

Soluble Epoxide Hydrolase Inhibition Attenuates Proteinuria by Alleviating Renal Inflammation and Podocyte Injuries in Adriamycin-Induced Nephropathy

Abstract
:1. Introduction
2. Results
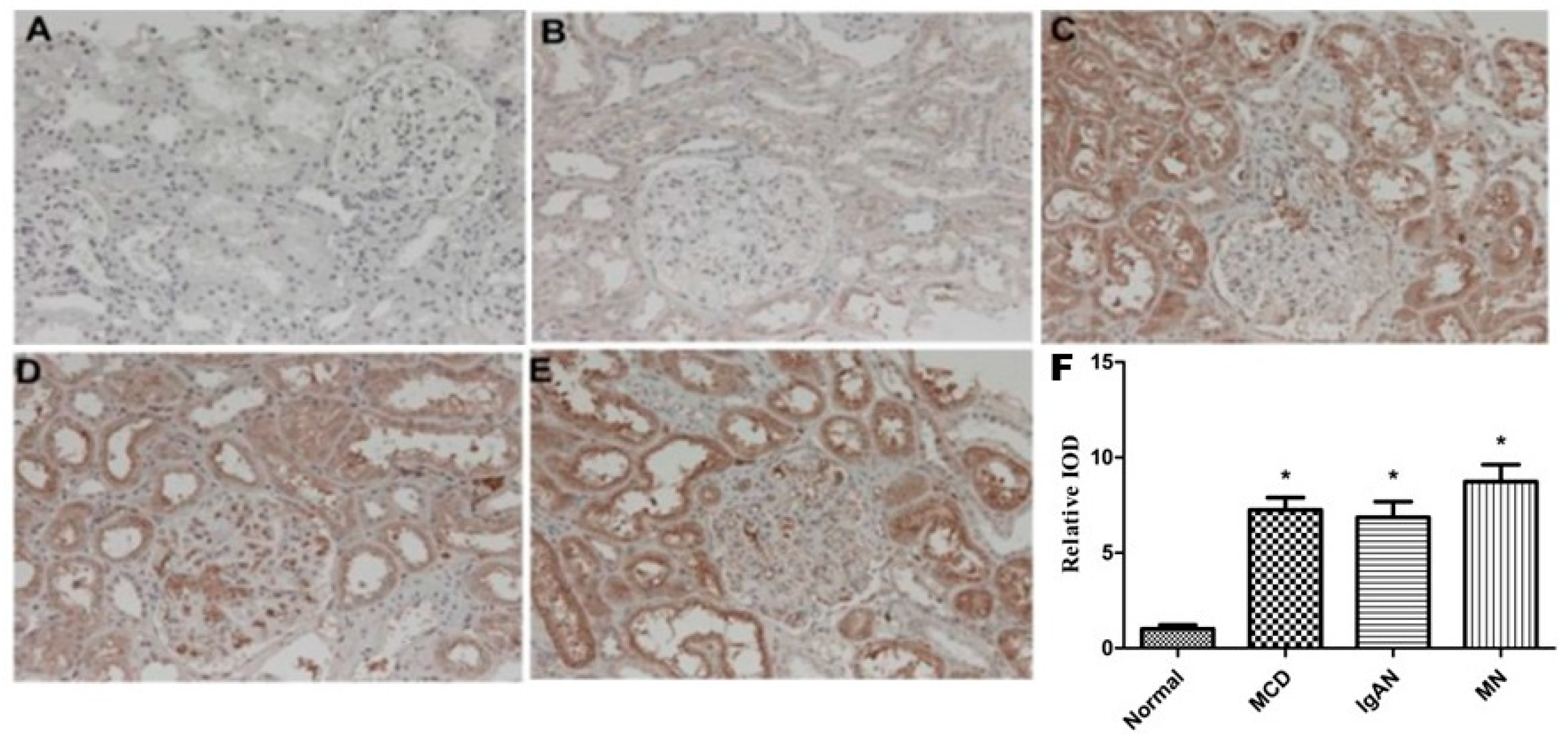
2.1. Expression of sEH in Renal Tissue of Patients with Primary Glomerular Diseases and Its Correlation with Clinical Indicators
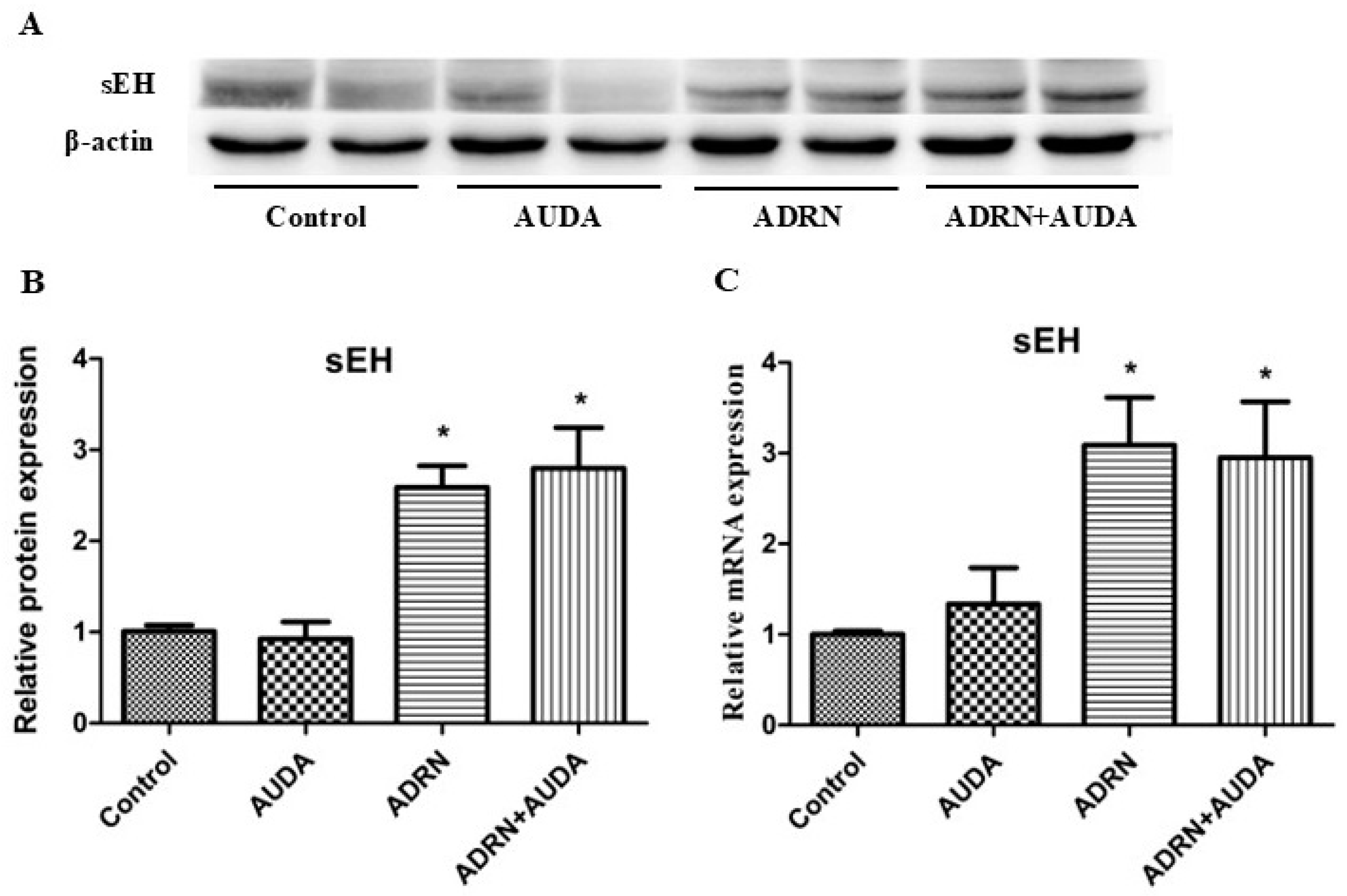
2.2. The Expression of Renal sEH Significantly Increased in Rats with ADRN
2.3. sEH Inhibition Reduced Urine Protein Excretion and Improved Renal Function in Rats with ADRN
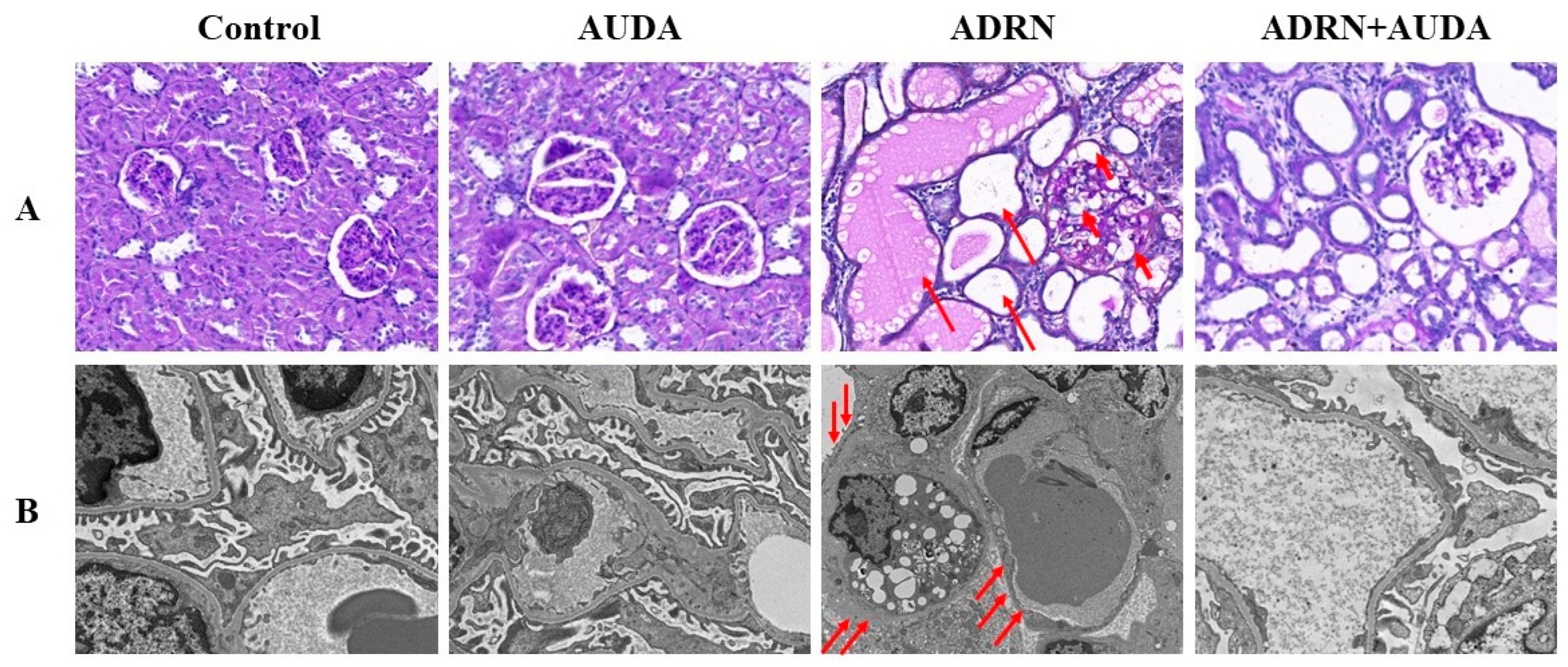
2.4. sEH Inhibition Alleviates Renal Pathological Damage in ADRN Rats
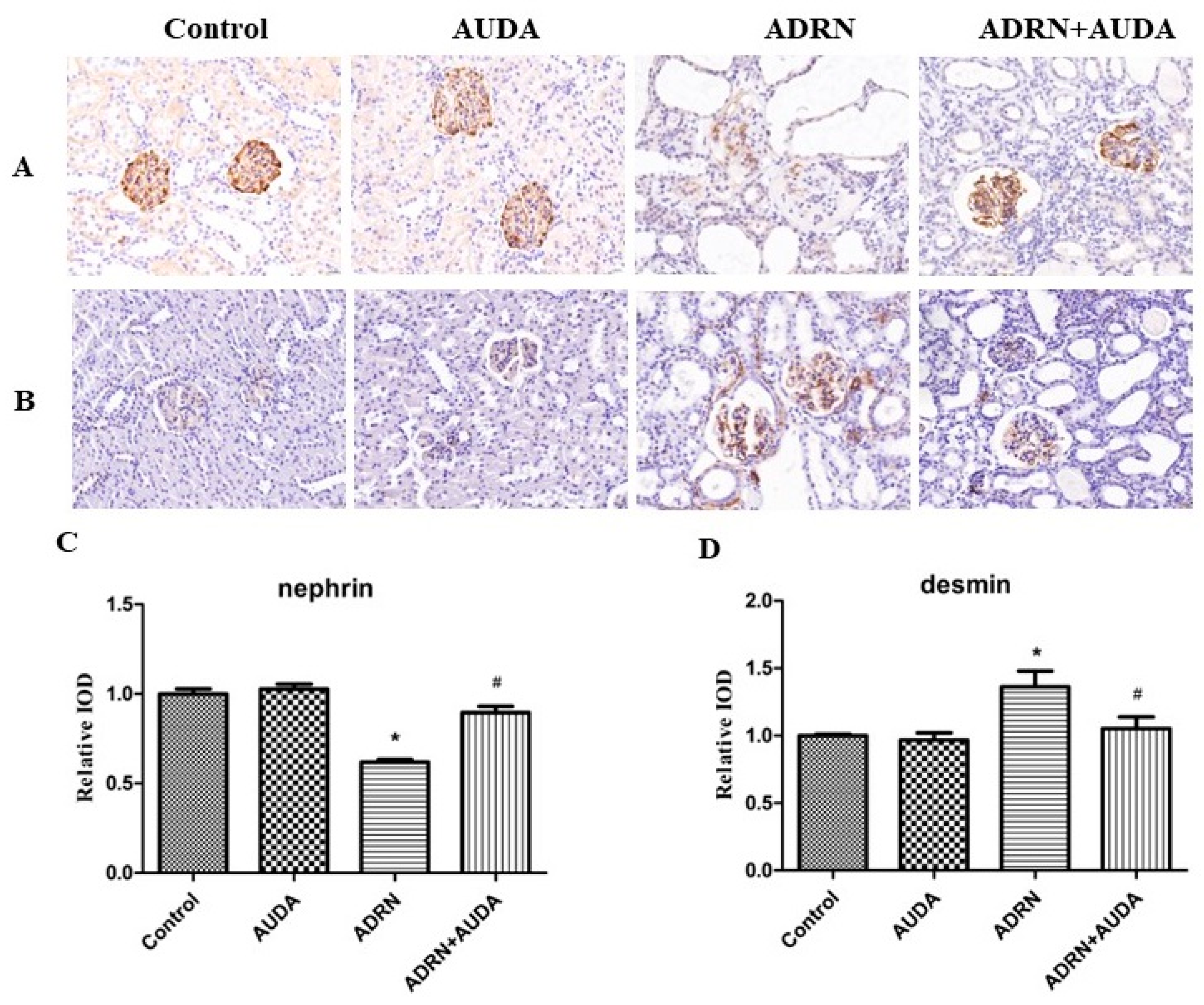
2.5. The Effect of sEH Inhibitor on the Expression of Nephrin and Desmin in Glomerular Podocytes
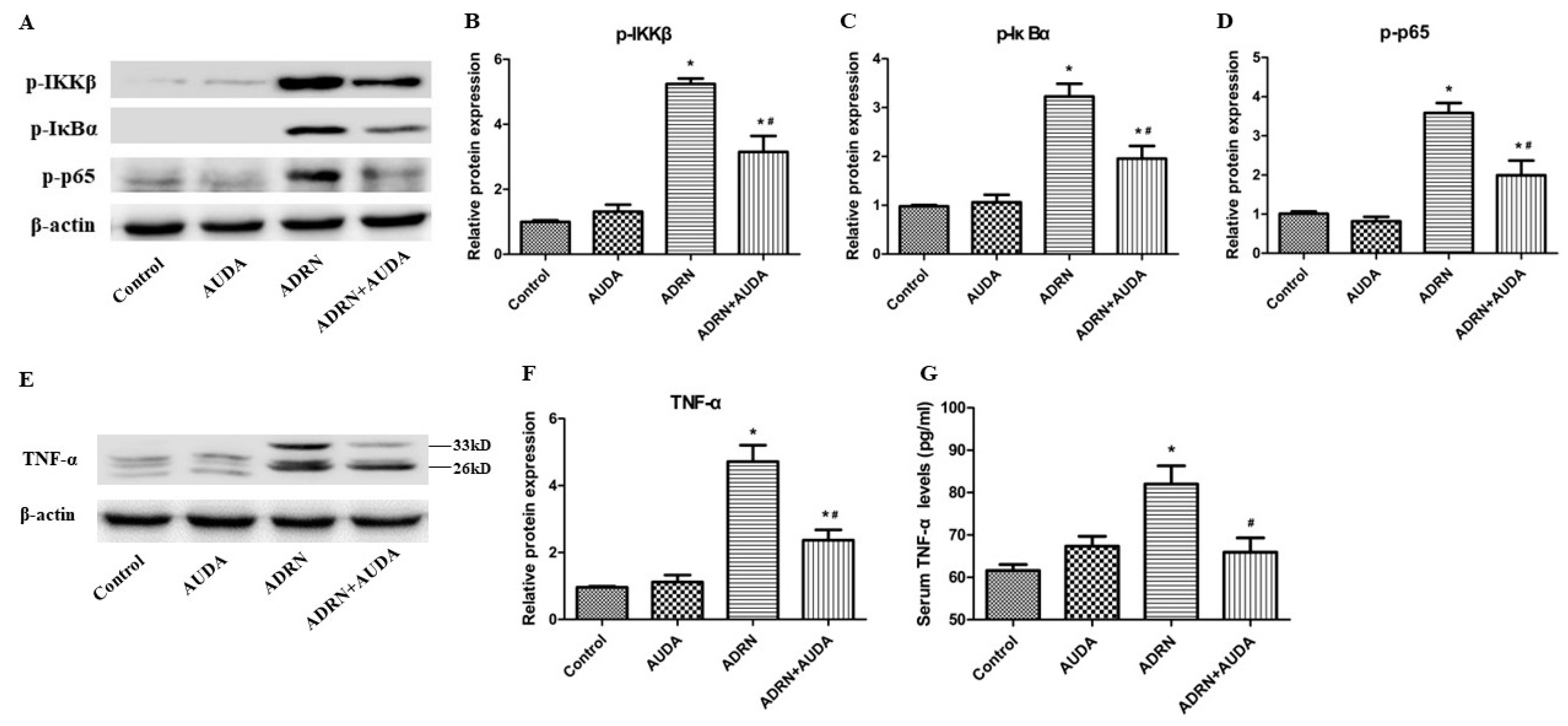
2.6. sEH Inhibitor Suppressed Renal NF-κB Activation and Reduced TNF-α Levels in Rats with ADRN
3. Discussion
4. Materials and Methods
4.1. Subjects
4.2. Animal Modeling and Grouping
4.3. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
4.4. Western Blot (WB)
4.5. Detection of Serum TNF-α Levels by ELISA
4.6. Immunohistochemical Examination and Morphological Observation
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rovin, B.H.; Adler, S.G.; Barratt, J.; Bridoux, F.; Burdge, K.A.; Chan, T.M.; Cook, H.T.; Fervenza, F.C.; Gibson, K.L.; Glassock, R.J.; et al. Kdigo 2021 Clinical Practice Guideline for the Management of Glomerular Diseases. Kidney Int. 2021, 100, S1–S276. [Google Scholar]
