
Structural Insights and Catalytic Mechanism of 3-Hydroxybutyryl-CoA Dehydrogenase from Faecalibacterium Prausnitzii A2-165

 , and
, and 
Abstract
1. Introduction
2. Results
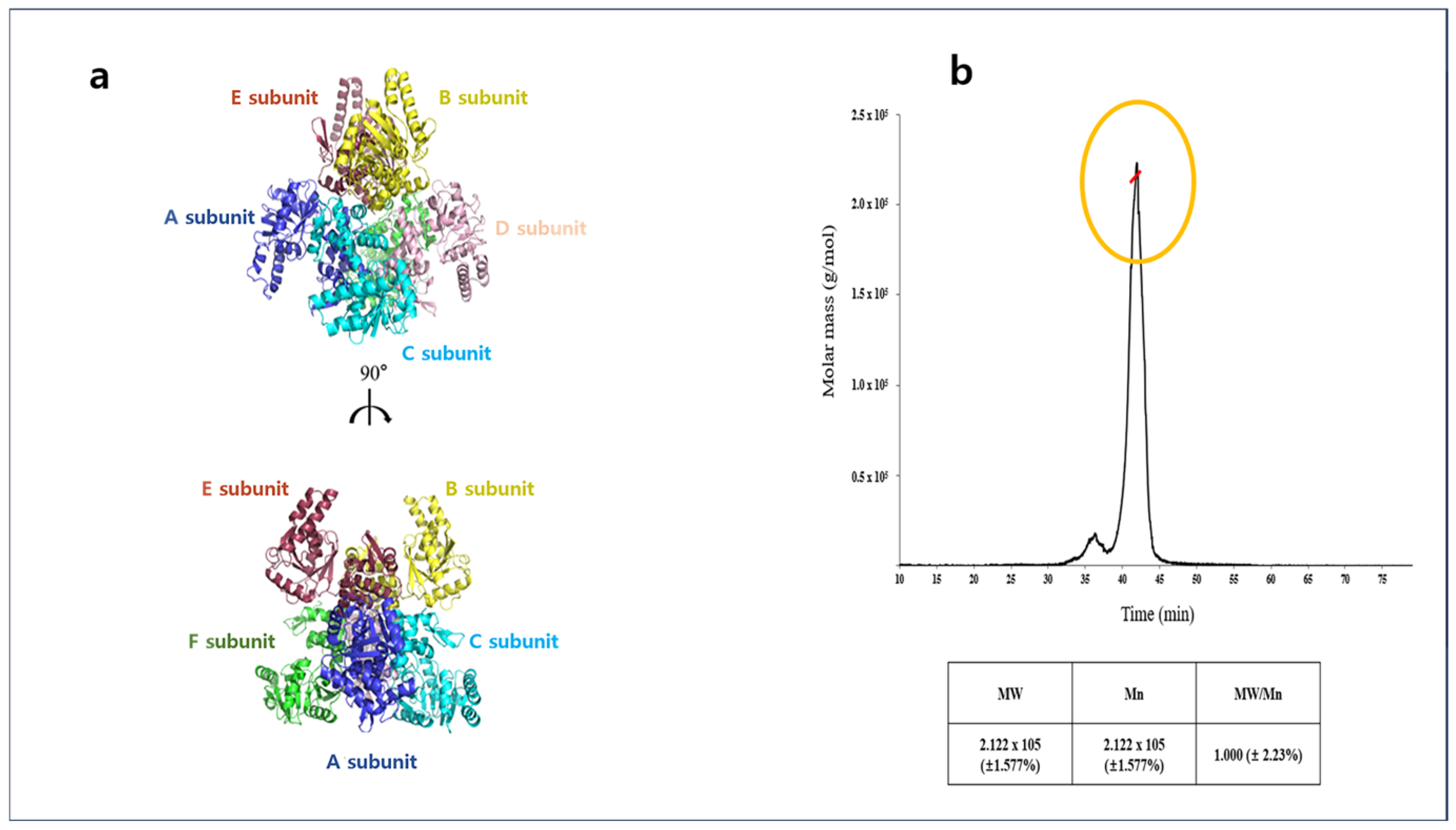
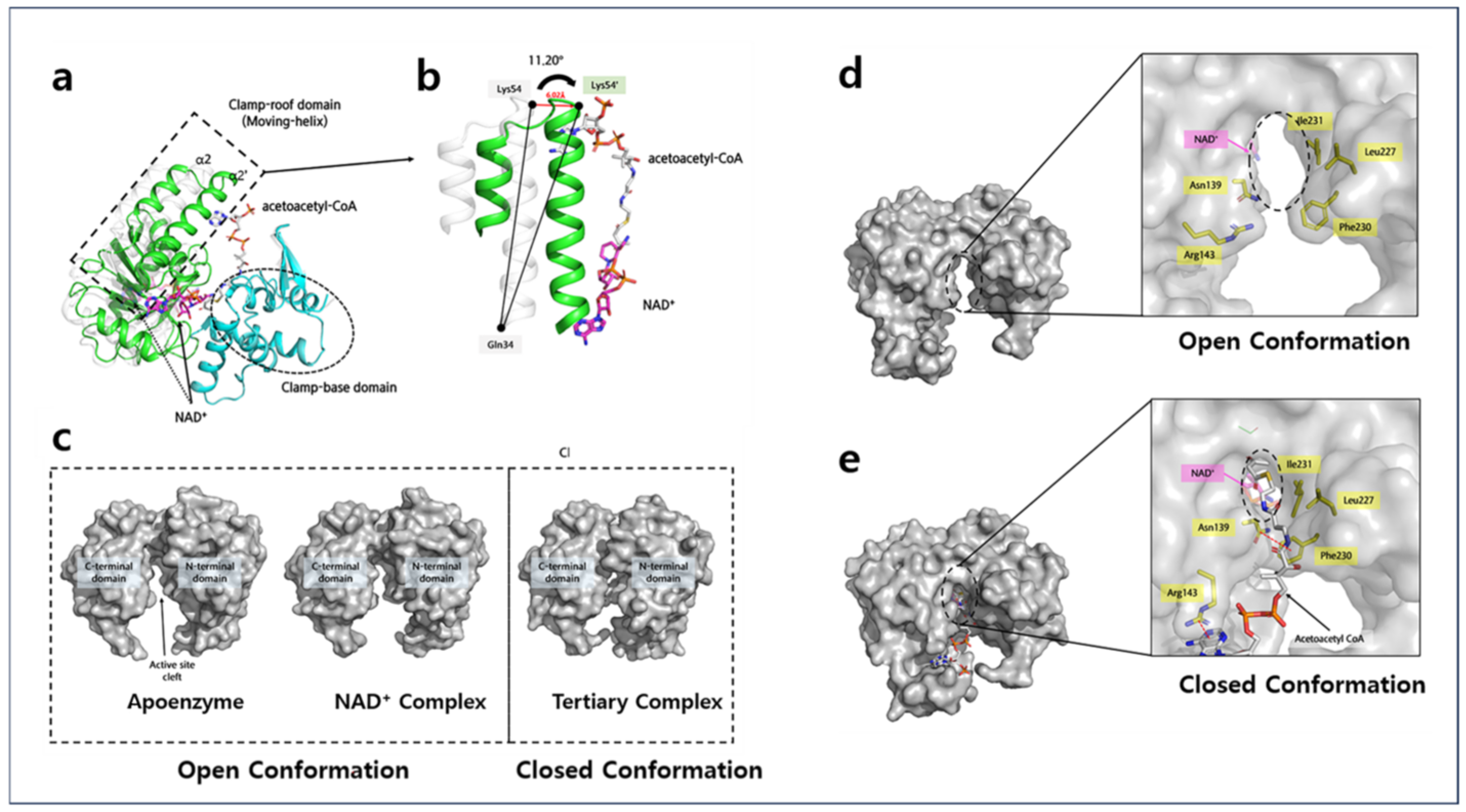
2.1. Overall Structures of Apo-A2HBD Complex with NAD+ and Acetoacetyl-CoA
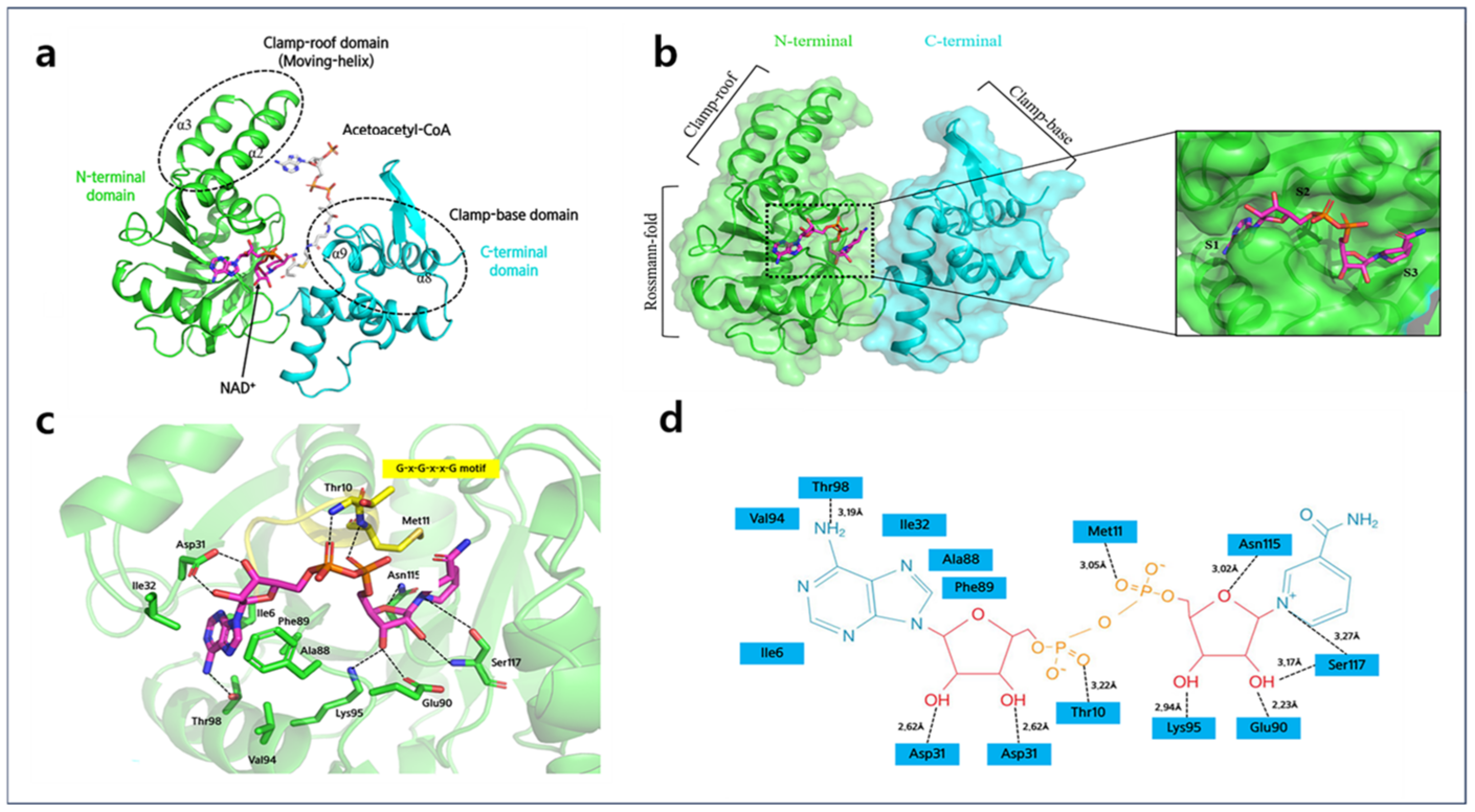
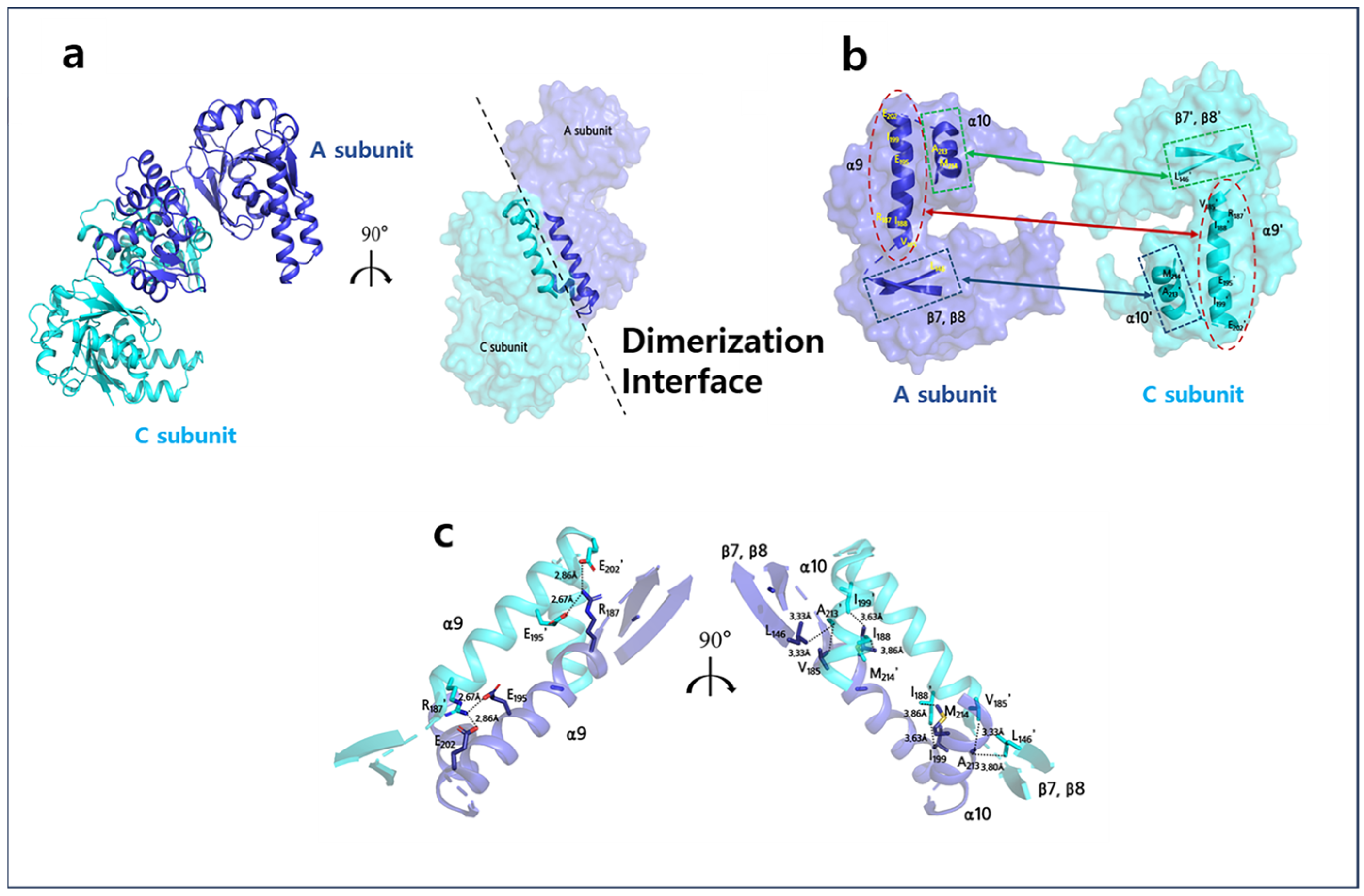
2.2. Monomer of A2HBD
2.3. NAD+ Binding Site of A2HBD
2.4. Acetoacetyl-CoA Binding Sites of A2HBD
3. Discussion
4. Materials and Methods
4.1. Cloning and Protein Expression
4.2. Protein Production and Crystallization
4.3. Data Collection for Apo, NAD+ Complex and Tertiary Comple
4.4. Structure Determination
4.5. Multi-Angle Light Scattering (MALS) Analysis
4.6. MicroScale Thermophoresis (MST) Measurement
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berke, R.; Singh, A.; Guralnick, M. Atopic dermatitis: An overview. Am. Fam. Physician 2012, 86, 35–42. [Google Scholar] [PubMed]
- Yu, J.-S.; Lee, C.-J.; Lee, H.-S.; Kim, J.; Han, Y.; Ahn, K.; Lee, S.-I. Prevalence of atopic dermatitis in Korea: Analysis by using national statistics. J. Korean Med. Sci. 2012, 27, 681. [Google Scholar] [CrossRef]
- Ricci, G.; Patrizi, A.; Baldi, E.; Menna, G.; Tabanelli, M.; Masi, M. Long-term follow-up of atopic dermatitis: Retrospective analysis of related risk factors and association with concomitant allergic diseases. J. Am. Acad. Dermatol. 