J.; Nguyen, T.; Rahman, M.; Er, J.; Li, J.; Li, K.; Lendvai, N.; Schecter, J.M.; Banerjee, A.; Roccia, T.; et al. CAR+ T-cell lymphoma post ciltacabtagene autoleucel therapy for relapsed refractory multiple myeloma. Blood 2023, 142, 6939. [Google Scholar] [CrossRef]
- European Medicines Agency. Pharmacovigilance Risk Assessment Committee (PRAC). Available online: https://www.ema.europa.eu/en/committees/pharmacovigilance-risk-assessment-committee-prac (accessed on 11 April 2024).
- Bouziana, S.; Bouzianas, D. The current landscape of secondary malignancies after CAR T-cell therapies: How could malignancies be prevented? Int. J. Mol. Sci. 2024, 25, 9518. [Google Scholar] [CrossRef]
- US Food & Drug Administration. BCMA-Directed or CD19-Directed Autologous Chimeric Antigen Receptor (CAR) T Cell Immunotherapies: FDA Safety Communication—FDA Investigating Serious Risk of T-Cell Malignancy. Available online: https://www.fda.gov/safety/medical-product-safety-information/bcma-directed-or-cd19-directed-autologous-chimeric-antigen-receptor-car-t-cell-immunotherapies-fda (accessed on 28 November 2023).
- Hu, L. Clinical Development of Chimeric Antigen Receptor (CAR)-T Cell Therapy in Cancer. Available online: https://www.fda.gov/media/167537/download (accessed on 2 December 2022).
- Bansal, R.; Vergidis, P.; Tosh, P.; Wilson, J.W.; Hathcock, M.; Bennani, N.N.; Paludo, J.; Villasboas, J.C.; Wang, Y.; Ansell, S.M.; et al. Vaccine titers in lymphoma patients receiving chimeric antigen receptor T-cell therapy. J. Clin. Oncol. 2021, 39, 7555. [Google Scholar] [CrossRef]
- Rahman, Z.A.; Gannon, N.; Melody, M.; Muniz, P.; Ayala, E.; Roy, V.; Brumble, L.; Ailawadhi, S.; Sher, T.; Foran, J.M.; et al. Impact of anti-CD19 CAR-T axicabtagene ciloleucel on vaccine titers of DTaP and MMR. Blood 2019, 134, 5610. [Google Scholar] [CrossRef]
- Shah, N.; Alarcon, A.; Palazzo, M.; Ruiz, J.D.; Batlevi, C.W.; Dahi, P.B.; Palomba, M.L.; Scordo, M.; Giralt, S.A.; Sauter, C.S. High rates of residual vaccine titers at 1-year post CD19 chimeric antigen receptor T cell therapy. Transplant. Cell. Ther. 2021, 27, S355. [Google Scholar] [CrossRef]
- Walti, C.S.; Krantz, E.M.; Maalouf, J.; Boonyaratanakornkit, J.; Keane-Candib, J.; Joncas-Schronce, L.; Stevens-Ayers, T.; Dasgupta, S.; Taylor, J.J.; Hirayama, A.V.; et al. Antibodies against vaccine-preventable infections after CAR-T cell therapy for B cell malignancies. JCI Insight 2021, 6, e146743. [Google Scholar] [CrossRef] [PubMed]
- Bhoj, V.G.; Arhontoulis, D.; Wertheim, G.; Capobianchi, J.; Callahan, C.A.; Ellebrecht, C.T.; Obstfeld, A.E.; Lacey, S.F.; Melenhorst, J.J.; Nazimuddin, F.; et al. Persistence of long-lived plasma cells and humoral immunity in individuals responding to CD19-directed CAR T-cell therapy. Blood 2016, 128, 360–370. [Google Scholar] [CrossRef]
- Walti, C.S.; Loes, A.N.; Shuey, K.; Krantz, E.M.; Boonyaratanakornkit, J.; Keane-Candib, J.; Loeffelholz, T.; Wolf, C.R.; Taylor, J.J.; Gardner, R.A.; et al. Humoral immunogenicity of the seasonal influenza vaccine before and after CAR-T-cell therapy: A prospective observational study. J. Immunother. Cancer 2021, 9, e003428. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Author | Year | Number of Patients | Age (Years) | Sex | Disease Activity Score | Organ/Tissue Involved | Previous Treatment | CAR-T-Cell Protocol | Response to CAR-T-Cell Therapy | Complications |
