
Microfluidic Affinity Selection of B-Lineage Cells from Peripheral Blood for Minimal Residual Disease Monitoring in Pediatric B-Type Acute Lymphoblastic Leukemia Patients

 and
and
Abstract
:
1. Introduction
2. Results
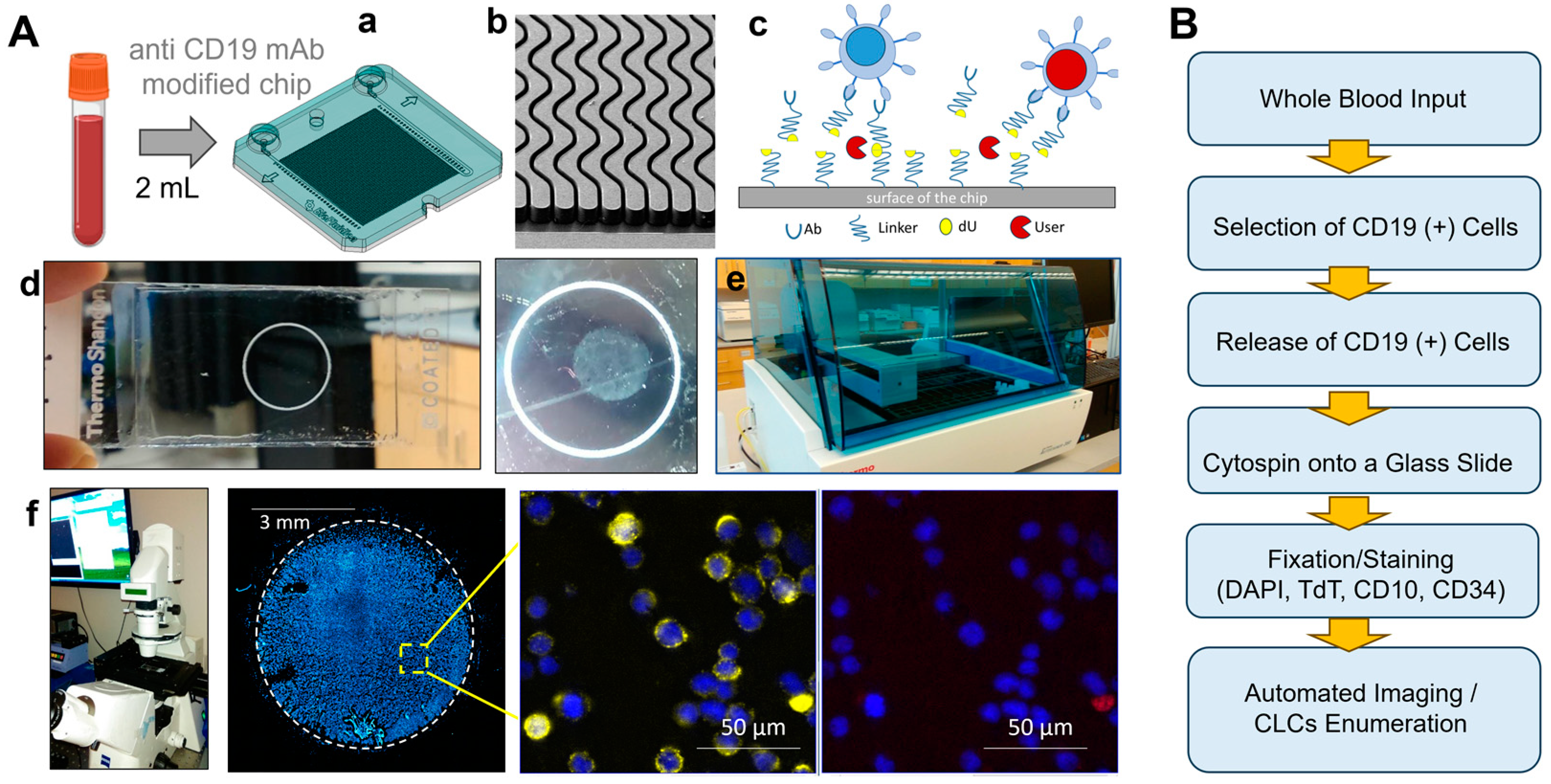
2.1. Evaluation of the Processing Pipeline for the Affinity Isolation of CLCs
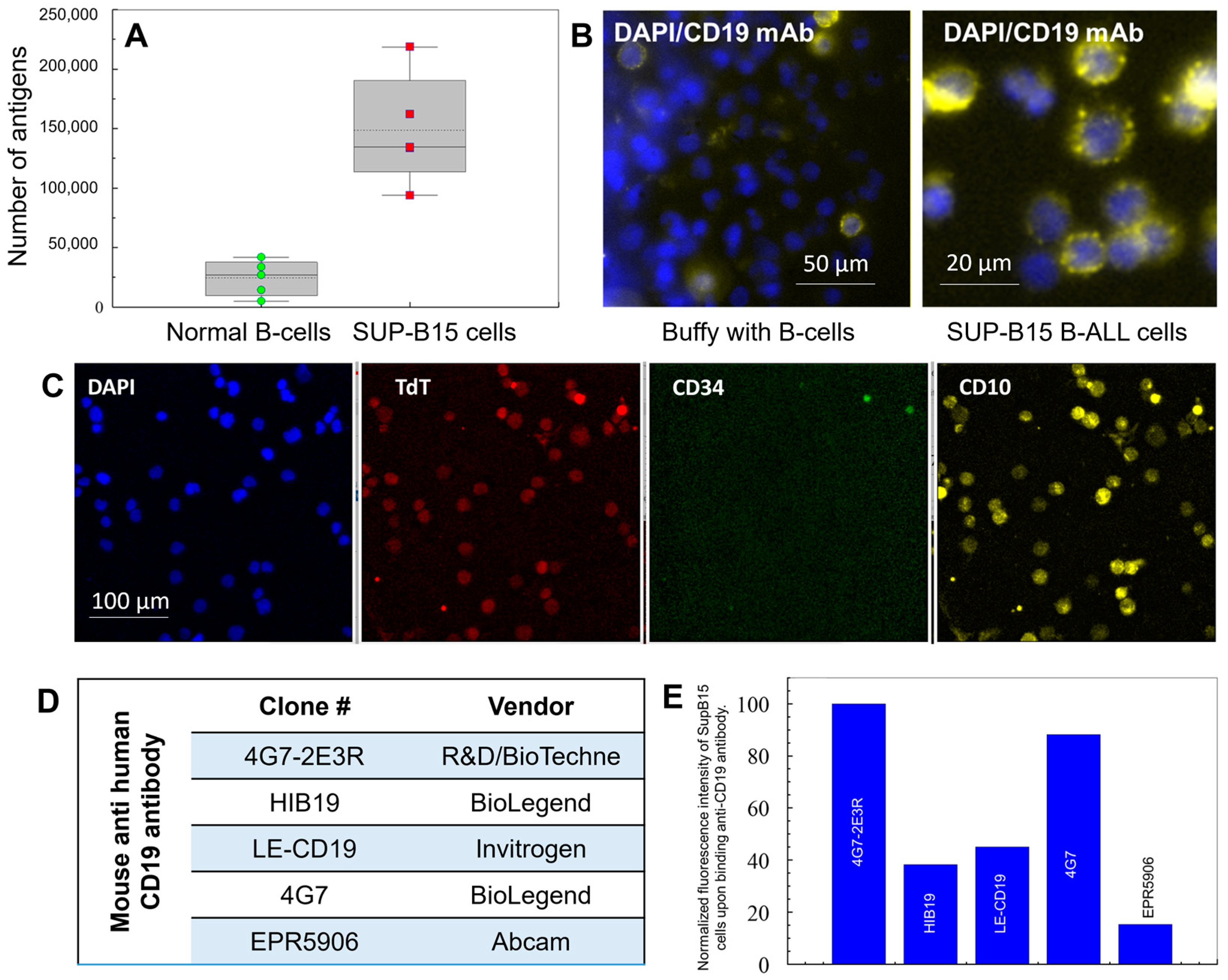
2.2. Choice of Antibodies for the Selection and Staining of CLCs
2.3. B-ALL Pediatric Patients
2.4. Treatment for B-ALL Pediatric Patients
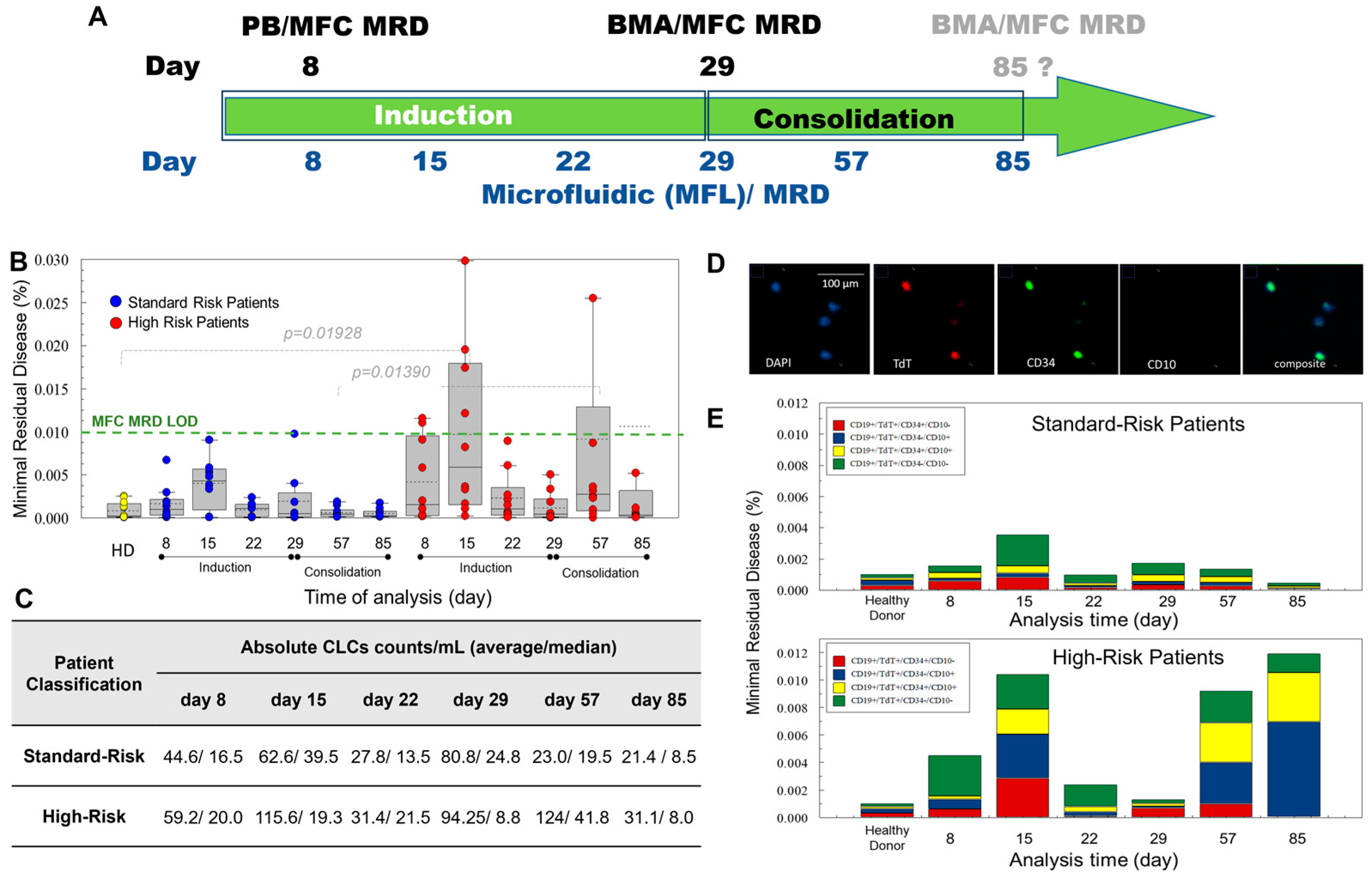
2.5. Improved Sensitivity of MF-MRD for Rare Cell Analysis Compared to MFC-MRD
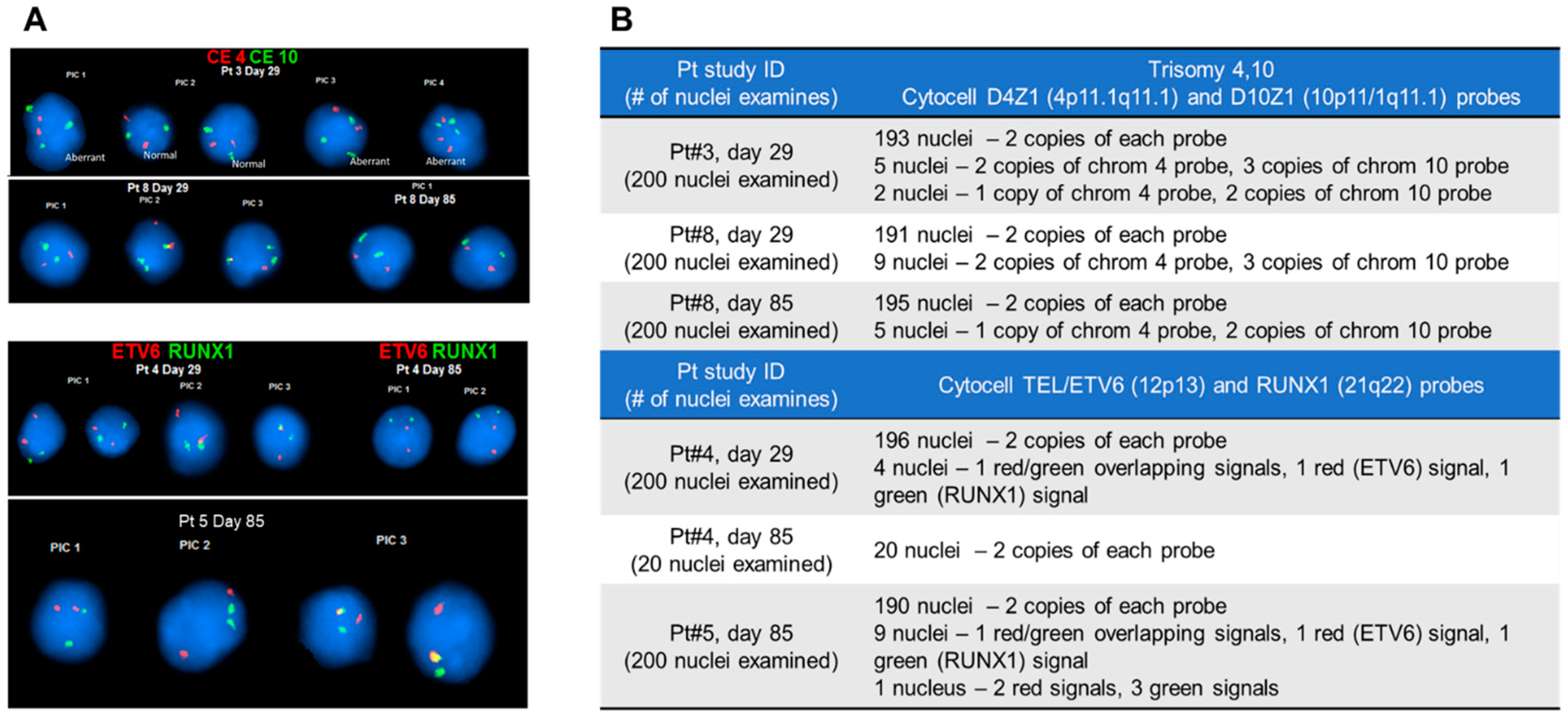
2.6. Fluorescence In Situ Hybridization (FISH) of CLCs
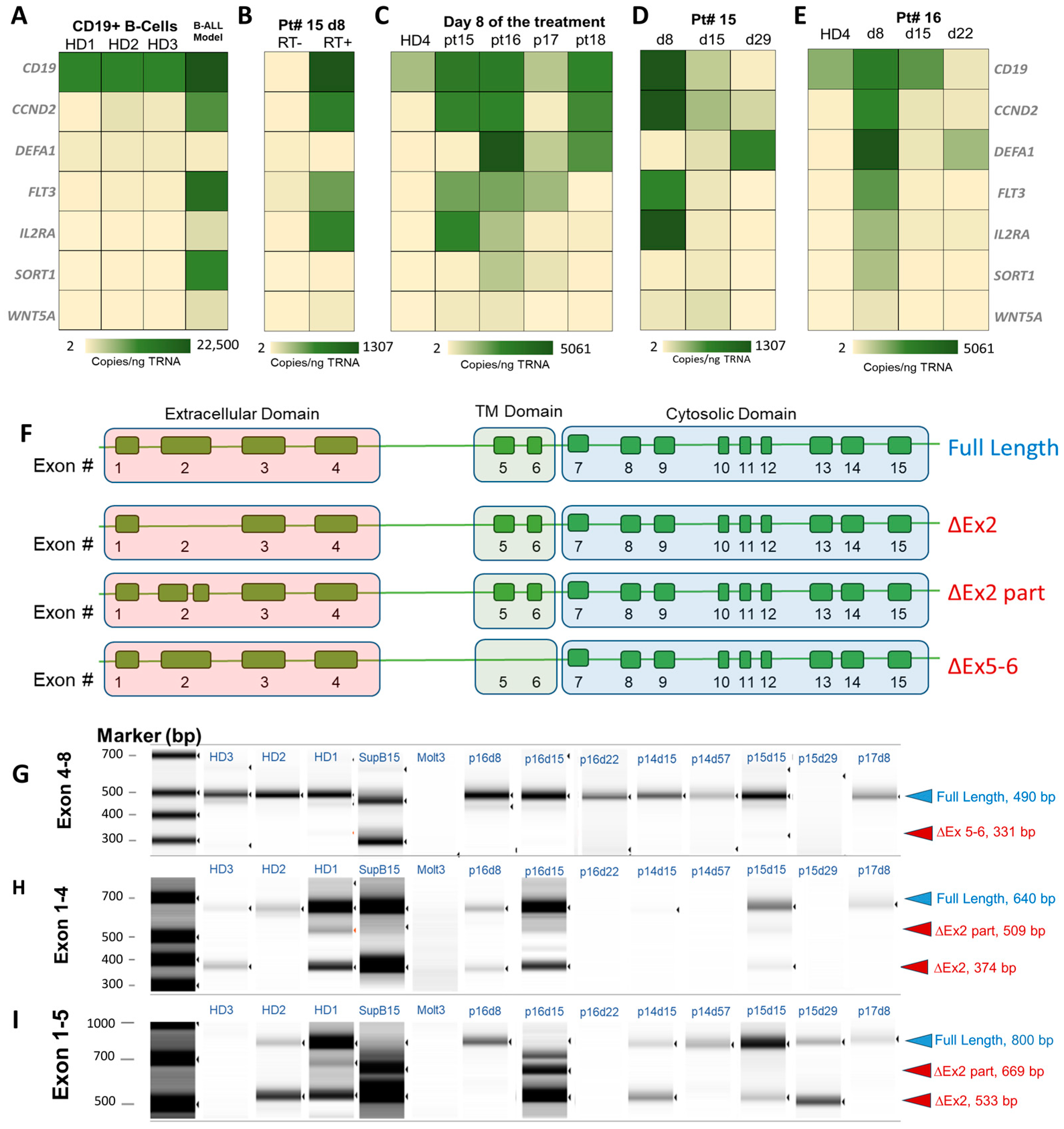
2.7. Molecular Profiling of CLCs
