
Behavioral and Amygdala Biochemical Damage Induced by Alternating Mild Stress and Ethanol Intoxication in Adolescent Rats: Reversal by Argan Oil Treatment?
 ,
,
Abstract
:
1. Introduction
2. Results
2.1. Body Weight Gain
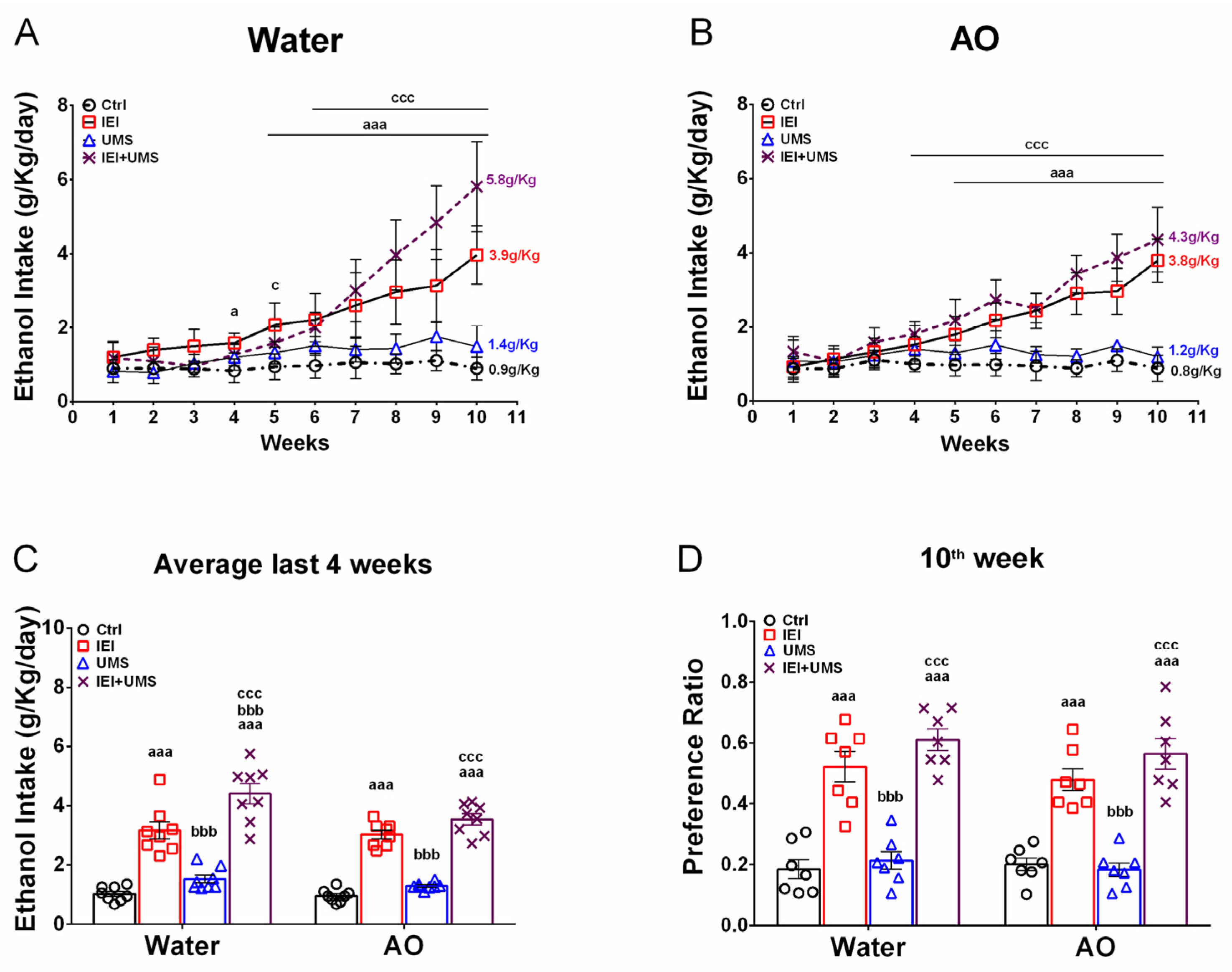
2.2. Home-Cage Voluntary Ethanol Consumption in a 2-Bottle Free Choice Test
2.3. EWS Score after 2–72 H of Abstinence
2.4. Binge-like Drinking in the DID Test
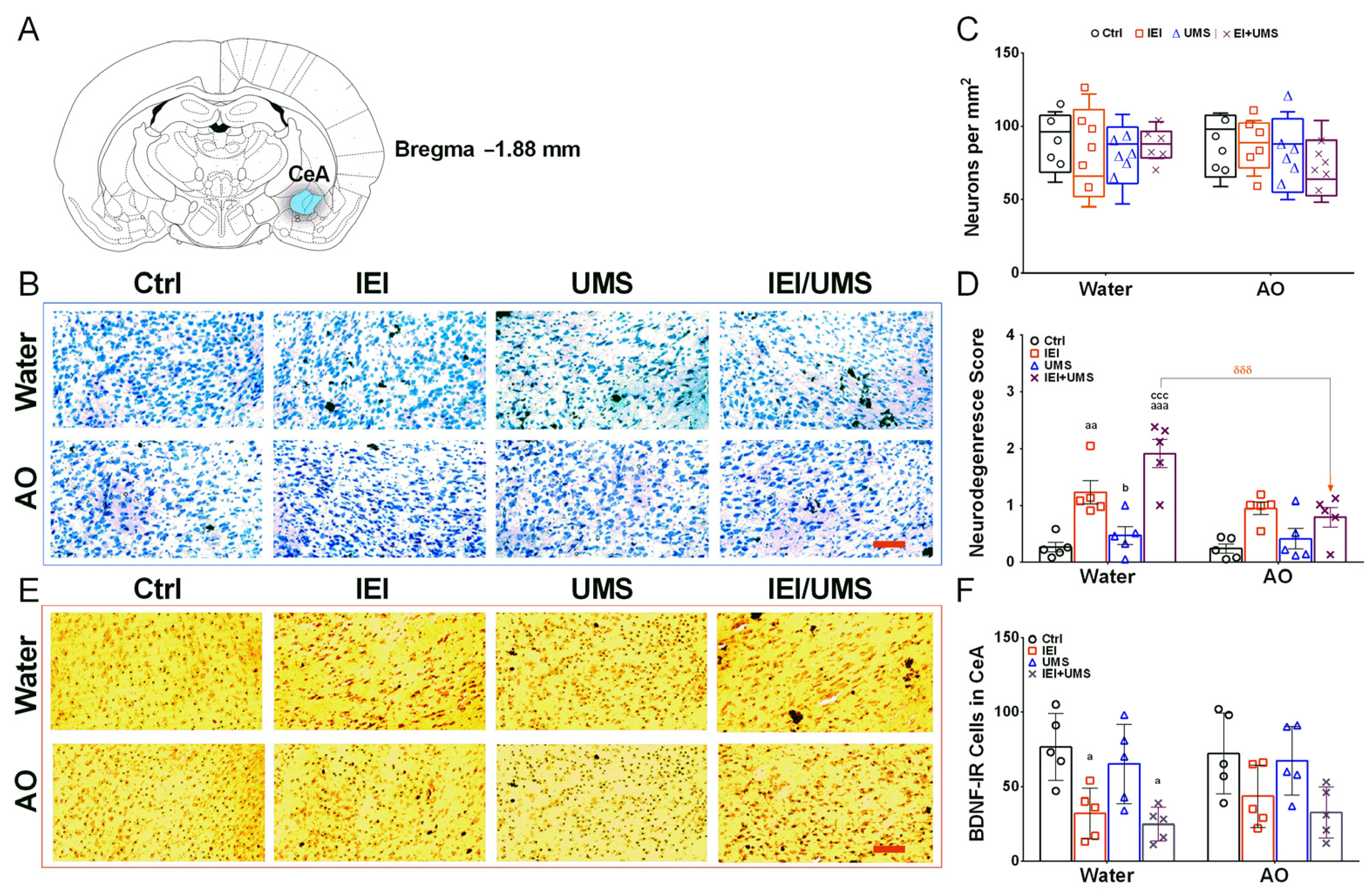
2.5. Neurodegeneration Histological Analysis in CeA
2.6. Immunohistochemistry Analysis of BDNF-IR Cells in CeA
2.7. CORT Levels in Plasma
2.8. Oxidative Stress in Amygdala
2.8.1. MDA Levels
2.8.2. NO Levels
2.8.3. SOD Activity
2.8.4. CAT Activity
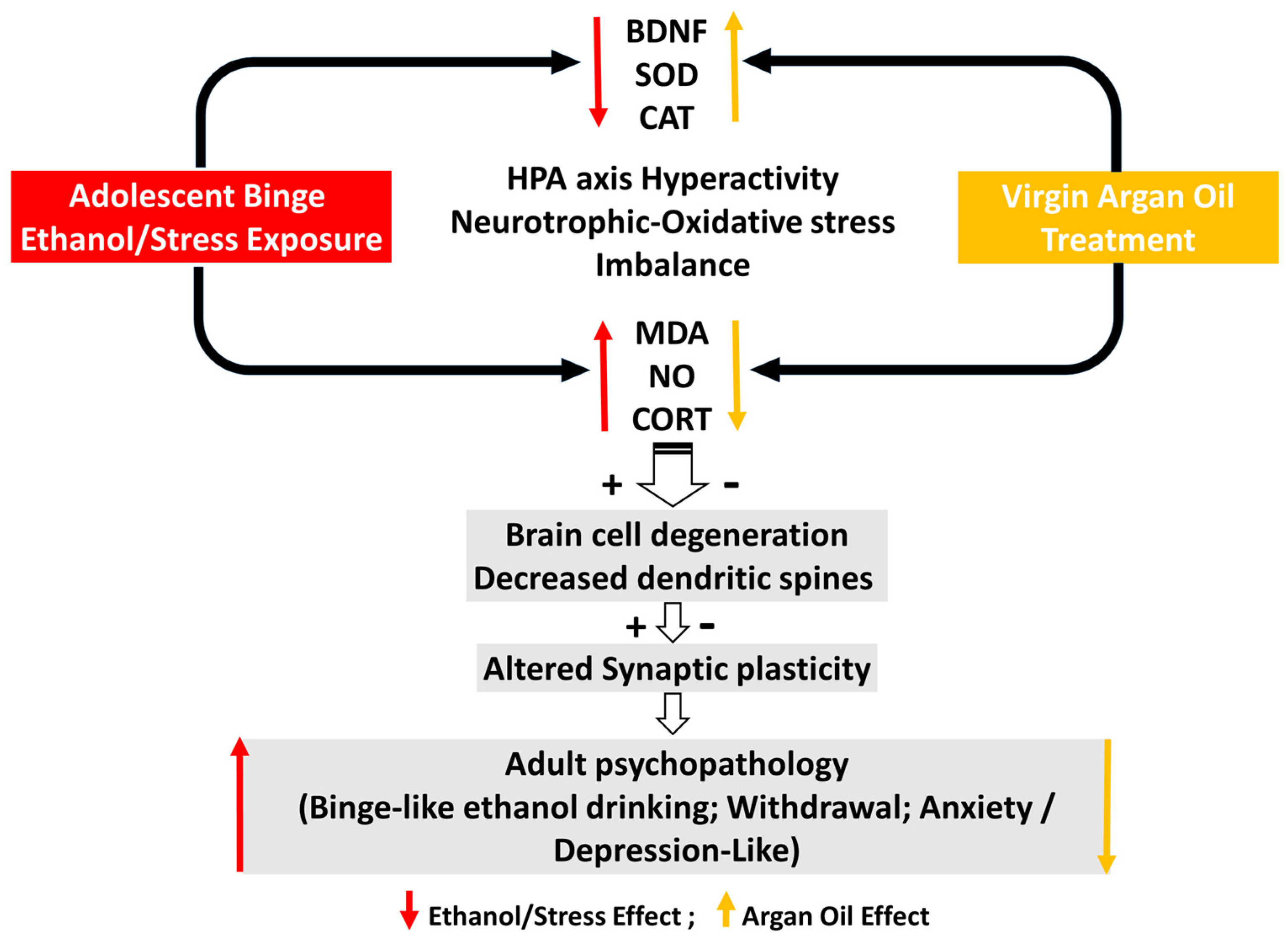
3. Discussion
- 1.1.
- Effectiveness of the forced binge-like ethanol and mild stress alternate exposure during adolescence
- 1.2.
- Effectiveness of AO treatment against binge-like ethanol/mild stress-induced behaviors, biochemical and histochemical disabilities
4. Materials and Methods
4.1. Ethics Statement and Animal Subjects
- Control group (Ctrl): untreated or supplemented with AO (n = 17);
- Ethanol intoxicated group (IEI): submitted to adolescent IEI with/without AO treatment; (n = 18)
- Stressed group (UMS): submitted to adolescent UMS with/without AO treatment (n = 17);
- Ethanol intoxicated and stressed group (IEI/UMS): submitted to adolescent IEI and UMS with/without AO treatment (n = 17).
4.2. Adolescent IEI Procedure
4.3. Adolescent UMS Procedure
4.4. AO Treatment Procedure
4.5. Ethanol Intake in Two-Bottle Intermittent Free Choice Test
4.6. Behavioral Testing
4.7. Histochemical Evaluation
4.7.1. Perfusion, Brain Isolation and Histology
4.7.2. Neurodegeneration in the CeA
4.7.3. BDNF Immunohistochemistry in the CeA
4.7.4. Corticosterone Blood Levels and Oxidative Stress in the Amygdala
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hauser, S.R.; Knight, C.P.; Truitt, W.A.; Waeiss, R.A.; Holt, I.S.; Carvajal, G.B.; Bell, R.L.; Rodd, Z.A. Adolescent Intermittent Ethanol Increases the Sensitivity to the Reinforcing Properties of Ethanol and the Expression of Select Cholinergic and Dopaminergic Genes within the Posterior Ventral Tegmental Area. Alcohol. Clin. Exp. Res. 2019, 43, 1937–1948. [Google Scholar] [CrossRef] [PubMed]
- Towner, T.T.; Varlinskaya, E.I. Adolescent Ethanol Exposure: Anxiety-Like Behavioral Alterations, Ethanol Intake, and Sensitivity. Front. Behav. Neurosci. 2020, 14, 45. [Google Scholar] [CrossRef] [PubMed]
- Kyzar, E.J.; Floreani, C.; Teppen, T.L.; Pandey, S.C. Adolescent Alcohol Exposure: Burden of Epigenetic Reprogramming, Synaptic Remodeling, and Adult Psychopathology. Front. Neurosci. 2016, 10, 222. [Google Scholar] [CrossRef] [PubMed]
- Spear, L.P. Adolescent alcohol exposure: Are there separable vulnerable periods within adolescence? Physiol. Behav. 2015, 148, 122–130. [Google Scholar] [CrossRef]