|---|---|---|---|---|---|---|---|---|---|---|
| Mougiakakos et al. [9] | 2021 | 1 | 20 | F | SLEDAI-2K score: 16 | Kidney, serous tissues, skin, joints, heart | GCs, HCQ, cyclophosphamide, MMF, tacrolimus, belimumab, RTX | Day −5 to day −3: fludarabine 25 mg/m2/d Day −3: cyclophosphamide 1000 mg/m2/d Day 0: 1.1 × 106 CD19-directed CAR-T cells/kg | -SLEDAI-2K score of 0 after 6 weeks -Proteinuria normalization (<250 mg protein/g creatinine) after 1 month -Complement level normalization in 1 month -Anti-dsDNA seroconversion in 1 month | None |
| Mackensen et al. [8] | 2022 | 5 | 18–22 | F: 4 M: 1 | SLEDAI-2K score: 8–16 | Skin, joints, kidneys, heart, serous tissues, muscle, bone marrow, lungs | GCs, HCQ, MMF, AZA, RTX, cyclophosphamide, tacrolimus, belimumab, MTX, leflunomide | Day −5 to day −3: fludarabine 25 mg/m2/d Day −3: cyclophosphamide 1000 mg/m2/d Day 0: 1.0 × 106 CD19-directed CAR-T cells/kg | -SLEDAI-2K score of 0 in 4/5 patients after 3 months -Proteinuria normalization (<300 mg protein/g creatinine) after 3 months -Complement level normalization -Anti-dsDNA seroconversion | CRS grade 1 in 3/5 patients |
| Müller et al. [10,76] | 2023 2024 | 15 SLE 8/15 | 18–38 | F: 7 M: 1 | SLEDAI-2K score: 9.3 to 16 | Kidney, skin, joints, bone marrow, lungs, heart | GCs, HCQ, MMF, AZA, RTX, cyclophosphamide, tacrolimus, belimumab, MTX, leflunomide, bortezomib, upadacitinib, ustekinumab, lenalidomide, thalidomide, interleukin-2 | Day −5 to day −3: fludarabine 25 mg/m2/d Day −3: cyclophosphamide 1000 mg/m2/d Day 0: 1.0 × 106 CD19-directed CAR-T cells/kg | -SLEDAI-2K score of 0 in 8/8 patients after 3 months -Proteinuria resolution after 3 months -Complement level normalization -Anti-dsDNA seroconversion | CRS grade 1 in 5/8 patients Hypogammaglobulinemia in 3/8 patients Pneumonia (requiring hospitalization) 1/8 patients |
| Wang et al. [77] | 2024 | 13 | 16–58 | F: 10 M: 3 | SLEDAI-2K score: 4° 16 | Kidneys, bone marrow, skin, joints, heart. Patient 1 and Patient 2 had DLBCL | GCs, HCQ, MMF, cyclophosphamide, belimumab, tacrolimus, thalidomide | Patient 1 and Patient 2: cyclophosphamide (0.3 g per m2) and fludarabine (0.03 g per m2) Patient 3-Patient 13: cyclophosphamide (0.3 g per m2) Day 0: 3 × 106 BCMA/CD19-directed CAR-T cells/kg except Patient 11 received a dose of 1.5 × 106 cells/kg | -Complete remission of DLBCL was achieved in Patient 1 and Patient 2 -SLEDAI-2K score of 0 in 11/13 patients -Complement level normalization -Anti-dsDNA seroconversion in 12/13 patients within 3 weeks Proteinuria normalization (<250 mg protein/g creatinine) after 6 months in 8/11 patients with lupus nephritis | CRS grade 1 in 9/13 patients Hypogammaglobulinemia in 10/13 patients |
| Hernández et al. [78] | 2024 | 3 | 38–50 | F 2 M 1 | SLEDAI-2K score: 12 to 22 | Kidneys, joints, skin, pleura, vasculature | N/A | Day −14: lymphodepletion with cyclophosphamide and fludarabine. Day 1: 12.5 × 106 CD19-directed CAR-T cells | -SLEDAI-2K score reduction by 50% at 2 months -Improvement in (PhGA) score, anti-dsDNA levels, proteinuria and complement levels | CRS grade 1 or 2 in 2/3 patients Hypogammaglobulinemia in 2/3 patients CMV reactivation in 1/3 patients |