2006, 55, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Beasley, R.; The International Study of Asthma and Allergies in Childhood Steering Committee. Worldwide variation in prevalence of symptoms of asthma allergic rhinoconjunctivitis, and atopic eczema: ISAAC. Lancet 1998, 351, 1225–1232. [Google Scholar] [CrossRef]
- Lee, J.Y.; Kim, J.; Ahn, K. Time trends in the prevalence of atopic dermatitis in Korean children according to age. Allergy Asthma Immunol. Res. 2022, 14, 123. [Google Scholar] [CrossRef] [PubMed]
- Ehlers, S.; Kaufmann, S.H. Infection, inflammation, and chronic diseases: Consequences of a modern lifestyle. Trends Immunol. 2010, 31, 184–190. [Google Scholar] [CrossRef]
- Lee, Y.K.; Mazmanian, S.K. Has the microbiota played a critical role in the evolution of the adaptive immune system? Science 2010, 330, 1768–1773. [Google Scholar] [CrossRef]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef]
- Trompette, A.; Pernot, J.; Perdijk, O.; Alqahtani, R.A.A.; Santo Domingo, J.; Camacho-Muñoz, D.; Wong, N.C.; Kendall, A.C.; Wiederkehr, A.; Nicod, L.P. Gut-derived short-chain fatty acids modulate skin barrier integrity by promoting keratinocyte metabolism and differentiation. Mucosal Immunol. 2022, 15, 908–926. [Google Scholar] [CrossRef]
- Song, H.; Yoo, Y.; Hwang, J.; Na, Y.-C.; Kim, H.S. Faecalibacterium prausnitzii subspecies–level dysbiosis in the human gut microbiome underlying atopic dermatitis. J. Allergy Clin. Immunol. 2016, 137, 852–860. [Google Scholar] [CrossRef]