- Fernandez, G.M.; Stewart, W.N.; Savage, L.M. Chronic Drinking During Adolescence Predisposes the Adult Rat for Continued Heavy Drinking: Neurotrophin and Behavioral Adaptation after Long-Term, Continuous Ethanol Exposure. PLoS ONE 2016, 11, e0149987. [Google Scholar] [CrossRef]
- Campbell, J.C.; Szumlinski, K.K.; Kippin, T.E. Contribution of early environmental stress to alcoholism vulnerability. Alcohol 2009, 43, 547–554. [Google Scholar] [CrossRef]
- Casement, M.D.; Shaw, D.S.; Sitnick, S.L.; Musselman, S.C.; Forbes, E.E. Life stress in adolescence predicts early adult reward-related brain function and alcohol dependence. Soc. Cogn. Affect. Neurosci. 2014, 10, 416–423. [Google Scholar] [CrossRef]
- Gilpin, N.W.; Weiner, J.L. Neurobiology of comorbid post-traumatic stress disorder and alcohol-use disorder. Genes Brain Behav. 2016, 16, 15–43. [Google Scholar] [CrossRef]
- Wille-Bille, A.; de Olmos, S.; Marengo, L.; Chiner, F.; Pautassi, R.M. Long-term ethanol self-administration induces ΔFosB in male and female adolescent, but not in adult, Wistar rats. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2017, 74, 15–30. [Google Scholar] [CrossRef]
- Koob, G.F.; Volkow, N.D. Neurobiology of addiction: A neurocircuitry analysis. Lancet Psychiatry 2016, 3, 760–773. [Google Scholar] [CrossRef]
- Koob, G.F. The dark side of emotion: The addiction perspective. Eur. J. Pharmacol. 2015, 753, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-Y.; Liu, T.-H.; He, Y.; Pan, H.-Q.; Zhang, W.-H.; Yin, X.-P.; Tian, X.-L.; Li, B.-M.; Wang, X.-D.; Holmes, A.; et al. Chronic Stress Remodels Synapses in an Amygdala Circuit–Specific Manner. Biol. Psychiatry 2018, 85, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Koob, G.F.; Volkow, N.D. Neurocircuitry of Addiction. Neuropsychopharmacology 2010, 35, 217–238. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.C.; Zhang, H.; Roy, A.; Misra, K. Central and Medial Amygdaloid Brain-Derived Neurotrophic Factor Signaling Plays a Critical Role in Alcohol-Drinking and Anxiety-Like Behaviors. J. Neurosci. 2006, 26, 8320–8331. [Google Scholar] [CrossRef]
- Flook, E.A.; Luchsinger, J.R.; Silveri, M.M.; Winder, D.G.; Benningfield, M.M.; Blackford, J.U. Anxiety during abstinence from alcohol: A systematic review of rodent and human evidence for the anterior insula’s role in the abstinence network. Addict. Biol. 2020, 26, e12861. [Google Scholar] [CrossRef]
- Silva-Peña, D.; Rivera, P.; Alén, F.; Vargas, A.; Rubio, L.; García-Marchena, N.; Pavón, F.J.; Serrano, A.; de Fonseca, F.R.; Suárez, J. Oleoylethanolamide Modulates BDNF-ERK Signaling and Neurogenesis in the Hippocampi of Rats Exposed to Δ9-THC and Ethanol Binge Drinking During Adolescence. Front. Mol. Neurosci. 2019, 12, 96. [Google Scholar] [CrossRef]