- Jiang, H.; Shen, Z.R.; Zhuang, J.; Lu, C.; Qu, Y.; Xu, C.R.; Yang, S.F.; Tian, X.F. Understanding the podocyte immune responses in proteinuric kidney diseases: From pathogenesis to therapy. Front. Immunol. 2024, 14, 1335936. [Google Scholar] [CrossRef]
- Ma, S.J.; Qiu, Y.; Zhang, C. Cytoskeleton Rearrangement in Podocytopathies: An Update. Int. J. Mol. Sci. 2024, 25, 647. [Google Scholar] [CrossRef]
- Kawachi, H.; Fukusumi, Y. New insight into podocyte slit diaphragm, a therapeutic target of proteinuria. Clin. Exp. Nephrol. 2020, 24, 193–204. [Google Scholar] [CrossRef]
- Imig, J.D.; Khan, M.A.H.; Burkhan, A.; Chen, G.; Adebesin, A.M.; Falck, J.R. Kidney-Targeted Epoxyeicosatrienoic Acid Analog, EET-F01, Reduces Inflammation, Oxidative Stress, and Cisplatin-Induced Nephrotoxicity. Int. J. Mol. Sci. 2021, 22, 2793. [Google Scholar] [CrossRef]
- Noh, M.R.; Jang, H.S.; Salem, F.E.; Ferrer, F.A.; Kim, J.; Padanilam, B.J. Epoxyeicosatrienoic acid administration or soluble epoxide hydrolase inhibition attenuates renal fibrogenesis in obstructive nephropathy. Am. J. Physiol. Ren. Physiol. 2023, 324, F138–F151. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.H.; Nolan, B.; Stavniichuk, A.; Merk, D.; Imig, J.D. Dual soluble epoxide hydrolase inhibitor—farnesoid X receptor agonist interventional treatment attenuates renal inflammation and fibrosis. Front. Immunol. 2024, 14, 1269261. [Google Scholar] [CrossRef]
