
Characterization and Hemocompatibility of α, β, and γ Cyclodextrin-Modified Magnetic Nano-Adsorbents

 , , and
, , and
Abstract
1. Introduction
2. Results and Discussion
2.1. Magnetic Nanoparticle Properties
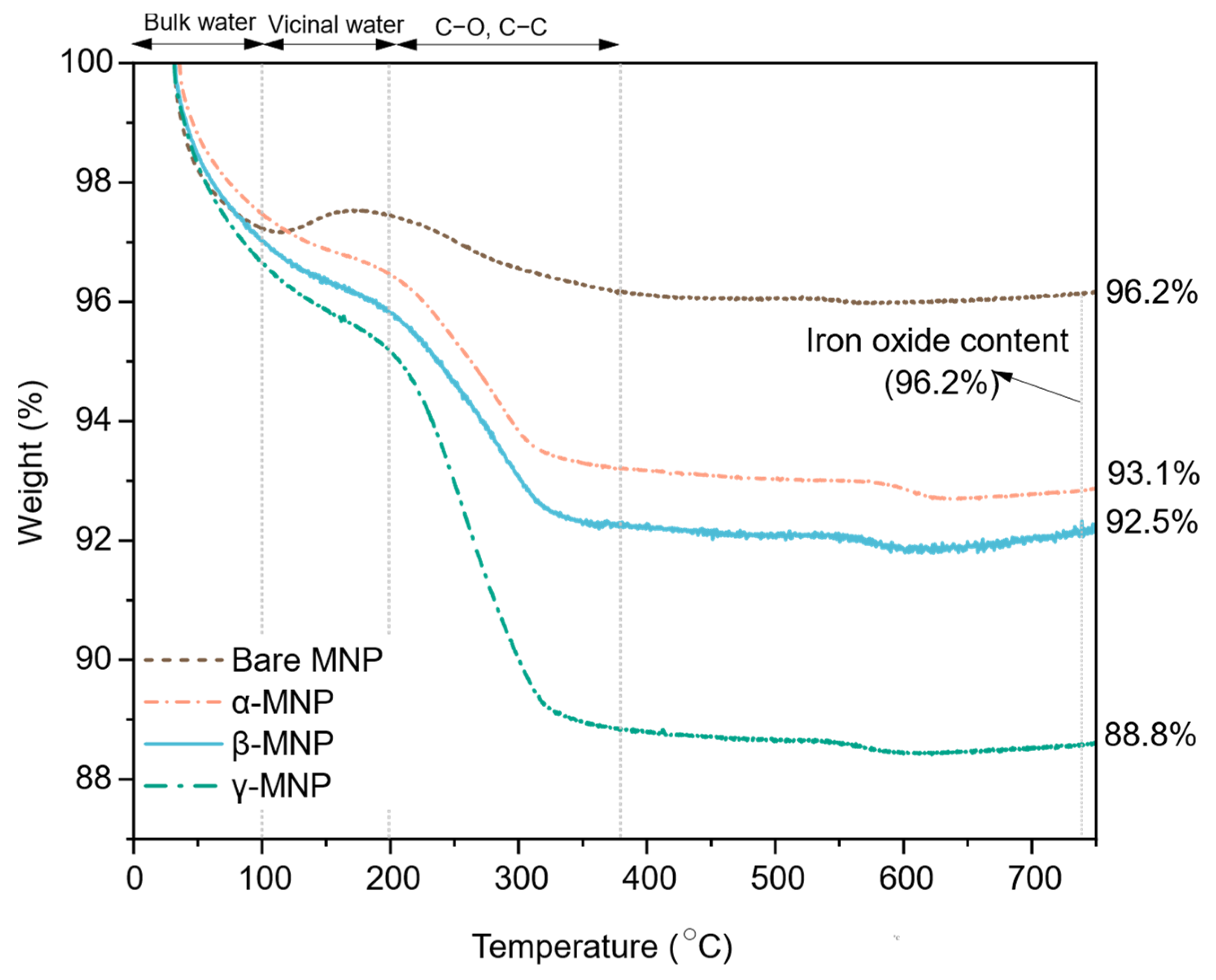
2.2. Film Hydration and Weight Loss
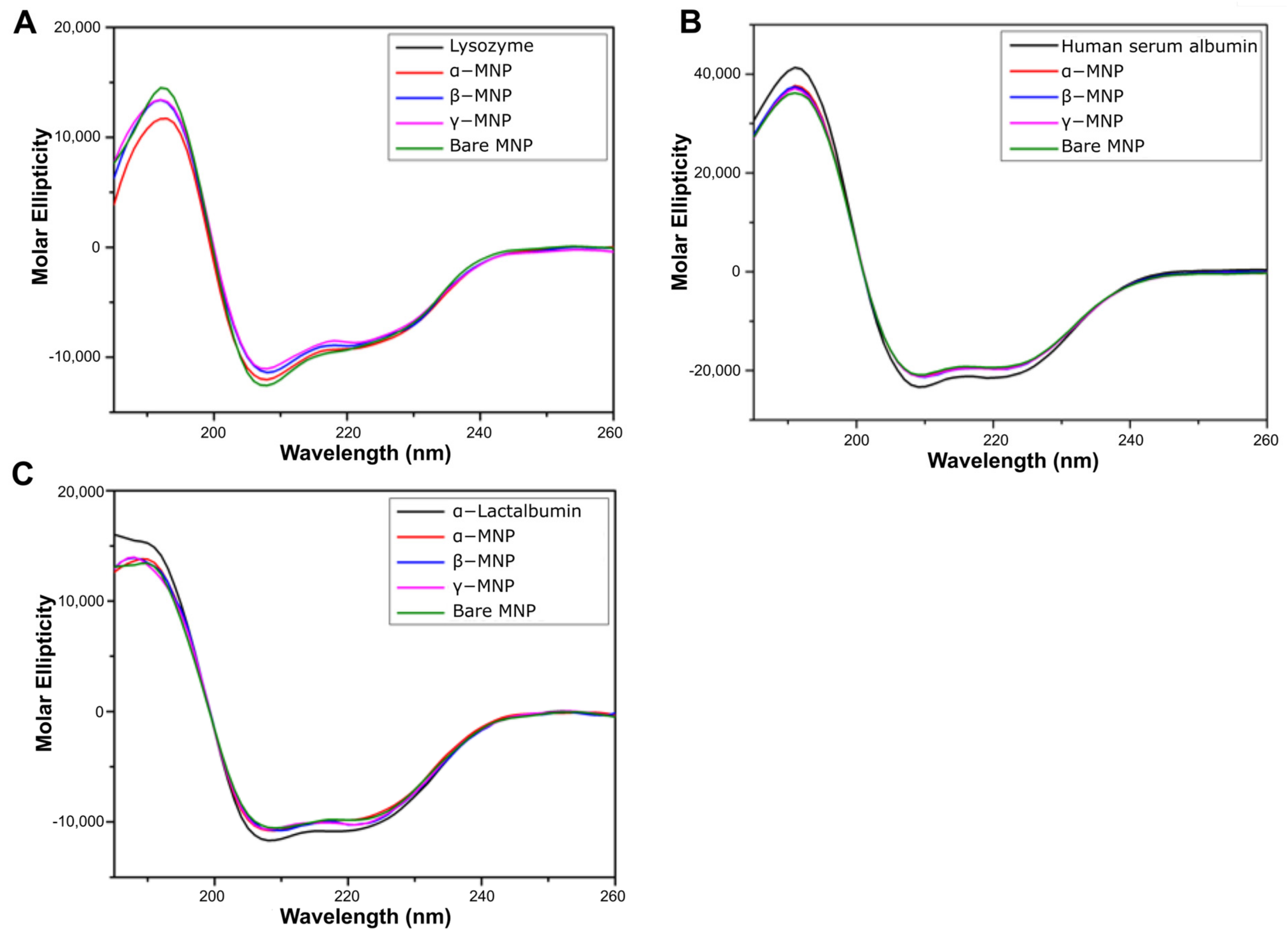
2.3. Effect of Coating on Adsorbed Protein Secondary Structure
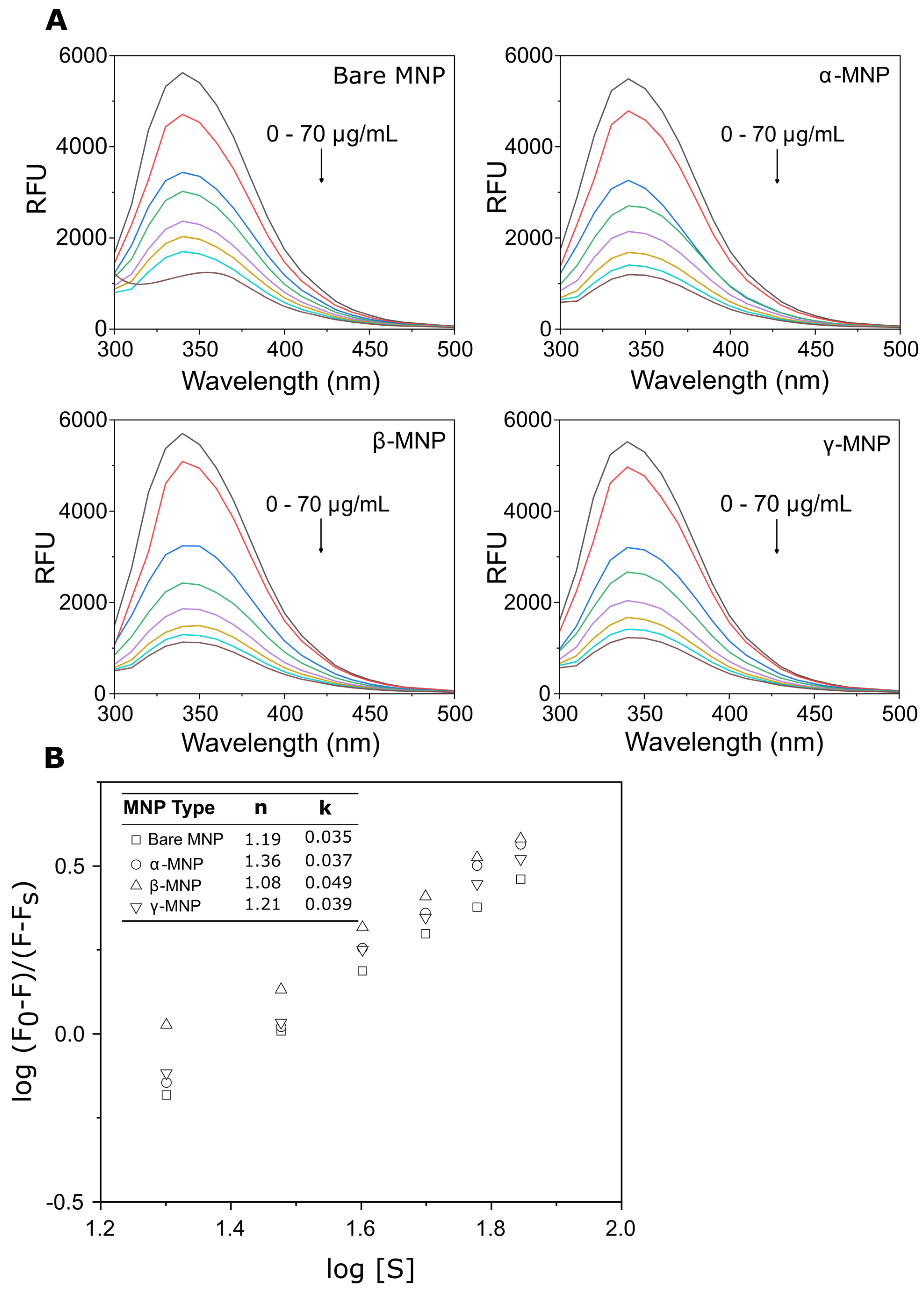
2.4. Effect of Coating on Adsorbed HSA Tertiary Structure
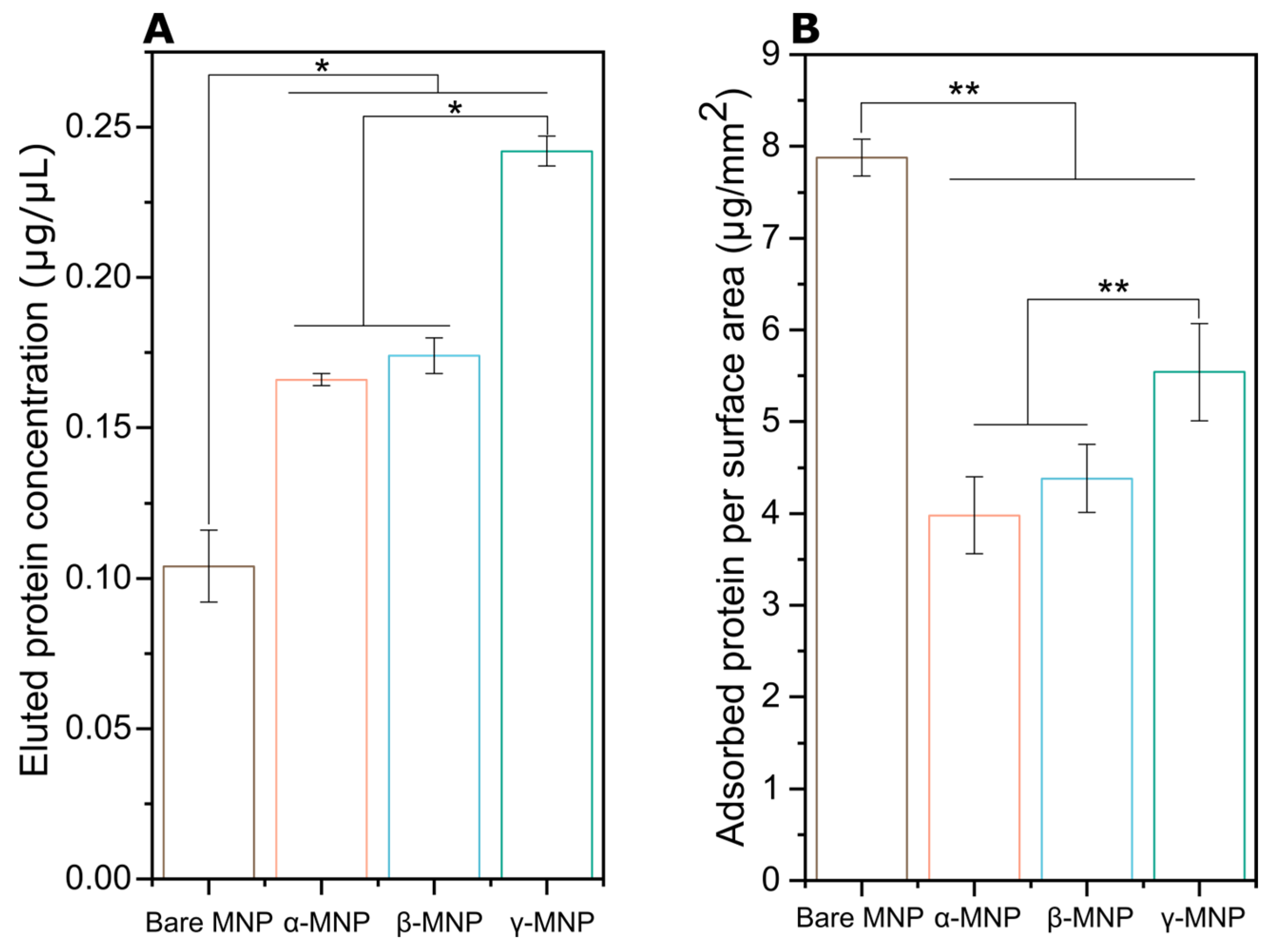
2.5. Total Adsorbed Protein
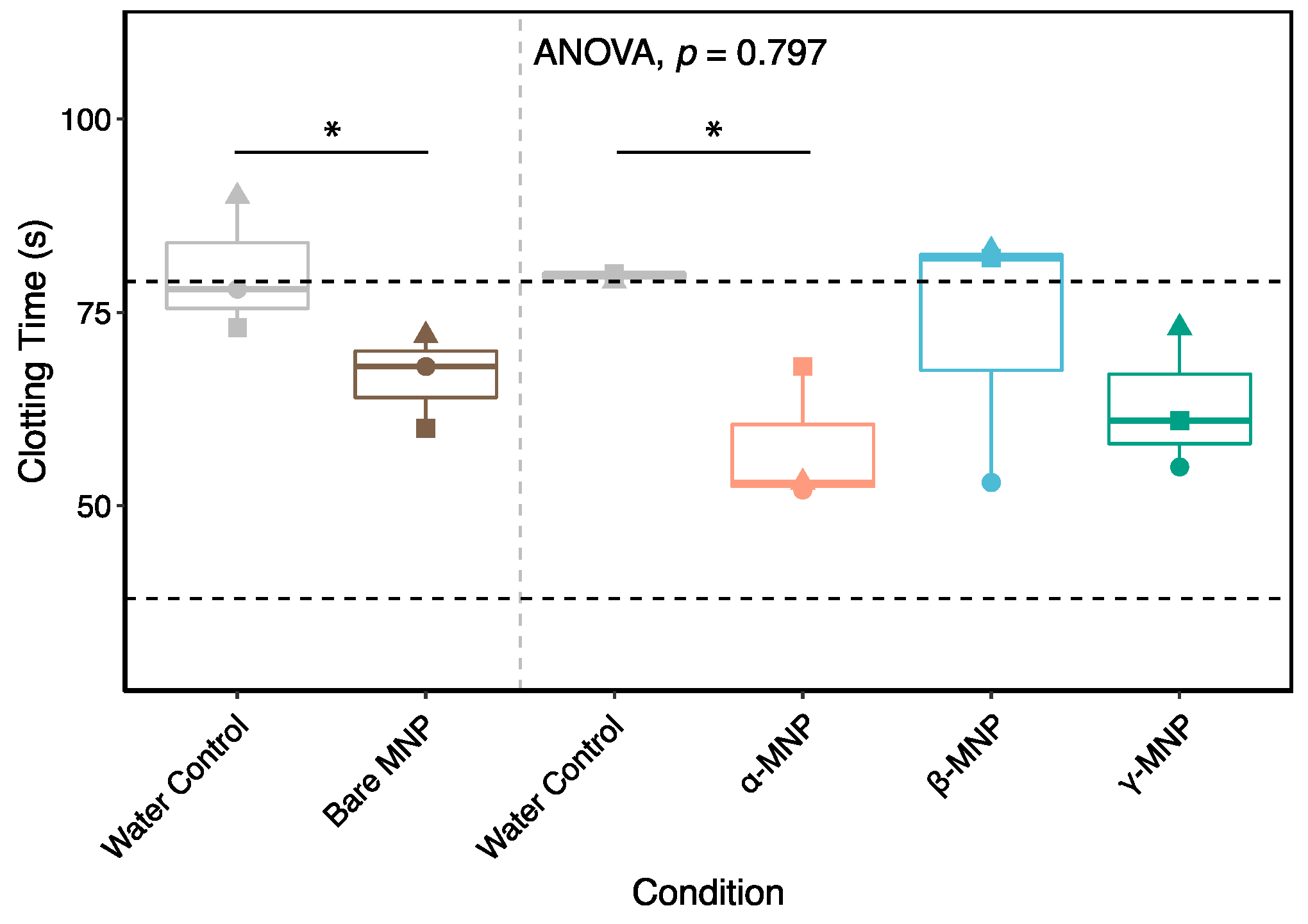
2.6. Coating Effect on Coagulation
2.7. Quantification of Plasma Proteins Adsorption
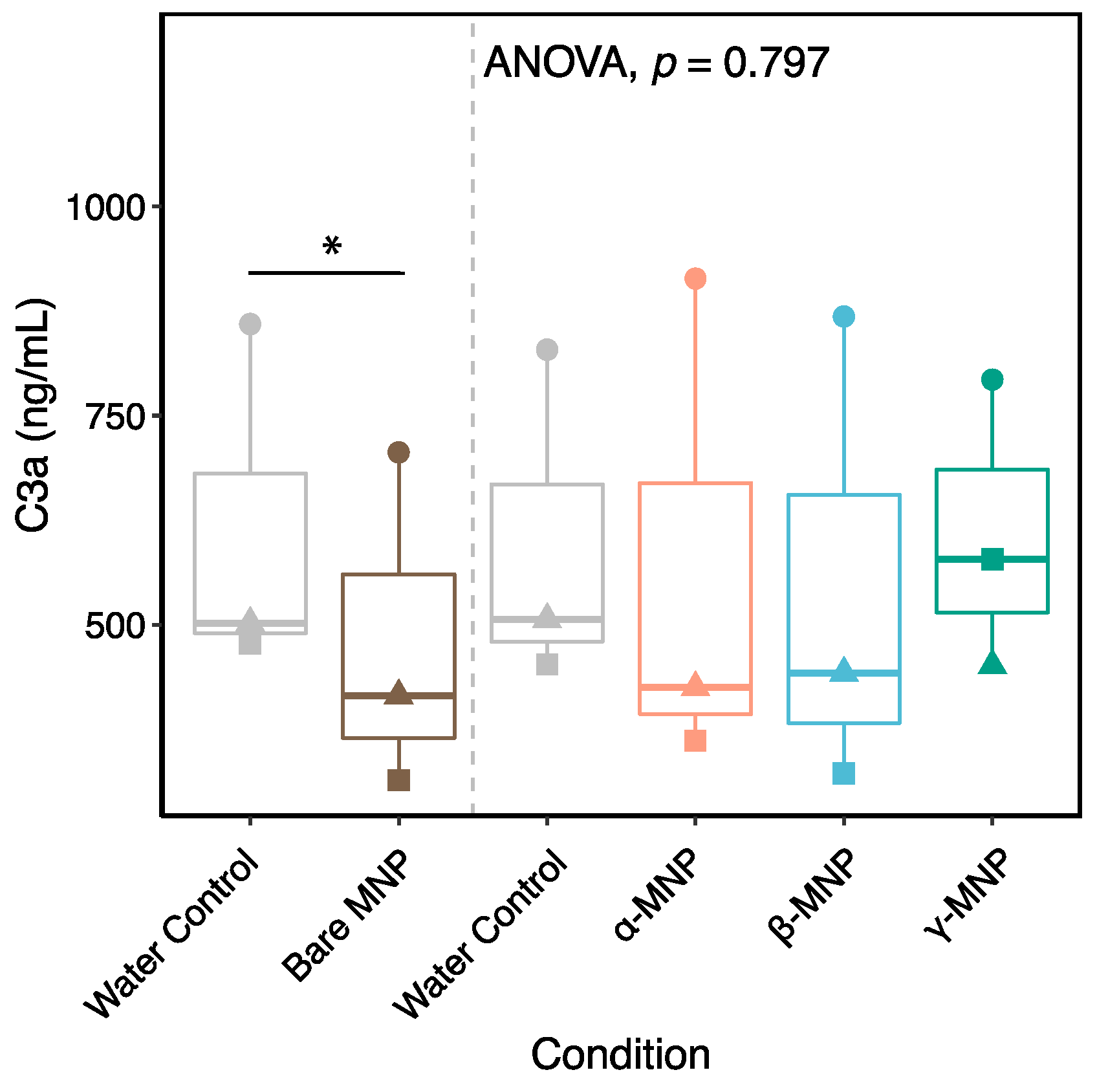
2.8. Immune Response-Related Proteins
2.9. Plasma Proteins Involved in Coagulation
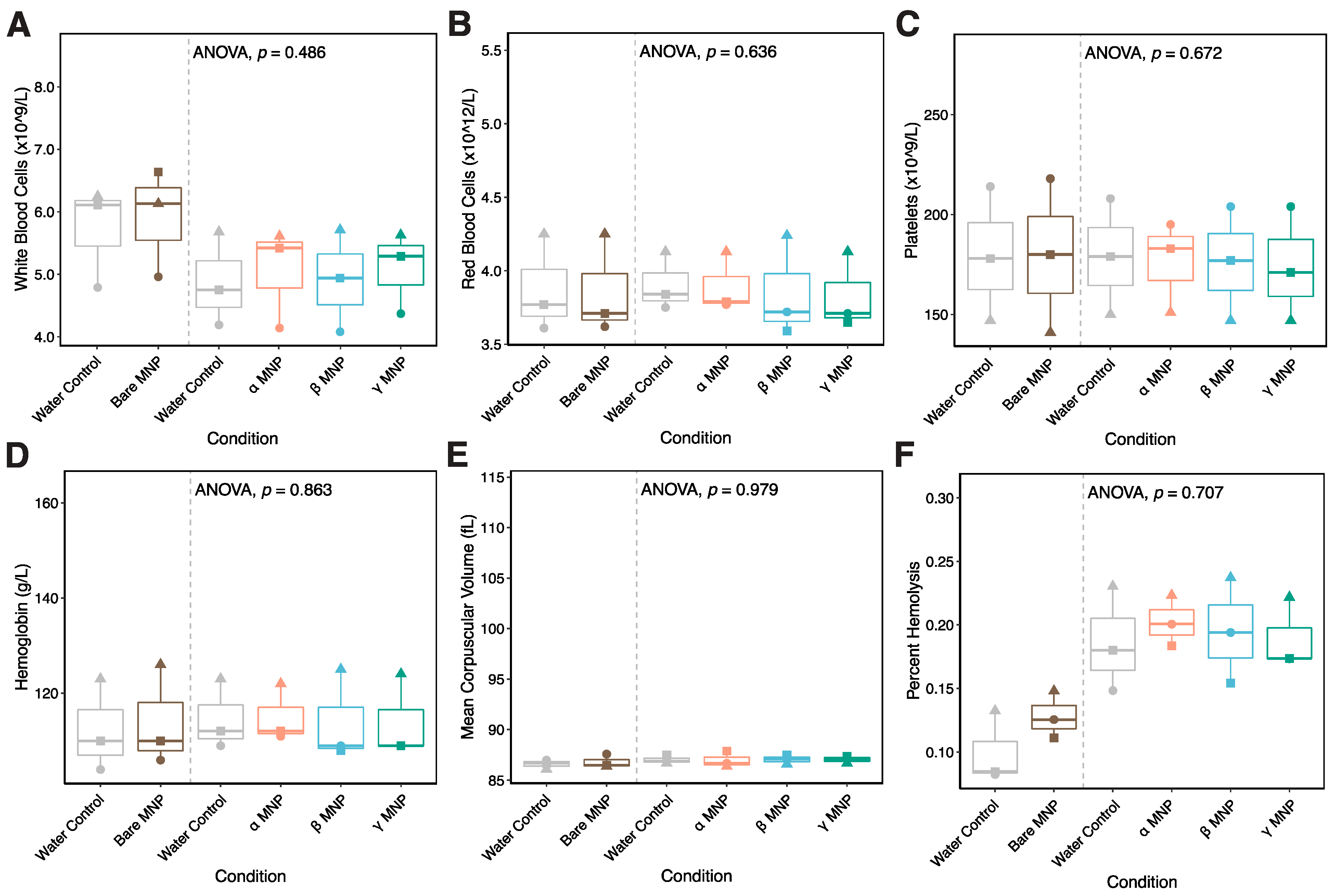
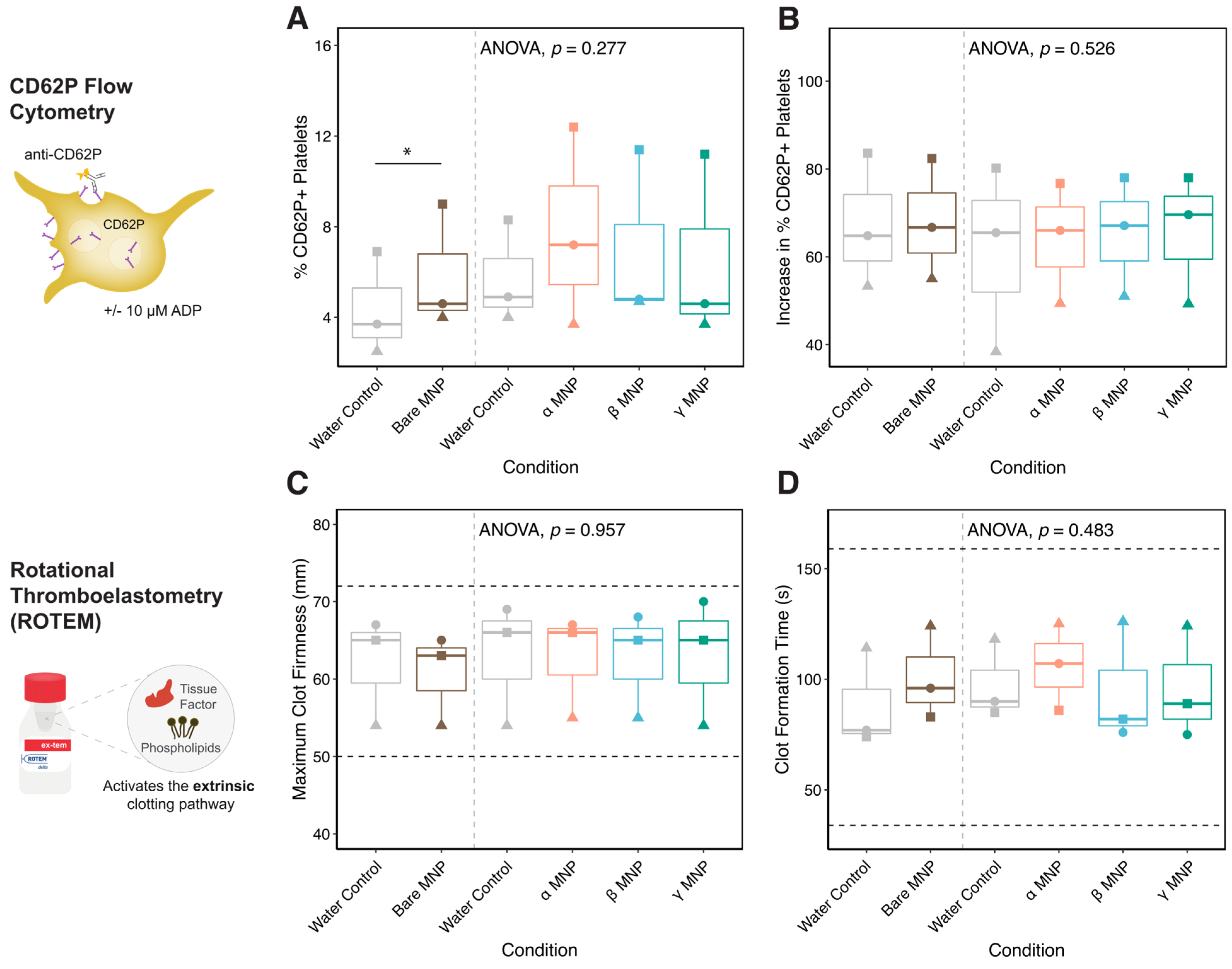
2.10. Whole Blood Hemocompatibility and Platelet Function
3. Conclusions
4. Materials and Methods
4.1. Materials