- Notaras, M.; van den Buuse, M. Neurobiology of BDNF in fear memory, sensitivity to stress, and stress-related disorders. Mol. Psychiatry 2020, 25, 2251–2274. [Google Scholar] [CrossRef]
- Li, Y.; Jia, Y.; Wang, D.; Zhuang, X.; Guo, C.; Chu, H.; Zhu, F.; Wang, J.; Wang, X.; Wang, Q.; et al. Programmed cell death 4 as an endogenous suppressor of BDNF translation is involved in stress-induced depression. Mol. Psychiatry 2020, 26, 2316–2333. [Google Scholar] [CrossRef]
- El Mostafi, H.; Elhessni, A.; Touil, T.; Ouichou, A.; Laaziz, A.; Doumar, H.; Mesfioui, A. Argan oil supplementation attenuates voluntary ethanol consumption and withdrawal syndrome promoted by adolescent intermittent ethanol in rat. Alcohol 2020, 87, 39–50. [Google Scholar] [CrossRef]
- El Mostafi, H.; Touil, T.; Abderrahim, L.; Bilal, E.K.; Ali, O.; Hessni, A.; Abdelhalim, M. Argan Oil Supplementation Reverses Anxiety and Depressive-Like Behaviors, Neurodegeneration and Oxidative Stress in Amygdala Induced by Chronic Mild Stress in Rats. J. Depress Anxiety 2018, 7, 319. [Google Scholar] [CrossRef]
- El Abbassi, A.; Khalid, N.; Zbakh, H.; Ahmad, A. Physicochemical Characteristics, Nutritional Properties, and Health Benefits of Argan Oil: A Review. Crit. Rev. Food Sci. Nutr. 2014, 54, 1401–1414. [Google Scholar] [CrossRef]
- López, L.C.; Cabrera-Vique, C.; Venegas, C.; García-Corzo, L.; Luna-Sánchez, M.; Acuña-Castroviejo, D.; Escames, G. Argan Oil-contained Antioxidants for Human Mitochondria. Nat. Prod. Commun. 2013, 8, 47–50. [Google Scholar] [CrossRef] [PubMed]
- Khallouki, F.; Younos, C.; Soulimani, R.; Oster, T.; Charrouf, Z.; Spiegelhalder, B.; Bartsch, H.; Owen, R.W. Consumption of argan oil (Morocco) with its unique profile of fatty acids, tocopherols, squalene, sterols and phenolic compounds should confer valuable cancer chemopreventive effects. Eur. J. Cancer Prev. 2003, 12, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Lizard, G.; Filali-Zegzouti, Y.; El Midaoui, A. Benefits of Argan Oil on Human Health—May 4–6 2017, Errachidia, Morocco. Int. J. Mol. Sci. 2017, 18, 1383. [Google Scholar] [CrossRef] [PubMed]
- El Midaoui, A.; Haddad, Y.; Couture, R. Beneficial effects of argan oil on blood pressure, insulin resistance, and oxidative stress in rat. Nutrition 2016, 32, 1132–1137. [Google Scholar] [CrossRef] [PubMed]
- El Monfalouti, H.; Guillaume, D.; Denhez, C.; Charrouf, Z. Therapeutic potential of argan oil: A review. J. Pharm. Pharmacol. 2010, 62, 1669–1675. [Google Scholar] [CrossRef]
- Ben Menni, H.; Belarbi, M.; Ben Menni, D.; Bendiab, H.; Kherraf, Y.; Ksouri, R.; Djebli, N.; Visioli, F. Anti-inflammatory activity of argan oil and its minor components. Int. J. Food Sci. Nutr. 2019, 71, 307–314. [Google Scholar] [CrossRef]
- Elmostafi, H.; Bahbiti, Y.; Elhessni, A.; Bousalham, R.; Doumar, H.; Ouichou, A.; Benmhammed, H.; Touil, T.; Mesfioui, A. Neuroprotective potential of Argan oil in neuropsychiatric disorders in rats: A review. J. Funct. Foods 2020, 75, 104233. [Google Scholar] [CrossRef]