- Liang, Y.X.; Jing, Z.Y.; Deng, H.; Li, Z.Q.; Zhuang, Z.; Wang, S.; Wang, Y. Soluble epoxide hydrolase inhibition ameliorates proteinuria-induced epithelial-mesenchymal transition by regulating the PI3K-Akt-GSK-3β signaling pathway. Biochem. Biophys. Res. Commun. 2015, 463, 70–75. [Google Scholar] [CrossRef]
- Wang, Q.; Pang, W.; Cui, Z.; Shi, J.B.; Liu, Y.; Liu, B.; Zhou, Y.F.; Guan, Y.F.; Hammock, B.D.; Wang, Y.; et al. Upregulation of soluble epoxide hydrolase in proximal tubular cells mediated proteinuria-induced renal damage. Am. J. Physiol. Renal 2013, 304, F168–F176. [Google Scholar] [CrossRef]
- Manhiani, M.; Quigley, J.E.; Knight, S.F.; Tasoobshirazi, S.; Moore, T.; Brands, M.W.; Hammock, B.D.; Imig, J.D. Soluble epoxide hydrolase gene deletion attenuates renal injury and inflammation with DOCA-salt hypertension. Am. J. Physiol. Renal 2009, 297, F740–F748. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Luan, Z.L.; Huo, X.K.; Zhang, M.; Morisseau, C.; Sun, C.P.; Hammock, B.D.; Ma, X.C. Direct targeting of sEH with alisol B alleviated the apoptosis, inflammation, and oxidative stress in cisplatin-induced acute kidney injury. Int. J. Biol. Sci. 2023, 19, 294–310. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.S.; Xiang, X.Y.; Chen, X.M.; He, J.L.; Liu, T.; Gan, H.; Du, X.G. Inhibition of soluble epoxide hydrolase attenuates renal tubular mitochondrial dysfunction and ER stress by restoring autophagic flux in diabetic nephropathy. Cell. Death Dis. 2020, 11, 385. [Google Scholar] [CrossRef]
- Favor, O.K.; Chauhan, P.S.; Pourmand, E.; Edwards, A.M.; Wagner, J.G.; Lewandowski, R.P.; Heine, L.K.; Harkema, J.R.; Lee, K.S.S.; Pestka, J.J. Lipidome modulation by dietary omega-3 polyunsaturated fatty acid supplementation or selective soluble epoxide hydrolase inhibition suppresses rough LPS-accelerated glomerulonephritis in lupus-prone mice. Front. Immunol. 2023, 14, 1124910. [Google Scholar] [CrossRef] [PubMed]
- Bettaieb, A.; Koike, S.; Chahed, S.; Zhao, Y.; Bachaalany, S.; Hashoush, N.; Graham, J.; Fatima, H.; Havel, P.J.; Gruzdev, A.; et al. Podocyte-specific soluble epoxide hydrolase deficiency in mice attenuates acute kidney injury. FEBS J. 2017, 284, 1970–1986. [Google Scholar] [CrossRef]
- Imig, J.D.; Zhao, X.Y.; Zaharis, C.Z.; Olearczyk, J.J.; Pollock, D.M.; Newman, J.W.; Kim, I.H.; Watanabe, T.; Hammock, B.D. An orally active epoxide hydrolase inhibitor lowers blood pressure and provides renal protection in salt-sensitive hypertension. Hypertension 2005, 46, 975–981. [Google Scholar] [CrossRef]