3. Discussion
3.1. Enumeration of CLCs
3.2. CLC Immunophenotype Modulation
3.3. CLCs and Chromosomal Aberrations
3.4. CLCs for Molecular Profiling
3.5. Consequences of Alternative Splicing of CD19
4. Materials and Methods
4.1. Microfluidic Chip Assembly and Capture Antibody Conjugation
4.2. Clinical Samples
4.3. Blood Sample Collection and Processing
4.4. Cell Culture
4.5. Immunophenotyping of Cells
4.6. Sample Preparation for FISH
4.7. RNA Isolation
4.8. Reverse Transcription (RT)
4.9. Droplet Digital PCR (ddPCR)
4.10. Image Collection and Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Terwilliger, T.; Abdul-Hay, M. Acute lymphoblastic leukemia: A comprehensive review and 2017 update. Blood Cancer J. 2017, 7, e577. [Google Scholar] [CrossRef]
- Chan, K.W. Acute lymphoblastic leukemia. Curr. Probl. Pediatr. Adolesc. Health Care 2002, 32, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Coustan-Smith, E.; Sancho, J.; Hancock, M.L.; Boyett, J.M.; Behm, F.G.; Raimondi, S.C.; Sandlund, J.T.; Rivera, G.K.; Rubnitz, J.E.; Ribeiro, R.C.; et al. Clinical importance of minimal residual disease in childhood acute lymphoblastic leukemia. Blood 2000, 96, 2691–2696. [Google Scholar] [CrossRef] [PubMed]
- Brüggemann, M.; Kotrova, M. Minimal residual disease in adult ALL: Technical aspects and implications for correct clinical interpretation. Blood Adv. 2017, 1, 2456–2466. [Google Scholar] [CrossRef] [PubMed]
- NCI Measurable Residual Disease. Available online: https://www.cancer.gov/publications/dictionaries/cancer-terms/def/measurable-residual-disease (accessed on 24 August 2024).