- El Mostafi, H.; Tariq, T.; Abderrahim, L.; Ali, O.; Aboubaker, E.; Abdelhalim, M. Cognitive Impairments Induced by Adolescent Binge-Like Ethanol in Rat: Neuroprotective Role of Argan Oil. Addict. Res. 2018, 2, 1–12. [Google Scholar] [CrossRef]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates; Hard Cover Edition; Academic Press: Cambridge, MA, USA, 2013; p. 466. [Google Scholar]
- Pascual, M.; Montesinos, J.; Guerri, C. Role of the innate immune system in the neuropathological consequences induced by adolescent binge drinking. J. Neurosci. Res. 2017, 96, 765–780. [Google Scholar] [CrossRef]
- Drevets, W.C. Prefrontal Cortical-Amygdalar Metabolism in Major Depression. Ann. N. Y. Acad. Sci. 1999, 877, 614–637. [Google Scholar] [CrossRef]
- Lee, K.M.; Coehlo, M.; McGregor, H.A.; Waltermire, R.S.; Szumlinski, K.K. Binge alcohol drinking elicits persistent negative affect in mice. Behav. Brain Res. 2015, 291, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Youn, K.; Jeong, W.-S.; Ho, C.-T.; Jun, M. Protective Effects of Red Ginseng Oil against Aβ25–35-Induced Neuronal Apoptosis and Inflammation in PC12 Cells. Int. J. Mol. Sci. 2017, 18, 2218. [Google Scholar] [CrossRef] [PubMed]
- Fernández, M.S.; Fabio, M.C.; Miranda-Morales, R.S.; Virgolini, M.B.; De Giovanni, L.N.; Hansen, C.; Wille-Bille, A.; Nizhnikov, M.E.; Spear, L.P.; Pautassi, R.M. Age-related effects of chronic restraint stress on ethanol drinking, ethanol-induced sedation, and on basal and stress-induced anxiety response. Alcohol 2016, 51, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Driessen, M.; Meier, S.; Hill, A.; Wetterling, T.; Lange, W.; Junghanns, K. The course of anxiety, depression and drinking behaviours after completed detoxification in alcoholics with and without comorbid anxiety and depressive disorders. Alcohol. Alcohol. 2001, 36, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Schuckit, M.A.; Hesseibrock, V. Alcohol Dependence and Anxiety Disorders: What Is the Relationship? Am. J. Psychiatry 1994, 151, 1723–1734. [Google Scholar] [CrossRef]
- Smith, R.J.; Aston-Jones, G. Noradrenergic transmission in the extended amygdala: Role in increased drug-seeking and relapse during protracted drug abstinence. Anat. Embryol. 2008, 213, 43–61. [Google Scholar] [CrossRef]
- Koob, G.F. Addiction is a Reward Deficit and Stress Surfeit Disorder. Front. Psychiatry 2013, 4, 72. [Google Scholar] [CrossRef]
- Silberman, Y.; Bajo, M.; Chappell, A.M.; Christian, D.T.; Cruz, M.; Diaz, M.R.; Kash, T.; Lack, A.K.; Messing, R.O.; Siggins, G.R.; et al. Neurobiological mechanisms contributing to alcohol–stress–anxiety interactions. Alcohol 2009, 43, 509–519. [Google Scholar] [CrossRef]
- Heilig, M.; Koob, G.F. A key role for corticotropin-releasing factor in alcohol dependence. Trends Neurosci. 2007, 30, 399–406. [Google Scholar] [CrossRef]