- Elmarakby, A.A.; Faulkner, J.; Al-Shabrawey, M.; Wang, M.H.; Maddipati, K.R.; Imig, J.D. Deletion of soluble epoxide hydrolase gene improves renal endothelial function and reduces renal inflammation and injury in streptozotocin-induced type 1 diabetes. Am. J. Physiol. Reg. I 2011, 301, R1307–R1317. [Google Scholar]
- Chen, G.Z.; Xu, R.F.; Wang, Y.N.; Wang, P.H.; Zhao, G.; Xu, X.Z.; Gruzdev, A.; Zeldin, D.C.; Wang, D.W. Genetic disruption of soluble epoxide hydrolase is protective against streptozotocin-induced diabetic nephropathy. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E563–E575. [Google Scholar] [CrossRef]
- Njeim, R.; Braych, K.; Ghadieh, H.E.; Azar, N.S.; Azar, W.S.; Dia, B.; Leone, A.; Cappello, F.; Kfoury, H.; Harb, F.; et al. VEGF-A: A Novel Mechanistic Link Between CYP2C-Derived EETs and Nox4 in Diabetic Kidney Disease. Diabetes 2023, 72, 947–957. [Google Scholar] [CrossRef]
- Klocke, J.; Ulu, A.; Wu, K.Y.; Rudolph, B.; Dragun, D.; Gollasch, M.; Schunck, W.H.; Hammock, B.D.; Riemekasten, G.; Enghard, P. Prophylactic inhibition of soluble epoxide hydrolase delays onset of nephritis and ameliorates kidney damage in NZB/W F1 mice. Sci. Rep. 2019, 9, 8993. [Google Scholar] [CrossRef]
- Kang, Y.S.; Li, Y.J.; Dai, C.S.; Kiss, L.P.; Wu, C.Y.; Liu, Y.H. Inhibition of integrin-linked kinase blocks podocyte epithelial-mesenchymal transition and ameliorates proteinuria. Kidney Int. 2010, 78, 363–373. [Google Scholar] [CrossRef]