- Nguyen, K.; Devidas, M.; Cheng, S.C.; La, M.; Raetz, E.A.; Carroll, W.L.; Winick, N.J.; Hunger, S.P.; Gaynon, P.S.; Loh, M.L. Factors influencing survival after relapse from acute lymphoblastic leukemia: A Children’s Oncology Group study. Leukemia 2008, 22, 2142–2150. [Google Scholar] [CrossRef]
- Seibel, N.L. Treatment of Acute Lymphoblastic Leukemia in Children and Adolescents: Peaks and Pitfalls. Hematology 2008, 2008, 374–380. [Google Scholar] [CrossRef]
- Abou Dalle, I.; Jabbour, E.; Short, N.J. Evaluation and management of measurable residual disease in acute lymphoblastic leukemia. Ther. Adv. Hematol. 2020, 11, 2040620720910023. [Google Scholar] [CrossRef]
- Borowitz, M.J.; Pullen, D.J.; Shuster, J.J.; Viswanatha, D.; Montgomery, K.; Willman, C.L.; Camitta, B. Minimal residual disease detection in childhood precursor–B-cell acute lymphoblastic leukemia: Relation to other risk factors. A Children’s Oncology Group study. Leukemia 2003, 17, 1566–1572. [Google Scholar] [CrossRef]
- Borowitz, M.J.; Wood, B.L.; Keeney, M.; Hedley, B.D. Measurable Residual Disease Detection in B-Acute Lymphoblastic Leukemia: The Children’s Oncology Group (COG) Method. Curr. Protoc. 2022, 2, e383. [Google Scholar] [CrossRef]
- Campana, D. Minimal residual disease in acute lymphoblastic leukemia. Semin. Hematol. 2009, 46, 100–106. [Google Scholar] [CrossRef]
- Chen, H.; Gu, M.; Liang, J.; Song, H.; Zhang, J.; Xu, W.; Zhao, F.; Shen, D.; Shen, H.; Liao, C.; et al. Minimal residual disease detection by next-generation sequencing of different immunoglobulin gene rearrangements in pediatric B-ALL. Nat. Commun. 2023, 14, 7468. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wood, B.L. Monitoring minimal residual disease in acute leukemia: Technical challenges and interpretive complexities. Blood Rev. 2017, 31, 63–75. [Google Scholar] [CrossRef]
- Della Starza, I.; Chiaretti, S.; De Propris, M.S.; Elia, L.; Cavalli, M.; De Novi, L.A.; Soscia, R.; Messina, M.; Vitale, A.; Guarini, A.; et al. Minimal Residual Disease in Acute Lymphoblastic Leukemia: Technical and Clinical Advances. Front. Oncol. 2019, 9, 726. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Othus, M.; Araki, D.; Wood, B.L.; Radich, J.P.; Halpern, A.B.; Mielcarek, M.; Estey, E.H.; Appelbaum, F.R.; Walter, R.B. Pre- and post-transplant quantification of measurable (‘minimal’) residual disease via multiparameter flow cytometry in adult acute myeloid leukemia. Leukemia 2016, 30, 1456–1464. [Google Scholar] [CrossRef] [PubMed]
- Van Dongen, J.J.M.; Van Der Velden, V.H.J.; Brüggemann, M.; Orfao, A. Minimal residual disease diagnostics in acute lymphoblastic leukemia: Need for sensitive, fast, and standardized technologies. Blood 2015, 125, 3996–4009. [Google Scholar] [CrossRef] [PubMed]
- Conter, V.; Bartram, C.R.; Valsecchi, M.G.; Schrauder, A.; Panzer-Grümayer, R.; Möricke, A.; Aricò, M.; Zimmermann, M.; Mann, G.; De Rossi, G.; et al. Molecular response to treatment redefines all prognostic factors in children and adolescents with B-cell precursor acute lymphoblastic leukemia: Results in 3184 patients of the AIEOP-BFM ALL 2000 study. Blood 2010, 115, 3206–3214. [Google Scholar] [CrossRef]
- Lane, J.; Ong, E.; Schreiber, B.; Habiba, U.; Leyton, A.; Giles, J.; Trahair, T.; Sutton, R.; Lee, L.; Stevenson, W.; et al. Measurable residual disease detection in acute lymphoblastic leukaemia—Comparison between NGS and ASO-qPCR. Pathology 2024, 56, S97. [Google Scholar] [CrossRef]
- Wood, B.; Wu, D.; Crossley, B.; Dai, Y.; Williamson, D.; Gawad, C.; Borowitz, M.J.; Devidas, M.; Maloney, K.W.; Larsen, E.; et al. Measurable residual disease detection by high-throughput sequencing improves risk stratification for pediatric B-ALL. Blood 2018, 131, 1350–1359. [Google Scholar] [CrossRef]
- Alexander, S. Clinically defining and managing high-risk pediatric patients with acute lymphoblastic leukemia. Hematology 2014, 2014, 181–189. [Google Scholar] [CrossRef]
- Amin, H.M.; Yang, Y.; Shen, Y.; Estey, E.H.; Giles, F.J.; Pierce, S.A.; Kantarjian, H.M.; O’Brien, S.M.; Jilani, I.; Albitar, M. Having a higher blast percentage in circulation than bone marrow: Clinical implications in myelodysplastic syndrome and acute lymphoid and myeloid leukemias. Leukemia 2005, 19, 1567–1572. [Google Scholar] [CrossRef]
- Bassan, R.; Intermesoli, T.; Scattolin, A.; Viero, P.; Maino, E.; Sancetta, R.; Carobolante, F.; Gianni, F.; Stefanoni, P.; Tosi, M.; et al. Minimal Residual Disease Assessment and Risk-based Therapy in Acute Lymphoblastic Leukemia. Clin. Lymphoma Myeloma Leuk. 2017, 17, S2–S9. [Google Scholar] [CrossRef] [PubMed]
- Bhojwani, D.; Howard, S.C.; Pui, C.-H. High-Risk Childhood Acute Lymphoblastic Leukemia. Clin. Lymphoma Myeloma 2009, 9, S222–S230. [Google Scholar] [CrossRef] [PubMed]
- Gajjar, A.; Ribeiro, R.; Hancock, M.; Rivera, G.; Mahmoud, H.; Sandlund, J.; Crist, W.; Pui, C. Persistence of circulating blasts after 1 week of multiagent chemotherapy confers a poor prognosis in childhood acute lymphoblastic leukemia. Blood 1995, 86, 1292–1295. [Google Scholar] [CrossRef]
- Jastaniah, W.; Elimam, N.; Abdalla, K.; Alazmi, A.A.; Elgaml, A.M.; Alkassar, A.; Daghistani, M.; Felimban, S. Early vs. late MRD response- and risk-based treatment intensification of childhood acute lymphoblastic leukemia: A prospective pilot study from Saudi Arabia. Exp. Hematol. Oncol. 2018, 7, 29. [Google Scholar] [CrossRef] [PubMed]