| Taubmann et al. [12] | 2024 | 1 | 32 | F | SLEDAI-2K score: 10 | Pericardium (effusion), kidneys, bone marrow, brain, skin | GCs, HCQ, MMF, tacrolimus, belimumab, cyclophosphamide, rituximab | Day −5 to day −3: fludarabine 12.5 mg/m2/d Day −3: cyclophosphamide 500 mg/m2/d. Day 0: CAR-T-cell volume not specified | -SLEDAI-2K score of 0 after 25 days -Proteinuria normalization (<300 mg protein/g creatinine) -Anti-dsDNA seroconversion | None |
| Krickau et al. [79] | 2024 | 1 | 15 | F | SLEDAI-2K score: 23 | Skin, joints, kidneys | GCs, HCQ, azathioprine, MMF, belimumab, cyclophosphamide, plasma exchange | Day −5 to day −3: fludarabine 12.5 mg/m2/d Day −3: cyclophosphamide 500 mg/m2/d. Day 0: 1.0 × 106 CAR-T cells/kg | -SLEDAI-2K score of 0 in 6 months -Hemodialysis free after 3 weeks -Complement level normalization in 6 weeks -Anti-dsDNA seroconversion in 6 weeks -Proteinuria decreased from 10,717 mg/g/day to 3400 mg/kg/day after 6 months | CRS grade 1 |
| Podoll et al. [80] | 2024 | 2 | 18, 28 | F | SLEDAI-2K score: 12–19 | Kidneys (class IV lupus nephritis), bone marrow | N/A | Day –7 to –5: -fludarabine (30 mg/m2/ day) -cyclophosphamide (300 mg/m2/day) Day 0: Patient-1: 0.5 × 108 Patient-2: 1.0 × 108 CD19-directed CAR-T cells | -SLEDAI-2K score reduction Patient 1: from 19 to 8 on Day 90 Patient 2: from 12 to 10 on Day 28 -Proteinuria improvement Patient 1: from 1.4 g/g/day to 0.5 g/g/day on Day 90 Patient 2: from 1.3 g/g/day to 0.6 g/g/day on Day 28 -Complement level elevation -Anti-dsDNA level reduction | CRS grade 1 in 2/2 patients |
| Marasco et al. [81] | 2024 | 1 | 15 | F | SLEDAI-2K score: 22 | Kidneys, serous tissues, skin, bone marrow, lungs (PAH). | GCs, HCQ, MMF, RTX, cyclophosphamide. | lymphodepletion: cyclophosphamide (1500 mg/m2). fludarabine (90 mg/m2), −1 × 10⁶ CD19-directed CAR-T cells/kg | -SLEDAI-2K Score: 2 at week 6 -Urinalysis: Normal results at week 3. -Complement levels: normalized at week 6. -ANA and anti-dsDNA titers: significantly decreased at week 8. -Right ventricular systolic pressure and NT-ProBNP levels: Normal at week 2. | -CRS grade 1 -Cytopenia (transient) |
| Abstract # | Title, Author, and Location | ||||||
|---|---|---|---|---|---|---|---|
| POS0054 | An open-label, single-arm, multicenter study to evaluate elmacabtagene autoleucel, the CD-19 directed CAR-T-cell therapy, for active systemic lupus erythematosus in china. Hu et al., China | ||||||
| # of patients | Disease/activity | Age | Sex | CAR-T-cell protocol/method | Previous medications | Response (follow-up duration) | Complications |