- Zorrilla, E.P.; Logrip, M.L.; Koob, G.F. Corticotropin releasing factor: A key role in the neurobiology of addiction. Front. Neuroendocr. 2014, 35, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Butler, T.R.; Ariwodola, O.J.; Weiner, J.L. The impact of social isolation on HPA axis function, anxiety-like behaviors, and ethanol drinking. Front. Integr. Neurosci. 2014, 7, 102. [Google Scholar] [CrossRef] [PubMed]
- Valdez, G.R.; Roberts, A.J.; Chan, K.; Davis, H.; Brennan, M.; Zorrilla, E.P.; Koob, G.F. Increased ethanol self-administration and anxiety-like behavior during acute ethanol withdrawal and protracted abstinence: Regulation by corticotropin-releasing factor. Alcohol. Clin. Exp. Res. 2002, 26, 1494–1501. [Google Scholar] [CrossRef] [PubMed]
- Likhtik, E.; Pelletier, J.G.; Paz, R.; Paré, D. Prefrontal Control of the Amygdala. J. Neurosci. 2005, 25, 7429–7437. [Google Scholar] [CrossRef]
- Bukalo, O.; Pinard, C.R.; Silverstein, S.; Brehm, C.; Hartley, N.D.; Whittle, N.; Colacicco, G.; Busch, E.; Patel, S.; Singewald, N.; et al. Prefrontal inputs to the amygdala instruct fear extinction memory formation. Sci. Adv. 2015, 1, e1500251. [Google Scholar] [CrossRef]
- Vyas, A.; Pillai, A.; Chattarji, S. Recovery after chronic stress fails to reverse amygdaloid neuronal hypertrophy and enhanced anxiety-like behavior. Neuroscience 2004, 128, 667–673. [Google Scholar] [CrossRef]
- Saito, M.; Chakraborty, G.; Hui, M.; Masiello, K.; Saito, M. Ethanol-Induced Neurodegeneration and Glial Activation in the Developing Brain. Brain Sci. 2016, 6, 31. [Google Scholar] [CrossRef]
- Zhou, M.; Liu, Z.; Yu, J.; Li, S.; Tang, M.; Zeng, L.; Wang, H.; Xie, H.; Peng, L.; Huang, H.; et al. Quantitative Proteomic Analysis Reveals Synaptic Dysfunction in the Amygdala of Rats Susceptible to Chronic Mild Stress. Neuroscience 2018, 376, 24–39. [Google Scholar] [CrossRef]
- Ernfors, P.; Ibanez, C.F.; Ebendalt, T.; Olsont, L.; Persson, H. Molecular cloning and neurotrophic activities of a protein with structural similarities to nerve growth factor: Developmental and topographical expression in the brain (nerve growth factor family/cDNA/neurotrophic factor/hippocampal neurons/nerve growth factor receptor binding). Proc. Natl. Acad. Sci. USA 1990, 87, 5454–5458. [Google Scholar]
- Xu, H.; Chen, Z.; He, J.; Haimanot, S.; Li, X.; Dyck, L.; Li, X. Synergetic effects of quetiapine and venlafaxine in preventing the chronic restraint stress-induced decrease in cell proliferation and BDNF expression in rat hippocampus. Hippocampus 2006, 16, 551–559. [Google Scholar] [CrossRef]
- Buján, G.E.; Serra, H.A.; Molina, S.J.; Guelman, L.R. Oxidative Stress-Induced Brain Damage Triggered by Voluntary Ethanol Consumption during Adolescence: A Potential Target for Neuroprotection? Curr. Pharm. Des. 2020, 25, 4782–4790. [Google Scholar] [CrossRef] [PubMed]