- Papadantonakis, N.; Advani, A.S. Recent advances and novel treatment paradigms in acute lymphocytic leukemia. Ther. Adv. Hematol. 2016, 7, 252–269. [Google Scholar] [CrossRef]
- Kamande, J.W.; Hupert, M.L.; Witek, M.A.; Wang, H.; Torphy, R.J.; Dharmasiri, U.; Njoroge, S.K.; Jackson, J.M.; Aufforth, R.D.; Snavely, A.; et al. Modular Microsystem for the Isolation, Enumeration, and Phenotyping of Circulating Tumor Cells in Patients with Pancreatic Cancer. Anal. Chem. 2013, 85, 9092–9100. [Google Scholar] [CrossRef]
- Witek, M.A.; Aufforth, R.D.; Wang, H.; Kamande, J.W.; Jackson, J.M.; Pullagurla, S.R.; Hupert, M.L.; Usary, J.; Wysham, W.Z.; Hilliard, D.; et al. Discrete microfluidics for the isolation of circulating tumor cell subpopulations targeting fibroblast activation protein alpha and epithelial cell adhesion molecule. npj Precis. Oncol. 2017, 1, 24. [Google Scholar] [CrossRef]
- Nair, S.V.; Witek, M.A.; Jackson, J.M.; Lindell, M.A.M.; Hunsucker, S.A.; Sapp, T.; Perry, C.E.; Hupert, M.L.; Bae-Jump, V.; Gehrig, P.A.; et al. Enzymatic cleavage of uracil-containing single-stranded DNA linkers for the efficient release of affinity-selected circulating tumor cells. Chem. Commun. 2015, 51, 3266–3269. [Google Scholar] [CrossRef]
- Kamande, J.W.; Lindell, M.A.M.; Witek, M.A.; Voorhees, P.M.; Soper, S.A. Isolation of circulating plasma cells from blood of patients diagnosed with clonal plasma cell disorders using cell selection microfluidics. Integr. Biol. 2018, 10, 82–91. [Google Scholar] [CrossRef]
- Jackson, J.M.; Taylor, J.B.; Witek, M.A.; Hunsucker, S.A.; Waugh, J.P.; Fedoriw, Y.; Shea, T.C.; Soper, S.A.; Armistead, P.M. Microfluidics for the detection of minimal residual disease in acute myeloid leukemia patients using circulating leukemic cells selected from blood. Analyst 2016, 141, 640–651. [Google Scholar] [CrossRef]
- Freed, I.M.; Kasi, A.; Fateru, O.; Hu, M.; Gonzalez, P.; Weatherington, N.; Pathak, H.; Hyter, S.; Sun, W.; Al-Rajabi, R.; et al. Circulating Tumor Cell Subpopulations Predict Treatment Outcome in Pancreatic Ductal Adenocarcinoma (PDAC) Patients. Cells 2023, 12, 2266. [Google Scholar] [CrossRef]
- Adams, A.A.; Okagbare, P.I.; Feng, J.; Hupert, M.L.; Patterson, D.; Göttert, J.; McCarley, R.L.; Nikitopoulos, D.; Murphy, M.C.; Soper, S.A. Highly Efficient Circulating Tumor Cell Isolation from Whole Blood and Label-Free Enumeration Using Polymer-Based Microfluidics with an Integrated Conductivity Sensor. J. Am. Chem. Soc. 2008, 130, 8633–8641. [Google Scholar] [CrossRef]
- Jackson, J.M.; Witek, M.A.; Soper, S.A. Circulating Tumor Cells: Isolation and Analysis. In Circulating Tumor Cells: Isolation and Analysis; Hugh Fan, Z., Vitha, M.F., Eds.; Wiley Publishing: Hoboken, NJ, USA, 2016. [Google Scholar]
- Hupert, M.L.; Jackson, J.M.; Wang, H.; Witek, M.A.; Kamande, J.; Milowsky, M.I.; Whang, Y.E.; Soper, S.A. Arrays of High-Aspect Ratio Microchannels for High-Throughput Isolation of Circulating Tumor Cells (CTCs). Microsyst. Technol. 2014, 20, 1815–1825. [Google Scholar] [CrossRef] [PubMed]
- Alves, G.V.; Fernandes, A.L.; Freire, J.M.; Paiva Ade, S.; Vasconcelos, R.C.; Sales, V.S.; Lemos, T.M.; Gil, E.A.; Freire, F.H.; Cavalcanti e Silva, D.G.; et al. Flow cytometry immunophenotyping evaluation in acute lymphoblastic leukemia: Correlation to factors affecting clinic outcome. J. Clin. Lab. Anal. 2012, 26, 431–440. [Google Scholar] [PubMed]
- Chantepie, S.P.; Cornet, E.; Salaün, V.; Reman, O. Hematogones: An overview. Leuk. Res. 2013, 37, 1404–1411. [Google Scholar] [CrossRef] [PubMed]
- Chiaretti, S.; Zini, G.; Bassan, R. Diagnosis and subclassification of acute lymphoblastic leukemia. Mediterr. J. Hematol. Infect. Dis. 2014, 6, e2014073. [Google Scholar] [CrossRef]
- Kulis, J.; Sędek, Ł.; Słota, Ł.; Perkowski, B.; Szczepański, T. Commonly Assessed Markers in Childhood BCP-ALL Diagnostic Panels and Their Association with Genetic Aberrations and Outcome Prediction. Genes 2022, 13, 1374. [Google Scholar] [CrossRef]
- Motea, E.A.; Berdis, A.J. Terminal deoxynucleotidyl transferase: The story of a misguided DNA polymerase. Biochim. Biophys. Acta 2010, 1804, 1151–1166. [Google Scholar] [CrossRef]
- Jackson, J.M.; Witek, M.A.; Kamande, J.W.; Soper, S.A. Materials and microfluidics: Enabling the efficient isolation and analysis of circulating tumour cells. Chem. Soc. Rev. 2017, 46, 4245–4280. [Google Scholar] [CrossRef]
- Hurford, M.T.; Altman, A.J.; DiGiuseppe, J.A.; Sherburne, B.J.; Rezuke, W.N. Unique pattern of nuclear TdT immunofluorescence distinguishes normal precursor B cells (Hematogones) from lymphoblasts of precursor B-lymphoblastic leukemia. Am. J. Clin. Pathol. 2008, 129, 700–705. [Google Scholar] [CrossRef]
- Salzer, W.L.; Burke, M.J.; Devidas, M.; Chen, S.; Gore, L.; Larsen, E.C.; Borowitz, M.; Wood, B.; Heerema, N.A.; Carroll, A.J.; et al. Toxicity associated with intensive postinduction therapy incorporating clofarabine in the very high-risk stratum of patients with newly diagnosed high-risk B-lymphoblastic leukemia: A report from the Children’s Oncology Group study AALL1131. Cancer 2018, 124, 1150–1159. [Google Scholar] [CrossRef] [PubMed]