| 3 | SLE Organs involved: skin, kidneys, bone marrow, joints | 21–36 years | F | 25 × 106 CAR T cells after lymphodepleting therapy (Cyclophosphamide and Fludarabine) | GCs, HCQ, MMF, tacrolimus, MTX, telitacicept, belimumab. | Clinical and Serological Responses: -SELENA-SLEDAI decreased to 0–1 -SRI-4 achieved in all patients -LLDAS achieved in two patients -Proteinuria improved -Autoantibodies decreased -C3 levels elevated Cellular Response: -Cellular expansion with median peak concentration (C_max) of 19.72 cells/μL between 8–22 days postinfusion. -Complete B-cell depletion was observed reaching the nadir between Day 8–11. | -CRS G1 in 1 patient and G3 in 1 patient -Cytopenia (1 patient) -Infection, MAS and effusion (1 patient) |
| POS0340 | Effects of CAR-T-cell treatment on b-cell immunity in systemic autoimmune diseases. Bucci et al., Germany | ||||||
| 12 | 8 SLE 2 IIM 2 SSc | N/A | N/A | CD19 CAR T-cell therapy | N/A | Clinical and Serological Responses: N/A Cellular response: -Reconstituted B cells had a naïve phenotype, with reduced CD19 + CD27+ memory B cells. -Minimal increase in memory B cells, mostly preswitched IgD+ CD27+. -Plasmablasts and activated CD11c+ memory B cells disappeared in SLE patients. -Increase in immature CD38+ B cells at 4 months, declining later. -Single-cell sequencing showed reduced expression of class-switched heavy chains and disease-associated chains, with increased IGHM and IGHD expression. | N/A |
| OP0027 | Long-term safety and efficacy of CAR-T-cell treatment in severe and refractory autoimmune disease cases. Taubmann et al., Germany | ||||||
| 15 | 8 SLE 4 SSc 3 IIM | 18–60 years | F: 10 M: 5 | 1.0 × 106 CD-19 CAR T cells after lymphodepleting therapy (Cyclophosphamide and Fludarabine) | N/A | Clinical and Serological Responses: SLE: DORIS: remission was achieved in all SLE patients IIM: ACR/EULAR: a major response was achieved in all patients SSc EUSTAR activity index: decreased in all patients. -Drug-free remission achieved in all patients Cellular Response: -N/A | -CRS (G1: 8 patients, G2: 1 patient) -ICANS (grade 1): 1 patient) -Late-stage neutropenia in 1 patient. -Infections (Pneumonia/upper respiratory tract infections.) |
| POS0046 | Preliminary results of an open-label, multicentre, phase 1/2 study to assess the safety, efficacy and cellular kinetics of ytb323 (rapcabtagene autoleucel), a rapidly manufactured CAR-T-cell therapy targeting CD19 on b cells, for severe refractory systemic lupus erythematosus. Cortés-Hernández, et al., Spain | ||||||
| 6 | SLE | N/A | N/A | YTB323 12.5 × 106 CD-19 CAR T cells after lymphodepleting therapy (Cyclophosphamide and Fludarabine) | N/A | Preliminary efficacy data for the first 3 patients showed: Clinical and Serological Responses: -Significant reductions in SLE Disease Activity Index (SLEDAI) and Physician’s Global Assessment (PhGA). -Improvements in disease biomarkers such as autoantibodies, complement levels, and proteinuria. Cellular response: -Peak CAR T-cell expansion 13–21 days postinfusion. -Deep B-cell depletion followed by B-cell recovery. | -CRS (G1 or G2 in 4 patient) -Cytopenia (G3 and G4) in all patients -Hypogammaglobulinemia. -Infection (pneumonia in 1 patient) |
| POS0030 | Safety and preliminary efficacy of CD19 CAR-T-cell treatment in rheumatic disease: data from the first part of the phase i/ii castle basket study (CASTLE study) Schett et al., Germany | ||||||