- Luo, C.-H.; Lai, A.C.-Y.; Chang, Y.-J. Butyrate inhibits Staphylococcus aureus-aggravated dermal IL-33 expression and skin inflammation through histone deacetylase inhibition. Front. Immunol. 2023, 14, 1114699. [Google Scholar] [CrossRef] [PubMed]
- Kiefer, D.; Ali-Akbarian, L. A brief evidence-based review of two gastrointestinal illnesses: Irritable bowel and leaky gut syndromes. Integr. Med. 2004, 3, 1–11. [Google Scholar]
- Takenoya, M.; Taguchi, S.; Yajima, S. Crystal structure and kinetic analyses of a hexameric form of (S)-3-hydroxybutyryl-CoA dehydrogenase from Clostridium acetobutylicum. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2018, 74, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.-J.; Kim, J.; Ahn, J.-W.; Kim, Y.-J.; Chang, J.H.; Kim, K.-J. Crystal structure of (S)-3-hydroxybutyryl-CoA dehydrogenase from Clostridium butyricum and its mutations that enhance reaction kinetics. J. Microbiol. Biotechnol. 2014, 24, 1636–1643. [Google Scholar] [CrossRef] [PubMed]
- Barycki, J.J.; O’Brien, L.K.; Bratt, J.M.; Zhang, R.; Sanishvili, R.; Strauss, A.W.; Banaszak, L.J. Biochemical characterization and crystal structure determination of human heart short chain L-3-hydroxyacyl-CoA dehydrogenase provide insights into catalytic mechanism. Biochemistry 1999, 38, 5786–5798. [Google Scholar] [CrossRef] [PubMed]
- Tóth, G.; Watts, C.R.; Murphy, R.F.; Lovas, S. Significance of aromatic-backbone amide interactions in protein structure. Proteins Struct. Funct. Bioinform. 2001, 43, 373–381. [Google Scholar] [CrossRef]