- Zuo, Y.Q.; Yang, H.C.; Potthoff, S.A.; Najafian, B.; Kon, V.; Ma, L.J.; Fogo, A.B. Protective effects of PPARγ agonist in acute nephrotic syndrome. Nephrol. Dial. Transpl. 2012, 27, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.J.; He, Y.F.; Wang, M.Z.; Lv, P.; Wang, J.X.; Liu, J.Z. Role of Protease-Activated Receptor 2 in Regulating Focal Segmental Glomerulosclerosis. Cell. Physiol. Biochem. 2017, 41, 1147–1155. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, M.; Sugiyama, H.; Sada, K.; Kobayashi, M.; Maeshima, Y.; Yamasaki, Y.; Makino, H. Desmin as a marker of proteinuria in early stages of membranous nephropathy in elderly patients. Clin. Nephrol. 2007, 68, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zou, L.X.; Han, Y.C.; Wu, L.; Chen, T.; Zhu, D.D.; Hu, P. A20 overexpression exerts protective effects on podocyte injury in lupus nephritis by downregulating UCH-L1. J. Cell. Physiol. 2019, 234, 16191–16204. [Google Scholar] [CrossRef]
- Huang, M.J.; Yang, Y.; Chen, Y.F.; Li, Y.; Qin, S.T.; Xiao, L.J.; Long, X.W.; Hu, K.; Li, Y.X.; Ying, H.M.; et al. Sweroside attenuates podocyte injury and proteinuria in part by activating Akt/BAD signaling in mice. J. Cell. Biochem. 2023, 124, 1749–1763. [Google Scholar] [CrossRef]
- He, M.J.; Li, Y.X.; Wang, L.; Guo, B.; Mei, W.; Zhu, B.; Zhang, J.J.; Ding, Y.; Meng, B.Y.; Zhang, L.M.; et al. MYDGF attenuates podocyte injury and proteinuria by activating Akt/BAD signal pathway in mice with diabetic kidney disease. Diabetologia 2020, 63, 1916–1931. [Google Scholar] [CrossRef]