| 8 (1st part) 16 (2nd part) | 5 SLE 3 SSc 1 IIM | 20–81 years | F: 6 M: 2 | 1.0 × 106 CD-19 CAR T cells/kg body weight after lymphodepleting therapy (Cyclophosphamide and Fludarabine) | N/A | Clinical and Serological Responses: -SLE: DORIS remission achieved in three patients -IIM: ACR moderate/major response achieved in one patient -SSc: lung function maintained in 1 patient. -Drug-free remission achieved in all patients Cellular response: -Complete B-cell depletion in all patients within 10 days. -CAR-T cells expanded in all patients. | -CRS (G1: 4 patients, G2: 1 patient. -Late-stage neutropenia: 2 patients. -Infections (pneumonia, SARS-CoV-2 and CMV) that resolved upon treatment: 2 patients. |
| POS0464 | Serum proteomic analysis identifies markers associated with anti-CD19 CAR-T therapeutic response in autoimmune diseases J. Chou et al., Germany | ||||||
| 8 | 3 SLE 3 diffuse SSc 2 DM Control: 10 HC 7 SLE 7 SSc | N/A | N/A | CD19-CAR T-cell therapy | N/A | Clinical and Serological Responses: N/A Cellular Response: -IgM, IgA, IgE: Significantly reduced at 3 months post-CD19-CAR T-cell infusion. -IgG: No significant change observed. -SLE Baseline: Elevated IFN signaling molecules (CXCL10, MX1). -SSc Baseline: Elevated markers of endothelial dysfunction (VEGF, ANG2). Downregulated Pathways Post-Therapy: HSF-1–mediated heat shock response (HSPA1A, DNAJA4), type I IFN signaling (IFIT3, ISG15). -Reduction in autoantigen PUF60, which is related to neutrophil degranulation and IL12 signaling. | N/A |
| POS1325 | Anti-CD19 CAR-T-cell therapy for refractory childhood-onset systemic lupus erythematosus Bracaglia et al., Italy | ||||||
| 2 | Childhood-Onset SLE Organs involved: Kidneys, lungs, heart, CNS | 15 and 17 years | F | 1 × 106 cells/kg body weight CD-19 CAR T cells | Patient 1: GC, MMF, RTX, CYC. Patient 2: GC, MMF, CYC pulses, plasmapheresis | Patient 1: Clinical and Serological Responses: -Pulmonary hypertension improved. -C3 and C4 normalized by week 6, -Proteinuria normalized by week 4. -Renal biopsy at month 6 showed no glomerular deposits. -SLEDAI-2K normalized at month 3 with sustained drug-free remission at month 6. Cellular response: -Peak CAR-T-Cell expansion on day 12 (52.4 cells/μL). -Complete B-cell Depletion by day 7. -B-cell recovery occurred at 4 months without SLE flare. Patient 2: Clinical and Serological Responses: -Normal C3 and C4. -Markedly decreasing proteinuria -Off immunosuppression. Cellular response: -N/A | Patient 1: -CRS (G1) -Transient anemia (G2) -Transient neutropenia (G3) Patient 2: -N/A |
| -What criteria, such as severe organ damage, life-threatening complications, or immune profiling, should be used to determine eligibility for CAR-T-cell therapy in SLE patients? -When should CAR-T-cell therapy be prescribed for SLE patients, particularly for those patients with early disease and poor predicted outcomes or patients with refractory disease or both? -How do different CAR-T-cell constructs targeting CD19 vs. BCMA or both compare in terms of efficacy and safety for treating SLE patients? -What are relative advantages/disadvantages of alternative cell-based B cell depleting strategies such as cd19CAR-NK or bispecific (CD3 × CD19) monoclonal reagents vs cd19CAR-T? -How important is seroconversion, specifically the