- Cooper, S.L.; Brown, P.A. Treatment of pediatric acute lymphoblastic leukemia. Pediatr. Clin. N. Am. 2015, 62, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Larson, R.A.; Dodge, R.K.; Burns, C.P.; Lee, E.J.; Stone, R.M.; Schulman, P.; Duggan, D.; Davey, F.R.; Sobol, R.E.; Frankel, S.R.; et al. A five-drug remission induction regimen with intensive consolidation for adults with acute lymphoblastic leukemia: Cancer and leukemia group B study 8811. Blood 1995, 85, 2025–2037. [Google Scholar] [CrossRef] [PubMed]
- Kruse, A.; Abdel-Azim, N.; Kim, H.N.; Ruan, Y.; Phan, V.; Ogana, H.; Wang, W.; Lee, R.; Gang, E.J.; Khazal, S.; et al. Minimal Residual Disease Detection in Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2020, 21, 1054. [Google Scholar] [CrossRef]
- Vrooman, L.M.; Blonquist, T.M.; Harris, M.H.; Stevenson, K.E.; Place, A.E.; Hunt, S.K.; O’Brien, J.E.; Asselin, B.L.; Athale, U.H.; Clavell, L.A.; et al. Refining risk classification in childhood B acute lymphoblastic leukemia: Results of DFCI ALL Consortium Protocol 05-001. Blood Adv. 2018, 2, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Garza-Veloz, I.; Martinez-Fierro, M.L.; Jaime-Perez, J.C.; Carrillo-Sanchez, K.; Ramos-Del Hoyo, M.G.; Lugo-Trampe, A.; Rojas-Martinez, A.; Gutierrez-Aguirre, C.H.; Gonzalez-Llano, O.; Salazar-Riojas, R.; et al. Identification of differentially expressed genes associated with prognosis of B acute lymphoblastic leukemia. Dis. Markers 2015, 2015, 828145. [Google Scholar] [CrossRef]
- Zhang, J.; Mullighan, C.G.; Harvey, R.C.; Wu, G.; Chen, X.; Edmonson, M.; Buetow, K.H.; Carroll, W.L.; Chen, I.M.; Devidas, M.; et al. Key pathways are frequently mutated in high-risk childhood acute lymphoblastic leukemia: A report from the Children’s Oncology Group. Blood 2011, 118, 3080–3087. [Google Scholar] [CrossRef]
- Carrà, G.; Cartellà, A.; Maffeo, B.; Circosta, P.; Cignetti, A.; Pautasso, M.; Familiari, U.; Volante, M.; Dragani, M.E.E.; Rege Cambrin, G.; et al. Interleukin-2 Receptor Alpha Chain, Also Called CD25, Is a Potential Target in Acute Lymphoblastic Leukemia. Blood 2020, 136, 11–12. [Google Scholar] [CrossRef]
- Chiles, T.C. Regulation and Function of Cyclin D2 in B Lymphocyte Subsets1. J. Immunol. 2004, 173, 2901–2907. [Google Scholar] [CrossRef]
- Farahi, L.; Ghaemimanesh, F.; Milani, S.; Razavi, S.M.; Akhondi, M.M.; Rabbani, H. Sortilin as a Novel Diagnostic and Therapeutic Biomarker in Chronic Lymphocytic Leukemia. Avicenna J. Med. Biotechnol. 2019, 11, 270–276. [Google Scholar]
- Gutierrez-Camino, A.; Richer, C.; Ouimet, M.; Fuchs, C.; Langlois, S.; Khater, F.; Caron, M.; Beaulieu, P.; St-Onge, P.; Bataille, A.R.; et al. Characterisation of FLT3 alterations in childhood acute lymphoblastic leukaemia. Br. J. Cancer 2024, 130, 317–326. [Google Scholar] [CrossRef]
- Liang, H.; Chen, Q.; Coles, A.H.; Anderson, S.J.; Pihan, G.; Bradley, A.; Gerstein, R.; Jurecic, R.; Jones, S.N. Wnt5a inhibits B cell proliferation and functions as a tumor suppressor in hematopoietic tissue. Cancer Cell 2003, 4, 349–360. [Google Scholar] [CrossRef] [PubMed]
- te Kronnie, G.; Bicciato, S.; Franceschini, L.; Accordi, B.; Delliorto, M.C.; Rinaldi, A.; Pession, A.; Barisone, E.; Conter, V.; Locatelli, F.; et al. Validation by RQ-PCR and flow cytometry of α-defensin1-3 (DEFA1-3) overexpression in relapsed and refractory acute lymphoblastic leukemia. Oncol. Rep. 2006, 15, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.J.; Ord, D.C.; Omori, S.A.; Tedder, T.F. Structure of the genes encoding the CD19 antigen of human and mouse B lymphocytes. Immunogenetics 1992, 35, 102–111. [Google Scholar] [CrossRef]
- Sotillo, E.; Barrett, D.M.; Black, K.L.; Bagashev, A.; Oldridge, D.; Wu, G.; Sussman, R.; Lanauze, C.; Ruella, M.; Gazzara, M.R.; et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015, 5, 1282–1295. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.H.; Evans, W.E. A 50-year journey to cure childhood acute lymphoblastic leukemia. Semin. Hematol. 2013, 50, 185–196. [Google Scholar] [CrossRef]
- Cloos, J.; Harris, J.R.; Janssen, J.; Kelder, A.; Huang, F.; Sijm, G.; Vonk, M.; Snel, A.N.; Scheick, J.R.; Scholten, W.J.; et al. Comprehensive Protocol to Sample and Process Bone Marrow for Measuring Measurable Residual Disease and Leukemic Stem Cells in Acute Myeloid Leukemia. J. Vis. Exp. 2018, 133, e56386. [Google Scholar]
- Bain, B.J.; Bailey, K. Pitfalls in obtaining and interpreting bone marrow aspirates: To err is human. J. Clin. Pathol. 2011, 64, 373–379. [Google Scholar] [CrossRef]
- Tembhare, P.R.; Subramanian PG, P.G.; Ghogale, S.; Chatterjee, G.; Patkar, N.V.; Gupta, A.; Shukla, R.; Badrinath, Y.; Deshpande, N.; Narula, G.; et al. A High-Sensitivity 10-Color Flow Cytometric Minimal Residual Disease Assay in B-Lymphoblastic Leukemia/Lymphoma Can Easily Achieve the Sensitivity of 2-in-106 and Is Superior to Standard Minimal Residual Disease Assay: A Study of 622 Patients. Cytom. Part B Clin. Cytom. 2020, 98, 57–67. [Google Scholar] [CrossRef]
- Kung, J.W.; Yablon, C.M.; Eisenberg, R.L. Bone Marrow Signal Alteration in the Extremities. Am. J. Roentgenol. 2011, 196, W492–W510. [Google Scholar] [CrossRef]
- Van Der Velden, V.; Jacobs, D.; Wijkhuijs, A.; Comans-Bitter, W.; Willemse, M.; Hählen, K.; Kamps, W.; Van Wering, E.; Van Dongen, J. Minimal residual disease levels in bone marrow and peripheral blood are comparable in children with T cell acute lymphoblastic leukemia (ALL), but not in precursor-B-ALL. Leukemia 2002, 16, 1432–1436. [Google Scholar] [CrossRef] [PubMed]