- Simms, J.A.; Steensland, P.; Medina, B.; Abernathy, K.E.; Chandler, L.J.; Wise, R.; Bartlett, S.E. Intermittent Access to 20% Ethanol Induces High Ethanol Consumption in Long–Evans and Wistar Rats. Alcohol. Clin. Exp. Res. 2008, 32, 1816–1823. [Google Scholar] [CrossRef] [PubMed]
- Hwa, L.S.; Chu, A.; Levinson, S.A.; Kayyali, T.M.; DeBold, J.F.; Miczek, K.A. Persistent Escalation of Alcohol Drinking in C57BL/6J Mice with Intermittent Access to 20% Ethanol. Alcohol. Clin. Exp. Res. 2011, 35, 1938–1947. [Google Scholar] [CrossRef] [PubMed]
- Wiss, D.A. The role of nutrition in addiction recovery: What we know and what we don’t. In The Assessment and Treatment of Addiction: Best Practices and New Frontiers; Elsevier: Amsterdam, The Netherlands, 2018; pp. 21–42. [Google Scholar] [CrossRef]
- Hasin, D.S.; Stinson, F.S.; Ogburn, E.; Grant, B.F. Prevalence, Correlates, Disability, and Comorbidity of DSM-IV Alcohol Abuse and Dependence in the United States Results from the National Epidemiologic Survey on Alcohol and Related Conditions. Arch. Gen. Psychiatry 2007, 64, 830–842. [Google Scholar] [CrossRef]
- Brigadski, T.; Leßmann, V. The physiology of regulated BDNF release. Cell Tissue Res. 2020, 382, 15–45. [Google Scholar] [CrossRef]
- Traber, M. Vitamins C and E: Beneficial effects from a mechanistic perspective. Free Radic Biol. Med. 2011, 51, 1000–1013. Available online: https://www.academia.edu/61328319/Vitamins_C_and_E_Beneficial_effects_from_a_mechanistic_perspective (accessed on 17 September 2024). [CrossRef]
- Wu, A.; Ying, Z.; Gomez-Pinilla, F. Dietary Omega-3 Fatty Acids Normalize BDNF Levels, Reduce Oxidative Damage, and Counteract Learning Disability after Traumatic Brain Injury in Rats. J. Neurotrauma 2004, 21, 1457–1467. [Google Scholar] [CrossRef]
- Naoi, M.; Inaba-Hasegawa, K.; Shamoto-Nagai, M.; Maruyama, W. Neurotrophic function of phytochemicals for neuroprotection in aging and neurodegenerative disorders: Modulation of intracellular signaling and gene expression. J. Neural Transm. 2017, 124, 1515–1527. [Google Scholar] [CrossRef]
- Badreddine, A.; Zarrouk, A.; Karym, E.M.; Debbabi, M.; Nury, T.; Meddeb, W.; Sghaier, R.; Bezine, M.; Vejux, A.; Martine, L.; et al. Argan Oil-Mediated Attenuation of Organelle Dysfunction, Oxidative Stress and Cell Death Induced by 7-Ketocholesterol in Murine Oligodendrocytes 158N. Int. J. Mol. Sci. 2017, 18, 2220. [Google Scholar] [CrossRef]
- Badreddine, A.; Zarrouk, A.; Meddeb, W.; Nury, T.; Rezig, L.; Debbabi, M.; Bessam, F.Z.; Brahmi, F.; Vejux, A.; Mejri, M.; et al. Antioxidant and neuroprotective properties of Mediterranean oil. In Oxidative Stress and Dietary Antioxidants in Neurological Diseases; Elsevier: Amsterdam, The Netherlands, 2020; pp. 143–154. [Google Scholar] [CrossRef]
- Angelova, P.R.; Abramov, A.Y. Role of mitochondrial ROS in the brain: From physiology to neurodegeneration. FEBS Lett. 2018, 592, 692–702. [Google Scholar] [CrossRef]