4.2. Methods
4.2.1. Synthesis of CD-Coated MNPs
4.2.2. Characterization of MNPs
4.2.3. Fluorescence Spectroscopy
4.2.4. SDS-PAGE and Immunoblot
4.2.5. Circular Dichroism
4.2.6. Whole Blood Hemocompatibility Testing
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kovesdy, C.P. Epidemiology of chronic kidney disease: An update 2022. Kidney Int. Suppl. 2022, 12, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Rosner, M.H.; Reis, T.; Husain-Syed, F.; Vanholder, R.; Hutchison, C.; Stenvinkel, P.; Blankestijn, P.J.; Cozzolino, M.; Juillard, L.; Kashani, K. Classification of uremic toxins and their role in kidney failure. Clin. J. Am. Soc. Nephrol. 2021, 16, 1918–1928. [Google Scholar] [CrossRef] [PubMed]
- Magnani, S.; Atti, M. Uremic Toxins and Blood Purification: A Review of Current Evidence and Future Perspectives. Toxins 2021, 13, 246. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Zhang, Q.; Yang, Y.; Li, Y.; Lin, W. Recent trends in therapeutic application of engineered blood purification materials for kidney disease. Biomater. Res. 2022, 26, 5. [Google Scholar] [CrossRef]
- Ma, Y.; Li, S.; Tonelli, M.; Unsworth, L.D. Adsorption-based strategies for removing uremic toxins from blood. Microporous Mesoporous Mater. 2021, 319, 111035. [Google Scholar] [CrossRef]
- Liu, Q.; Zhou, Y.; Lu, J.; Zhou, Y. Novel cyclodextrin-based adsorbents for removing pollutants from wastewater: A critical review. Chemosphere 2020, 241, 125043. [Google Scholar] [CrossRef]
- Wankar, J.; Kotla, N.G.; Gera, S.; Rasala, S.; Pandit, A.; Rochev, Y.A. Recent advances in host–guest self-assembled cyclodextrin carriers: Implications for responsive drug delivery and biomedical engineering. Adv. Funct. Mater. 2020, 30, 1909049. [Google Scholar] [CrossRef]
- Zhang, Y.M.; Liu, Y.H.; Liu, Y. Cyclodextrin-based multistimuli-responsive supramolecular assemblies and their biological functions. Adv. Mater. 2020, 32, 1806158. [Google Scholar] [CrossRef]