- Infield, D.T.; Rasouli, A.; Galles, G.D.; Chipot, C.; Tajkhorshid, E.; Ahern, C.A. Cation-π interactions and their functional roles in membrane proteins. J. Mol. Biol. 2021, 433, 167035. [Google Scholar] [CrossRef]
- Yang, S.-Y.; He, X.-Y. Molecular mechanisms of fatty acid β-oxidation enzyme catalysis. In Current Views of Fatty Acid Oxidation and Ketogenesis: From Organelles to Point Mutations; Quant, P.A., Eaton, S., Eds.; Springer: Boston, MA, USA, 1999; pp. 133–143. [Google Scholar]
- He, X.-Y.; Zhang, G.; Blecha, F.; Yang, S.-Y. Identity of heart and liver L-3-hydroxyacyl coenzyme A dehydrogenase. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 1999, 1437, 119–123. [Google Scholar] [CrossRef]
- Barycki, J.J.; O’BRIEN, L.K.; Birktoft, J.J.; Strauss, A.W.; Banaszak, L.J. Pig heart short chain L-3-hydroxyacyl-CoA dehydrogenase revisited: Sequence analysis and crystal structure determination. Protein Sci. 1999, 8, 2010–2018. [Google Scholar] [CrossRef]
- Barycki, J.J.; O’Brien, L.K.; Strauss, A.W.; Banaszak, L.J. Glutamate 170 of human l-3-hydroxyacyl-CoA dehydrogenase is required for proper orientation of the catalytic histidine and structural integrity of the enzyme. J. Biol. Chem. 2001, 276, 36718–36726. [Google Scholar] [CrossRef]
- Park, J. Structural Analysis of 3-Hydroxybutyryl-CoA Dehyrogenase from Faecalibacterium prausnitzii A2-165. Master’s Thesis, Korea University, Seoul, Republic of Korea, 2021. [Google Scholar]