- Wilkins, H.M.; Weidling, I.W.; Ji, Y.; Swerdlow, R.H. Mitochondria-Derived Damage-Associated Molecular Patterns in Neurodegeneration. Front. Immunol. 2017, 8, 508. [Google Scholar] [CrossRef] [PubMed]
- Zsurka, G.; Kunz, W.S. Mitochondrial involvement in neurodegenerative diseases. IUBMB Life 2013, 65, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Bahbiti, Y.; Ammouri, H.; Berkiks, I.; El Hessni, A.; Ouichou, A.; Nakache, R.; Chakit, M.; Bikjdaouene, L.; Mesfioui, A. Anticonvulsant effect of argan oil on pilocarpine model induced status epilepticus in wistar rats. Nutr. Neurosci. 2016, 21, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Alaux-Cantin, S.; Warnault, V.; Legastelois, R.; Botia, B.; Pierrefiche, O.; Vilpoux, C.; Naassila, M. Alcohol intoxications during adolescence increase motivation for alcohol in adult rats and induce neuroadaptations in the nucleus accumbens. Neuropharmacology 2013, 67, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Pascual, M.; Blanco, A.M.; Cauli, O.; Miñarro, J.; Guerri, C. Intermittent ethanol exposure induces inflammatory brain damage and causes long-term behavioural alterations in adolescent rats. Eur. J. Neurosci. 2007, 25, 541–550. [Google Scholar] [CrossRef]
- Kyzar, E.J.; Zhang, H.; Pandey, S.C. Adolescent Alcohol Exposure Epigenetically Suppresses Amygdala Arc Enhancer RNA Expression to Confer Adult Anxiety Susceptibility. Biol. Psychiatry 2019, 85, 904–914. [Google Scholar] [CrossRef]
- Willner, P. The validity of animal models of depression. Psychopharmacology 1984, 83, 1–16. [Google Scholar] [CrossRef]
- Bousalham, R.; Rhazali, L.J.; Harmouch, A.; Lotfi, H.; Benazzouz, B.; El Hessni, A.; Ouichou, A.; Akhouayri, O.; Mesfioui, A. Does Argan Oil Supplementation Affect Metabolic Parameters and Behavior in Wistar Rats? Food Nutr. Sci. 2015, 6, 816–824. [Google Scholar] [CrossRef]
- Khallouki, F.; Voggel, J.; Breuer, A.; Klika, K.D.; Ulrich, C.M.; Owen, R.W. Comparison of the major polyphenols in mature Argan fruits from two regions of Morocco. Food Chem. 2017, 221, 1034–1040. [Google Scholar] [CrossRef]
- Venegas, C.; Cabrera-Vique, C.; García-Corzo, L.; Escames, G.; Acuña-Castroviejo, D.; López, L.C. Determination of Coenzyme Q10, Coenzyme Q9, and Melatonin Contents in Virgin Argan Oils: Comparison with Other Edible Vegetable Oils. J. Agric. Food Chem. 2011, 59, 12102–12108. [Google Scholar] [CrossRef]
- Erden, B.; Ozdemirci, S.; Yildiran, G.; Utkan, T.; Gacar, N.; Ulak, G. Dextromethorphan Attenuates Ethanol Withdrawal Syndrome in Rats. Pharmacol. Biochem. Behav. 1999, 62, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Giardino, W.; Ryabinin, A.E. CRF1 receptor signaling regulates food and fluid intake in the drinking-in-the-dark model of binge alcohol consumption. Alcohol. Clin. Exp. Res. 2013, 37, 1161–1170. [Google Scholar] [CrossRef] [PubMed]