absolute resolution of all autoantibodies? -What is the risk–benefit ratio regarding the persistence of CAR-T cells and the duration of B-cell depletion? -What are the optimal management strategies for SLE patients receiving CAR-T-cell therapy, including the use of hydroxychloroquine, immunosuppressive agents, and biologics? -In cases of relapse after CAR-T-cell therapy, which treatments should be used? -What type of concomitant immunosuppression is appropriate given the B-cell aplasia induced by CAR-T-cell therapy? -What are the optimal requirements and methods for achieving lymphodepletion, including the extent and intensity of the chemotherapy regimen? -What risk factors, including infections and malignancies, should be assessed before CAR-T-cell therapy is initiated in SLE patients? -How can the risk of malignancy be mitigated when CAR-T-cell therapy is the best option for SLE treatment? -How should the efficacy of vaccination be evaluated, how should vaccination be scheduled for SLE patients receiving CAR-T-cell therapy, and what vaccines are needed before and after treatment? -How should antimicrobial prophylaxis be managed in SLE patients with a history of severe infections receiving CAR-T-cell therapy? -What are the safety profiles of specific CAR-T-cell therapies for high-risk SLE patients? -What neurological side effects could arise from CAR-T-cell therapy in SLE patients, and how can these side effects be monitored and managed? |
| Clinical Trial | Number of Participants | CAR-T-Cell Therapy Target | Title | Study Phase | Location | Status |
|---|---|---|---|---|---|---|
| NCT06106906 | 15 | CD19 | A Clinical Study of CD19 CAR-T in Refractory/Moderate-to-Severe Systemic Lupus Erythematosus | Phase I Phase II | China | Not yet recruiting |
| NCT06340750 | 18 | BAFF-ligand | BAFF CAR-T Cells (LMY-920) for Systemic Lupus Erythematosus | Phase I | N/A | Not yet recruiting |
| NCT0610689 | 15 | CD19 | A Clinical Study of CD19 Universal CAR-γδT Cells in Active Systemic Lupus Erythematosus | Phase I Phase II | China | Recruiting |
| NCT06150651 | 6 | CD19 | Safety of PiggyBac Transposon CAR-T cells Targeting CD-19 in Refractory Lupus. | Phase I | Thailand | Recruiting |
| NCT06428188 | 60 | BCMA CD19 | Sequential CAR-T Cells Targeting BCMA/CD19 in Patients with Relapsed/Refractory Autoimmune Diseases (BAH247) | Phase I Phase II | China | Recruiting |
| NCT06340490 | 24 | CD19 | A Study of RJMty19 in Refractory Systemic Lupus Erythematosus (SLE) | Phase I | China | Not yet recruiting |
| NCT05030779 | 9 | CD19 BCMA | A Study of CD19/BCMA Chimeric Antigen Receptor T Cells Therapy for Patients with Refractory Systemic Lupus Erythematosus | Early phase I | China | Unknown |
| NCT05988216 | 12 | CD19 | Universal CAR-T Cells (BRL-301) in Refractory Systemic Lupus Erythematosus | N/A | China | Recruiting |
| NCT03030976 | 5 | CD19 | A Study of CD19 Redirected Autologous T Cells for CD19 Positive Systemic Lupus Erythematosus (SLE) | Phase I | China | Unknown |
| NCT06350110 | 75 | CD19 BCMA | Fourth-gen CAR-T Cells Targeting BCMA/CD19 for Refractory Systemic Lupus Erythematosus (SLE) (BAH242) | Phase I Phase II | China | Not yet recruiting |