- Davis, M.E.; Brewster, M.E. Cyclodextrin-based pharmaceutics: Past, present and future. Nat. Rev. Drug Discov. 2004, 3, 1023–1035. [Google Scholar] [CrossRef]
- Tran, N.; Webster, T.J. Magnetic nanoparticles: Biomedical applications and challenges. J. Mater. Chem. 2010, 20, 8760–8767. [Google Scholar] [CrossRef]
- Cai, K.; Li, J.; Luo, Z.; Hu, Y.; Hou, Y.; Ding, X. β-Cyclodextrin conjugated magnetic nanoparticles for diazepam removal from blood. Chem. Commun. 2011, 47, 7719–7721. [Google Scholar] [CrossRef] [PubMed]
- Shelat, R.; Chandra, S.; Khanna, A. Detailed toxicity evaluation of β-cyclodextrin coated iron oxide nanoparticles for biomedical applications. Int. J. Biol. Macromol. 2018, 110, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Jaffer, I.H.; Weitz, J.I. The blood compatibility challenge. Part 1: Blood-contacting medical devices: The scope of the problem. Acta Biomater. 2019, 94, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Mu, Q.; Zhang, B.; Yan, B. Analytical strategies for detecting nanoparticle–protein interactions. Analyst 2010, 135, 1519–1530. [Google Scholar] [CrossRef]