- Boutet, S.; Lomb, L.; Williams, G.J.; Barends, T.R.; Aquila, A.; Doak, R.B.; Weierstall, U.; DePonte, D.P.; Steinbrener, J.; Shoeman, R.L. High-resolution protein structure determination by serial femtosecond crystallography. Science 2012, 337, 362–364. [Google Scholar] [CrossRef] [PubMed]
- Rayment, I. Small-scale batch crystallization of proteins revisited: An underutilized way to grow large protein crystals. Structure 2002, 10, 147–151. [Google Scholar] [CrossRef] [PubMed]
- ZaM, W.O. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276 Pt A, 20. [Google Scholar]
- Martiel, I.; Müller-Werkmeister, H.M.; Cohen, A.E. Strategies for sample delivery for femtosecond crystallography. Biol. Crystallogr. 2019, 75, 160–177. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Nam, K.H. Sample delivery systems for serial femtosecond crystallography at the PAL-XFEL. Photonics 2023, 10, 557. [Google Scholar] [CrossRef]
- Barty, A.; Kirian, R.A.; Maia, F.R.; Hantke, M.; Yoon, C.H.; White, T.A.; Chapman, H. Cheetah: Software for high-throughput reduction and analysis of serial femtosecond X-ray diffraction data. J. Appl. Crystallogr. 2014, 47, 1118–1131. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with phenix. refine. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012, 68, 352–367. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef]
- DeLano, W.L.; Lam, J.W. PyMOL: A communications tool for computational models. In Abstracts of Papers of the American Chemical Society; American Chemical Society: Washington, DC, USA, 2005; Volume 230, pp. U1371–U1372. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Apo | Native with NAD | S117A with AACoA | S117A with AACoA&NAD | |