| NCT06347718 | 24 | CD19 | CAR-T Cells in Systemic B-Cell Mediated Autoimmune Disease (CASTLE) | Phase I Phase II | Germany | Recruiting |
| NCT06189157 | 29 | CD19 | MB-CART19.1 in Refractory SLE | Phase I Phase II | Germany | Not yet recruiting |
| NCT05858684 | 18 | CD19 BCMA | Dual Target CAR-T-Cell Treatment for Refractory Systemic Lupus Erythematosus (SLE) Patients | Early phase I | China | Recruiting |
| NCT06153095 | 30 | CD19 CD20 | A Study of IMPT-514 in Active Refractory Systemic Lupus Erythematosus (SLE) | Phase I Phase II | United States | Recruiting |
| NCT06342960 | 32 | CD19 | A Study of Anti-CD19 Chimeric Antigen Receptor T-Cell (CD19 CAR-T) Therapy in Subjects with Refractory Lupus Nephritis (KYSA-3) | Phase I Phase II | Germany | Recruiting |
| NCT06429800 | 26 | CD19 | A Study to Evaluate the Safety and Preliminary Efficacy of ATA3219 in Participants with Lupus Nephritis | Phase I | Unknown | Not yet recruiting |
| NCT05474885 | 15 | CD19 BCMA | BCMA-CD19 cCAR-T-Cell Treatment of Relapsed/Refractory Systemic Lupus Erythematosus (SLE) | Phase I | China | Recruiting |
| NCT05938725 | 32 | CD19 | A Study of Anti-CD19 Chimeric Antigen Receptor T-Cell (CD19 CAR-T) Therapy, in Subjects with Refractory Lupus Nephritis | Phase I Phase II | United States | Recruiting |
| NCT0627742 | 24 | BCMA | Refractory ANCA Associated Vasculitis and Lupus Nephritis Treated With BCMA-targeting CAR-T Cells | N/A | China | Recruiting |
| NCT06373081 | 6 | CD19 CD3E | Anti-CD19-CD3E-CAR-T Cells in Relapsed/Refractory Autoimmune Disease | N/A | China | Recruiting |
| NCT06316791 | 24 | CD19 | Exploratory Clinical Study of CNCT19 Anti CD19 Cell Therapy in the Treatment of Refractory Autoimmune Diseases | Early phase I | China | Recruiting |
| NCT06222853 | 19 | CD19 | Study of Therapeutic Efficacy of Anti-CD19 CAR-T Cells in Children with Refractory Systemic Lupus Erythematosus | Phase I | China | Recruiting |
| NCT05765006 | 24 | CD19 | CD19-CART(Relma-cel) for Moderate to Severe Active Systemic Lupus Erythematosus | Phase I | China | Recruiting |
| NCT05085418 | 9 | CD19 BCMA | A Study of CD19/BCMA Chimeric Antigen Receptor T Cells Therapy for Patients with Refractory Immune Nephritis | Early phase I | China | Recruiting |
| NCT05846347 | 15 | CD19 BCMA | Phase I Clinical Study of GC012F Injection in Treatment of Refractory Systemic Lupus Erythematosus | Phase I | China | Recruiting |
| NCT05859997 | 15 | CD19 | Universal CAR-T Cells (BRL-301) in Relapse or Refractory Autoimmune Diseases | N/A | China | Recruiting |
| NCT06420154 | 9 | CD19 | The Safety and Efficacy of Anti-CD19 CAR-T Cells in Patients with Relapsed/Refractory Autoimmune Diseases | Early phase I | China | Not yet recruiting |
| NCT06297408 | 24 | CD19 | Relma-cel for Moderate to Severe Active Systemic Lupus Erythematosus | Phase I | Unknown | Not yet recruiting |
| NCT06038474 | 30 | BCMA | Descartes-08 for Patients with Systemic Lupus Erythematosus (SLE-001) | Phase II | United States | Recruiting |
| NCT06294236 | 36 | CD19 | Study Evaluating SC291 in Subjects with Severe r/r B-cell Mediated Autoimmune Diseases (GLEAM) | Phase I | United States | Recruiting |