- Deitch, A.D.; Moses, M.J. The Nissl Substance of Living and Fixed Spinal Ganglion Cells. J. Cell Biol. 1957, 3, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Bankstahl, M.; Breuer, H.; Leiter, I.; Märkel, M.; Bascuñana, P.; Michalski, D.; Bengel, F.M.; Löscher, W.; Meier, M.; Bankstahl, J.P.; et al. Blood–Brain Barrier Leakage during Early Epileptogenesis Is Associated with Rapid Remodeling of the Neurovascular Unit. eNeuro 2018, 5. [Google Scholar] [CrossRef] [PubMed]
- Polascheck, N.; Bankstahl, M.; Löscher, W. The COX-2 inhibitor parecoxib is neuroprotective but not antiepileptogenic in the pilocarpine model of temporal lobe epilepsy. Exp. Neurol. 2010, 224, 219–233. [Google Scholar] [CrossRef]
- Berman, A.K.; Lott, R.B.; Donaldson, S.T. Periodic maternal deprivation may modulate offspring anxiety-like behavior through mechanisms involving neuroplasticity in the amygdala. Brain Res. Bull. 2013, 101, 7–11. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conditions | Water | AO | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Ctrl | IEI | UMS | IEI/UMS | Ctrl | IEI | UMS | IEI/UMS | ||
| CORT in plasma (ng/mL) | 90.34 ± 14.89 | 140.21 ± 9.56 a | 105.83 ± 11.09 | 147.23 ± 5.74 aa | 84.34 ± 12.11 | 108.81 ± 6.27 | 106.23 ± 9.77 | 127.72 ± 10.07 | |
| OS in Amygdala | MDA | 5.24 ± 0.67 | 11.34 ± 1.81 a | 6.59 ± 0.95 | 12.34 ± 1.19 aac | 4.43 ± 0.80 | 8.74 ± 1.54 | 5.39 ± 0.60 | 7.34 ± 0.94 δ |
| NO | 73.07 ± 12.09 | 119.09 ± 9.74 a | 86.32 ± 7.59 | 125.89 ± 9.58 aa | 78.67 ± 13.03 | 101.09 ± 6.86 | 76.32 ± 7.59 | 109.69 ± 8.35 | |
| SOD | 67.09 ± 9.64 | 31.19 ± 6.67 a | 54.06 ± 5.58 | 21.39 ± 5.45 aa | 65.29 ± 8.37 | 55.19 ± 8.19 | 60.06 ± 10.27 | 58.19 ± 6.26 δ | |
| CAT | 7.41 ± 0.85 | 4.12 ± 0.99 | 6.62 ± 1.04 | 2.72 ± 0.61 a | 8.01 ± 1.24 | 5.92 ± 0.93 | 7.22 ± 1.02 | 6.12 ± 0.67 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Mostafi, H.; Elhessni, A.; Doumar, H.; Touil, T.; Mesfioui, A. Behavioral and Amygdala Biochemical Damage Induced by Alternating Mild Stress and Ethanol Intoxication in Adolescent Rats: Reversal by Argan Oil Treatment? Int. J. Mol. Sci. 2024, 25, 10529. https://doi.org/10.3390/ijms251910529
El Mostafi H, Elhessni A, Doumar H, Touil T, Mesfioui A. Behavioral and Amygdala Biochemical Damage Induced by Alternating Mild Stress and Ethanol Intoxication in Adolescent Rats: Reversal by Argan Oil Treatment? International Journal of Molecular Sciences. 2024; 25(19):10529. https://doi.org/10.3390/ijms251910529
Chicago/Turabian StyleEl Mostafi, Hicham, Aboubaker Elhessni, Hanane Doumar, Tarik Touil, and Abdelhalem Mesfioui. 2024. "Behavioral and Amygdala Biochemical Damage Induced by Alternating Mild Stress and Ethanol Intoxication in Adolescent Rats: Reversal by Argan Oil Treatment?" International Journal of Molecular Sciences 25, no. 19: 10529. https://doi.org/10.3390/ijms251910529