|---|---|---|---|---|
| Data collection | ||||
| X-ray source | PAL11C | PAL11C | SFX | PAL11C |
| Wavelength (Å) | 1.000 | 1.000 | 1.000 | 1.000 |
| Space group | P21 | P21 | P3212 | P3212 |
| a, b, c (Å) | 87.66, 126.61, 108.09 | 90.82, 80.48, 128.35 | 91.01, 91.01, 212.7 | 90.56, 90.56,212.50 |
| α, β, γ (°) | 90.0, 110.2, 90.0 | 90.0, 102.2, 90.0 | 90, 90, 120 | 90, 90, 120 |
| Resolution (Å) | 48.04–2.69 (2.72–2.69) | 46.64–2.55 (2.58–2.55) | 45.51–1.90 (1.97–1.90) | 45.28–2.20 (2.23–2.2) |
| Unique reflections | 60,712 (1807) | 54,530 (1522) | 190,068 (7874) | 50,509 (1392) |
| Completeness | 96.7 (78.3) | 91.7 (77.2) | 99.99 (100.0) | 99.0 (83.6) |
| Redundancy | 6.8 (5.5) | 4.1 (2.3) | 3794 (189.7) | 10.2 (3.8) |
| I/σ | 10 | 23.1 | 26.1 | 15.6 |
| Rmerge (%) | 0.22 (0.75) | 0.08 (0.25) | 0.09 (0.26) | 0.19 (0.70) |
| Refinement statistics | ||||
| Resolution (A) | 48.04–2.69 | 46.64–2.55 | 45.51–1.9 | 45.28–2.20 |
| Reflections used in refinement | 60,712 | 54,530 | 190,068 | 50,509 |
| Rwork/Rfree | 0.2458/0.2943 | 0.2058/0.2661 | 0.1795/0.2224 | 0.1988/0.2442 |
| R.M.S. deviations | ||||
| Bond lengths (A) | 0.011 | 0.017 | 0.010 | 0.022 |
| Bond angles (°) | 1.18 | 1.24 | 1.26 | 1.4 |
| Ramachandran plot (%) | ||||
| Favored | 92.28 | 93.94 | 96.26 | 99.06 |
| Allowed | 6.16 | 5.65 | 3.51 | 0.94 |
| No. atoms | ||||
| Protein | 12,930 | 12,939 | 6423 | 6423 |
| Ligands | 0 | 264 | 98 | 98 |