- Shimo, T.; Adachi, Y.; Yamanouchi, S.; Tsuji, S.; Kimata, T.; Umezawa, K.; Okigaki, M.; Takaya, J.; Ikehara, S.; Kaneko, K. A Novel Nuclear Factor κB Inhibitor, Dehydroxymethylepoxyquinomicin, Ameliorates Puromycin Aminonucleoside-Induced Nephrosis in Mice. Am. J. Nephrol. 2013, 37, 302–309. [Google Scholar] [CrossRef]
- Ozkan, S.; Isildar, B.; Ercin, M.; Gezginci-Oktayoglu, S.; Konukoglu, D.; Nesetoglu, N.; Oncul, M.; Koyuturk, M. Therapeutic potential of conditioned medium obtained from deferoxamine preconditioned umbilical cord mesenchymal stem cells on diabetic nephropathy model. Stem Cell Res. Ther. 2022, 13, 438. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control (n = 5) | IgAN (n = 7) | MCD (n = 7) | MN (n = 6) | |
|---|---|---|---|---|
| Age (years) | 43.7 ± 16.5 | 36.4 ± 2.0 | 36.8 ± 11.1 | 42.2 ± 11.0 |
| SBP (mmHg) | 123.1 ± 4.9 | 127.6 ± 5.5 | 120.5 ± 6. 6 | 127.0 ± 6.7 |
| DBP (mmHg) | 75.2 ± 6.7 | 81.4 ± 6.3 | 78.8 ± 7.5 | 80.0 ± 7.1 |
| UP (g/24 h) | 0.116 ± 0.028 | 3.141 ± 1.850 * | 6.205 ± 3.921 *# | 9.529 ± 3.899 *# |
| ALB (g/L) | 44.98 ± 4.41 | 39.92 ± 8.56 | 26.83 ± 10.13 *# | 29.64 ± 7.00 *# |
| SCr (μmol/L) | 58.72 ± 8.35 | 76.40 ± 13.95 | 63.25 ± 14.38 | 63.20 ± 13.91 |
| Control (n = 6) | AUDA (n = 6) | ADRN (n = 6) | ADRN + AUDA (n = 6) | |