| NCT06462144 | 36 | CD19 CD20 | IMPT-514 in Systemic Lupus Erythematosus, Anca-associated Vasculitis, and Idiopathic Inflammatory Myopathy | Early phase I | China | Not yet recruiting |
| NCT06333483 | 12 | CD19 | A Study of CD19 Targeted CAR-T-Cell Therapy in Patients with Severe, Refractory Systemic Lupus Erythematosus (SLE) (CARLYSE) | Phase I | United Kingdom | Recruiting |
| NCT06249438 | 30 | BCMA CD20 | A Study of C-CAR168 in the Treatment of Autoimmune Diseases Refractory to Standard Therapy (CAR-AID) | Phase I | China | Recruiting |
| NCT05930314 | 12 | CD19 | CNCT19 Cell Injection for Refractory Systemic Lupus Erythematosus | Early phase I | China | Enrolling by invitation |
| NCT06465147 | 12 | CD19 | REACT-01: Reversing Autoimmunity Through Cell Therapy | Phase I | United States | Not yet recruiting |
| NCT05798117 | 24 | CD19 | An Open-label, Study to Assess Safety, Efficacy and Cellular Kinetics of YTB323 in Severe, Refractory Systemic Lupus Erythematosus | Phase I Phase II | United States | Recruiting |
| NCT06121297 | 12 | CD19 | RESET-SLE: A Phase 1/2 Open-Label Study to Evaluate the Safety and Efficacy of CABA-201 in Subjects with Active Systemic Lupus Erythematosus | Phase I Phase II | United States | Recruiting |
| NCT06310811 | 12 | CD19 | Anti-CD19 CAR-T-Cell Therapy in Participants with Moderate to Severe Active Systemic Lupus Erythematosus | N/A | China | Recruiting |
| NCT06375993 | 40 | CD20 | A Phase 1 Study of ADI-001 in Lupus Nephritis | Phase I | Unknown | Not yet recruiting |
| NCT05869955 | 129 | CD19 | A Study of CC-97540, CD-19-Targeted Nex-T CAR-T Cells, in Participants with Severe, Refractory Autoimmune Diseases | Phase I | United States | Recruiting |
| NCT06417398 | 10 | CD19 | Preliminary Clinical Study of UTAA09 Injection in the Treatment of Relapsed/Refractory Autoimmune Diseases | Early phase I | Unknown | Not yet recruiting |
| NCT06361745 | 10 | CD19 | Early Clinical Study of UTAA09 Injection in the Treatment of Relapsed/Refractory Autoimmune Diseases | N/A | China | Recruiting |
| NCT06285279 | 24 | BCMA CD19 | FKC288 in Participants with Autoimmune Kidney Diseases | Phase I | China | Recruiting |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdalhadi, H.M.; Chatham, W.W.; Alduraibi, F.K. CAR-T-Cell Therapy for Systemic Lupus Erythematosus: A Comprehensive Overview. Int. J. Mol. Sci. 2024, 25, 10511. https://doi.org/10.3390/ijms251910511
Abdalhadi HM, Chatham WW, Alduraibi FK. CAR-T-Cell Therapy for Systemic Lupus Erythematosus: A Comprehensive Overview. International Journal of Molecular Sciences. 2024; 25(19):10511. https://doi.org/10.3390/ijms251910511
Chicago/Turabian StyleAbdalhadi, Haneen M., Walter W. Chatham, and Fatima K. Alduraibi. 2024. "CAR-T-Cell Therapy for Systemic Lupus Erythematosus: A Comprehensive Overview" International Journal of Molecular Sciences 25, no. 19: 10511. https://doi.org/10.3390/ijms251910511
APA StyleAbdalhadi, H. M., Chatham, W. W., & Alduraibi, F. K. (2024). CAR-T-Cell Therapy for Systemic Lupus Erythematosus: A Comprehensive Overview. International Journal of Molecular Sciences, 25(19), 10511. https://doi.org/10.3390/ijms251910511