- Cedervall, T.; Lynch, I.; Lindman, S.; Berggård, T.; Thulin, E.; Nilsson, H.; Dawson, K.A.; Linse, S. Understanding the nanoparticle–protein corona using methods to quantify exchange rates and affinities of proteins for nanoparticles. Proc. Natl. Acad. Sci. USA 2007, 104, 2050–2055. [Google Scholar] [CrossRef]
- Brash, J.L.; Horbett, T.A.; Latour, R.A.; Tengvall, P. The blood compatibility challenge. Part 2: Protein adsorption phenomena governing blood reactivity. Acta Biomater. 2019, 94, 11–24. [Google Scholar] [CrossRef]
- Burns, N.L.; Holmberg, K.; Brink, C. Influence of surface charge on protein adsorption at an amphoteric surface: Effects of varying acid to base ratio. J. Colloid Interface Sci. 1996, 178, 116–122. [Google Scholar] [CrossRef]
- Phillips, S.; Wilson, L.; Borkman, R. Acrylamide and iodide fluorescence quenching as a structural probe of tryptophan microenvironment in bovine lens crystallins. Curr. Eye Res. 1986, 5, 611–620. [Google Scholar] [CrossRef]
- Dabaghi, M.; Hilger, I. Magnetic nanoparticles behavior in biological solutions; the impact of clustering tendency on sedimentation velocity and cell uptake. Materials 2020, 13, 1644. [Google Scholar] [CrossRef]
- Yallapu, M.M.; Othman, S.F.; Curtis, E.T.; Gupta, B.K.; Jaggi, M.; Chauhan, S.C. Multi-functional magnetic nanoparticles for magnetic resonance imaging and cancer therapy. Biomaterials 2011, 32, 1890–1905. [Google Scholar] [CrossRef]
- Monteiro, A.P.; Caminhas, L.D.; Ardisson, J.D.; Paniago, R.; Cortés, M.E.; Sinisterra, R.D. Magnetic nanoparticles coated with cyclodextrins and citrate for irinotecan delivery. Carbohydr. Polym. 2017, 163, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.-W.; Lee, H.; Song, Y.; Sohn, D. Colloidal stability of iron oxide nanoparticles with multivalent polymer surfactants. J. Colloid Interface Sci. 2015, 443, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Poulson, B.G.; Alsulami, Q.A.; Sharfalddin, A.; El Agammy, E.F.; Mouffouk, F.; Emwas, A.