- Szczepański, T.; Willemse, M.J.; Brinkhof, B.; van Wering, E.R.; van der Burg, M.; van Dongen, J.J.M. Comparative analysis of Ig and TCR gene rearrangements at diagnosis and at relapse of childhood precursor-B–ALL provides improved strategies for selection of stable PCR targets for monitoring of minimal residual disease. Blood 2002, 99, 2315–2323. [Google Scholar] [CrossRef]
- Campana, D. Role of minimal residual disease monitoring in adult and pediatric acute lymphoblastic leukemia. Hematol. Oncol. Clin. N. Am. 2009, 23, 1083–1098. [Google Scholar] [CrossRef]
- Mendoza, H.; Tormey, C.; Siddon, A.J. Clinical Utility of B- and T-Cell Gene Rearrangement Studies in Blood and Bone Marrow Samples. Blood 2019, 134 (Suppl. S1), 5223. [Google Scholar] [CrossRef]
- Wood, B.L.; Arroz, M.; Barnett, D.; DiGiuseppe, J.; Greig, B.; Kussick, S.J.; Oldaker, T.; Shenkin, M.; Stone, E.; Wallace, P. 2006 Bethesda International Consensus recommendations on the immunophenotypic analysis of hematolymphoid neoplasia by flow cytometry: Optimal reagents and reporting for the flow cytometric diagnosis of hematopoietic neoplasia. Cytom. Part B Clin. Cytom. 2007, 72B (Suppl. S1), S14–S22. [Google Scholar] [CrossRef]
- Jackson, J.M.; Witek, M.A.; Hupert, M.L.; Brady, C.; Pullagurla, S.; Kamande, J.; Aufforth, R.D.; Tignanelli, C.J.; Torphy, R.J.; Yeh, J.J.; et al. UV activation of polymeric high aspect ratio microstructures: Ramifications in antibody surface loading for circulating tumor cell selection. Lab Chip 2014, 14, 106–117. [Google Scholar] [CrossRef]
- Allard, W.J.; Matera, J.; Miller, M.C.; Repollet, M.; Connelly, M.C.; Rao, C.; Tibbe, A.G.; Uhr, J.W.; Terstappen, L.W. Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clin. Cancer Res. 2004, 10, 6897–6904. [Google Scholar] [CrossRef] [PubMed]
- Nagrath, S.; Sequist, L.V.; Maheswaran, S.; Bell, D.W.; Irimia, D.; Ulkus, L.; Smith, M.R.; Kwak, E.L.; Digumarthy, S.; Muzikansky, A.; et al. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature 2007, 450, 1235–1239. [Google Scholar] [CrossRef] [PubMed]
- Pearl, M.L.; Zhao, Q.; Yang, J.; Dong, H.; Tulley, S.; Zhang, Q.; Golightly, M.; Zucker, S.; Chen, W.T. Prognostic analysis of invasive circulating tumor cells (iCTCs) in epithelial ovarian cancer. Gynecol. Oncol. 2014, 134, 581–590. [Google Scholar] [CrossRef]
- Pecot, C.V.; Bischoff, F.Z.; Mayer, J.A.; Wong, K.L.; Pham, T.; Bottsford-Miller, J.; Stone, R.L.; Lin, Y.G.; Jaladurgam, P.; Roh, J.W.; et al. A novel platform for detection of CK+ and CK- CTCs. Cancer Discov. 2011, 1, 580–586. [Google Scholar] [CrossRef]
- Stott, S.L.; Hsu, C.-H.; Tsukrov, D.I.; Yu, M.; Miyamoto, D.T.; Waltman, B.A.; Rothenberg, S.M.; Shah, A.M.; Smas, M.E.; Korir, G.K.; et al. Isolation of circulating tumor cells using a microvortex-generating herringbone-chip. Proc. Natl. Acad. Sci. USA 2010, 107, 18392–18397. [Google Scholar] [CrossRef] [PubMed]
- Schultz, K.R.; Pullen, D.J.; Sather, H.N.; Shuster, J.J.; Devidas, M.; Borowitz, M.J.; Carroll, A.J.; Heerema, N.A.; Rubnitz, J.E.; Loh, M.L.; et al. Risk- and response-based classification of childhood B-precursor acute lymphoblastic leukemia: A combined analysis of prognostic markers from the Pediatric Oncology Group (POG) and Children’s Cancer Group (CCG). Blood 2007, 109, 926–935. [Google Scholar] [CrossRef]
- Chatterjee, G.; Sriram, H.; Ghogale, S.; Deshpande, N.; Khanka, T.; Panda, D.; Pradhan, S.N.; Girase, K.; Narula, G.; Dhamane, C.; et al. Immunophenotypic shift in the B-cell precursors from regenerating bone marrow samples: A critical consideration for measurable residual disease assessment in B-lymphoblastic leukemia. Cytom. Part B Clin. Cytom. 2021, 100, 434–445. [Google Scholar] [CrossRef]
- Tembhare, P.R.; Ghogale, S.; Ghatwai, N.; Badrinath, Y.; Kunder, N.; Patkar, N.V.; Bibi, A.R.; Chatterjee, G.; Arora, B.; Narula, G.; et al. Evaluation of new markers for minimal residual disease monitoring in B-cell precursor acute lymphoblastic leukemia: CD73 and CD86 are the most relevant new markers to increase the efficacy of MRD 2016; 00B: 000–000. Cytom. Part B Clin. Cytom. 2018, 94, 100–111. [Google Scholar] [CrossRef]
- Dworzak, M.N.; Gaipa, G.; Schumich, A.; Maglia, O.; Ratei, R.; Veltroni, M.; Husak, Z.; Basso, G.; Karawajew, L.; Gadner, H.; et al. Modulation of antigen expression in B-cell precursor acute lymphoblastic leukemia during induction therapy is partly transient: Evidence for a drug-induced regulatory phenomenon. Results of the AIEOP-BFM-ALL-FLOW-MRD-Study Group. Cytom. Part B Clin. Cytom. 2010, 78B, 147–153. [Google Scholar] [CrossRef]
- Modvig, S.; Wernersson, R.; Øbro, N.F.; Olsen, L.R.; Christensen, C.; Rosthøj, S.; Degn, M.; Jürgensen, G.W.; Madsen, H.O.; Albertsen, B.K.; et al. High CD34 surface expression in BCP-ALL predicts poor induction therapy response and is associated with altered expression of genes related to cell migration and adhesion. Mol. Oncol. 2022, 16, 2015–2030. [Google Scholar] [CrossRef] [PubMed]
- Uckun, F.M.; Sather, H.; Gaynon, P.; Arthur, D.; Nachman, J.; Sensel, M.; Steinherz, P.; Hutchinson, R.; Trigg, M.; Reaman, G. Prognostic significance of the CD10+CD19+CD34+ B-progenitor immunophenotype in children with acute lymphoblastic leukemia: A report from the Children’s Cancer Group. Leuk. Lymphoma 1997, 27, 445–457. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.G. Genetics and prognosis of ALL in children vs adults. Hematol. Am. Soc. Hematol. Educ. Program 2018, 2018, 137–145. [Google Scholar] [CrossRef]