|---|---|---|---|---|
| UP (mg/24 h) | 18.83 ± 4.56 | 13.44 ± 2.74 | 649.49 ± 53.86 * | 489.76 ± 87.12 *# |
| ALB (g/L) | 40.85 ± 2.96 | 40.35 ± 2.18 | 17.81 ± 2.67 * | 19.63 ± 2.28 * |
| SCr (μmol/L) | 26.67 ± 3.93 | 25.50 ± 3.83 | 123.50 ± 15.05 * | 73.17 ± 8.98 *# |
| TC (mmol/L) | 1.96 ± 0.21 | 1.99 ± 0.26 | 10.67 ± 1.87 * | 10.50 ± 1.57 * |
| FBG (mmol/L) | 9.39 ± 1.44 | 9.85 ± 1.20 | 8.51 ± 0.96 | 8.32 ± 2.71 |
| ALT (U/L) | 40.93 ± 6.48 | 39.17 ± 10.40 | 44.20 ± 6.61 | 36.25 ± 8.19 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niu, Q.; Guo, Z.; Liang, Y.; Zuo, L. Soluble Epoxide Hydrolase Inhibition Attenuates Proteinuria by Alleviating Renal Inflammation and Podocyte Injuries in Adriamycin-Induced Nephropathy. Int. J. Mol. Sci. 2024, 25, 10629. https://doi.org/10.3390/ijms251910629
Niu Q, Guo Z, Liang Y, Zuo L. Soluble Epoxide Hydrolase Inhibition Attenuates Proteinuria by Alleviating Renal Inflammation and Podocyte Injuries in Adriamycin-Induced Nephropathy. International Journal of Molecular Sciences. 2024; 25(19):10629. https://doi.org/10.3390/ijms251910629
Chicago/Turabian StyleNiu, Qingyu, Ziyu Guo, Yaoxian Liang, and Li Zuo. 2024. "Soluble Epoxide Hydrolase Inhibition Attenuates Proteinuria by Alleviating Renal Inflammation and Podocyte Injuries in Adriamycin-Induced Nephropathy" International Journal of Molecular Sciences 25, no. 19: 10629. https://doi.org/10.3390/ijms251910629