| Solvent | 0 | 79 | 250 | 250 |
| B-factors | 52 | 62.3 | 26.2 | 37.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.; Jeon, H.J.; Park, S.; Park, J.; Jang, S.; Shin, B.; Bang, K.; Hawkes, H.-J.K.; Park, S.; Kim, S.; et al. Structural Insights and Catalytic Mechanism of 3-Hydroxybutyryl-CoA Dehydrogenase from Faecalibacterium Prausnitzii A2-165. Int. J. Mol. Sci. 2024, 25, 10711. https://doi.org/10.3390/ijms251910711
Yang J, Jeon HJ, Park S, Park J, Jang S, Shin B, Bang K, Hawkes H-JK, Park S, Kim S, et al. Structural Insights and Catalytic Mechanism of 3-Hydroxybutyryl-CoA Dehydrogenase from Faecalibacterium Prausnitzii A2-165. International Journal of Molecular Sciences. 2024; 25(19):10711. https://doi.org/10.3390/ijms251910711
Chicago/Turabian StyleYang, Jaewon, Hyung Jin Jeon, Seonha Park, Junga Park, Seonhye Jang, Byeongmin Shin, Kyuhyeon Bang, Hye-Jin Kim Hawkes, Sungha Park, Sulhee Kim, and et al. 2024. "Structural Insights and Catalytic Mechanism of 3-Hydroxybutyryl-CoA Dehydrogenase from Faecalibacterium Prausnitzii A2-165" International Journal of Molecular Sciences 25, no. 19: 10711. https://doi.org/10.3390/ijms251910711
APA StyleYang, J., Jeon, H. J., Park, S., Park, J., Jang, S., Shin, B., Bang, K., Hawkes, H.-J. K., Park, S., Kim, S., & Hwang, K. Y. (2024). Structural Insights and Catalytic Mechanism of 3-Hydroxybutyryl-CoA Dehydrogenase from Faecalibacterium Prausnitzii A2-165. International Journal of Molecular Sciences, 25(19), 10711. https://doi.org/10.3390/ijms251910711