- Vardiman, J.W.; Thiele, J.; Arber, D.A.; Brunning, R.D.; Borowitz, M.J.; Porwit, A.; Harris, N.L.; Le Beau, M.M.; Hellström-Lindberg, E.; Tefferi, A.; et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: Rationale and important changes. Blood 2009, 114, 937–951. [Google Scholar] [CrossRef]
- Beder, T.; Hansen, B.T.; Hartmann, A.M.; Zimmermann, J.; Amelunxen, E.; Wolgast, N.; Walter, W.; Zaliova, M.; Antić, Ž.; Chouvarine, P.; et al. The Gene Expression Classifier ALLCatchR Identifies B-cell Precursor ALL Subtypes and Underlying Developmental Trajectories Across Age. Hemasphere 2023, 7, e939. [Google Scholar] [CrossRef]
- Rabilloud, T.; Potier, D.; Pankaew, S.; Nozais, M.; Loosveld, M.; Payet-Bornet, D. Single-cell profiling identifies pre-existing CD19-negative subclones in a B-ALL patient with CD19-negative relapse after CAR-T therapy. Nat. Commun. 2021, 12, 865. [Google Scholar] [CrossRef] [PubMed]
- Orlando, E.J.; Han, X.; Tribouley, C.; Wood, P.A.; Leary, R.J.; Riester, M.; Levine, J.E.; Qayed, M.; Grupp, S.A.; Boyer, M.; et al. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat. Med. 2018, 24, 1504–1506. [Google Scholar] [CrossRef] [PubMed]
- Pal Singh, S.; Dammeijer, F.; Hendriks, R.W. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol. Cancer 2018, 17, 57. [Google Scholar] [CrossRef] [PubMed]
- Harris, P.A.; Taylor, R.; Minor, B.L.; Elliott, V.; Fernandez, M.; O’Neal, L.; McLeod, L.; Delacqua, G.; Delacqua, F.; Kirby, J.; et al. The REDCap consortium: Building an international community of software platform partners. J. Biomed. Inf. 2019, 95, 103208. [Google Scholar] [CrossRef]
- Harris, P.A.; Taylor, R.; Thielke, R.; Payne, J.; Gonzalez, N.; Conde, J.G. Research electronic data capture (REDCap)—A metadata-driven methodology and workflow process for providing translational research informatics support. J. Biomed. Inf. 2009, 42, 377–381. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Merits | Method for Minimal Residual Disease Detection in B-ALL | ||
|---|---|---|---|
| Multiparameter Flow Cytometry | Quantitative PCR or RT-qPCR | Next Generation Sequencing | |
| Material tested | fresh cells | gDNA/mRNA | gDNA |
| Target | ALL immunophenotypes |
|
|
| Analytical Sensitivity | 10−4 (0.01%) | 10−4–10−5 (0.01–0.001%) | 10−6 (0.0001%) |
| Clinical Sensitivity | 61–74% * | ≤50% (see challenges) | 26–39% more than MFC ** |
| Advantages |
|
|
|
| Challenges |
|
|
|
| Patient and Disease Characteristics | n (%) |
|---|---|
| Gender | |
| Female | 9 (45%) |
| Male | 11 (55%) |
| Age | |
| <1 year-old | 0 (0%) |
| 1–10-year-old | 17 (85%) |
| >10-year-old | 3 (15%) |
| Cytogenetics | |
| t(9;22) | 0 (0%) |
| Double trisomies (+4,+10) | 7 (35%) |
| KMT2A rearrangement | 0 (0%) |
| t(12;21) | 5 (25%) |
| iAMP21 | 0 (0%) |
| WBC count | |
| <50 × 106/mL | 18 (90%) |
| >50 × 106/mL | 2 (10%) |
| CNS Involvement | |
| Yes | 0 (0%) |
| No | 20 (100%) |
| Risk Classification after Consolidation Therapy | |
| Standard Risk | 11 (55%) |
| High Risk | 9 (45%) |
| Microfluidic (MF)-MRD Testing | |
|---|---|
| Characteristic | Metric |
| Biological Material Tested | Peripheral blood/CD19(+) cells |
| Blood sample consumption | 2 mL for enumeration, 2–5 mL for molecular profiling |
| Processing throughput | 1.5 mL/h |
| LOD | 5 × 10−4% |
| Specificity | 85.7% |
| Sensitivity | 71.0% |
| NPV | 100% |
| Cost per analysis | $130 |
| Molecular analysis possible? | Yes |
| FISH possible? | Yes |
| Can observe blasts clonal evolution? | Yes |
| Clinical significance of low levels of MRD established | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Witek, M.A.; Larkey, N.E.; Bartakova, A.; Hupert, M.L.; Mog, S.; Cronin, J.K.; Vun, J.; August, K.J.; Soper, S.A. Microfluidic Affinity Selection of B-Lineage Cells from Peripheral Blood for Minimal Residual Disease Monitoring in Pediatric B-Type Acute Lymphoblastic Leukemia Patients. Int. J. Mol. Sci. 2024, 25, 10619. https://doi.org/10.3390/ijms251910619
Witek MA, Larkey NE, Bartakova A, Hupert ML, Mog S, Cronin JK, Vun J, August KJ, Soper SA. Microfluidic Affinity Selection of B-Lineage Cells from Peripheral Blood for Minimal Residual Disease Monitoring in Pediatric B-Type Acute Lymphoblastic Leukemia Patients. International Journal of Molecular Sciences. 2024; 25(19):10619. https://doi.org/10.3390/ijms251910619
Chicago/Turabian StyleWitek, Malgorzata A., Nicholas E. Larkey, Alena Bartakova, Mateusz L. Hupert, Shalee Mog, Jami K. Cronin, Judy Vun, Keith J. August, and Steven A. Soper. 2024. "Microfluidic Affinity Selection of B-Lineage Cells from Peripheral Blood for Minimal Residual Disease Monitoring in Pediatric B-Type Acute Lymphoblastic Leukemia Patients" International Journal of Molecular Sciences 25, no. 19: 10619. https://doi.org/10.3390/ijms251910619
APA StyleWitek, M. A., Larkey, N. E., Bartakova, A., Hupert, M. L., Mog, S., Cronin, J. K., Vun, J., August, K. J., & Soper, S. A. (2024). Microfluidic Affinity Selection of B-Lineage Cells from Peripheral Blood for Minimal Residual Disease Monitoring in Pediatric B-Type Acute Lymphoblastic Leukemia Patients. International Journal of Molecular Sciences, 25(19), 10619. https://doi.org/10.3390/ijms251910619