-H.; Jaremko, L.; Jaremko, M. Cyclodextrins: Structural, chemical, and physical properties, and applications. Polysaccharides 2021, 3, 1–31. [Google Scholar] [CrossRef]
- Hădărugă, N.G.; Bandur, G.N.; David, I.; Hădărugă, D.I. A review on thermal analyses of cyclodextrins and cyclodextrin complexes. Environ. Chem. Lett. 2019, 17, 349–373. [Google Scholar] [CrossRef]
- Ghosh, S.; Paul, B.K.; Chattopadhyay, N. Interaction of cyclodextrins with human and bovine serum albumins: A combined spectroscopic and computational investigation. J. Chem. Sci. 2014, 126, 931–944. [Google Scholar] [CrossRef]
- Tayeh, N.; Rungassamy, T.; Albani, J.R. Fluorescence spectral resolution of tryptophan residues in bovine and human serum albumins. J. Pharm. Biomed. Anal. 2009, 50, 107–116. [Google Scholar] [CrossRef]
- Li, S.; Sharaf, M.G.; Rowe, E.M.; Serrano, K.; Devine, D.V.; Unsworth, L.D. Hemocompatibility of β-Cyclodextrin-Modified (Methacryloyloxy) ethyl Phosphorylcholine Coated Magnetic Nanoparticles. Biomolecules 2023, 13, 1165. [Google Scholar] [CrossRef]
- Dobrovolskaia, M.A.; Patri, A.K.; Zheng, J.; Clogston, J.D.; Ayub, N.; Aggarwal, P.; Neun, B.W.; Hall, J.B.; McNeil, S.E. Interaction of colloidal gold nanoparticles with human blood: Effects on particle size and analysis of plasma protein binding profiles. Nanomed. Nanotechnol. Biol. Med. 2009, 5, 106–117. [Google Scholar] [CrossRef]
- Kowalczyńska, H.M.; Nowak-Wyrzykowska, M.; Szczepankiewicz, A.A.; Dobkowski, J.; Dyda, M.; Kamiński, J.; Kołos, R. Albumin adsorption on unmodified and sulfonated polystyrene surfaces, in relation to cell–substratum adhesion. Colloids Surf. B Biointerfaces 2011, 84, 536–544. [Google Scholar] [CrossRef]
- Dabare, P.R.L.; Bachhuka, A.; Parkinson-Lawrence, E.; Vasilev, K. Surface nanotopography mediated albumin adsorption, unfolding and modulation of early innate immune responses. Mater. Today Adv. 2021, 12, 100187. [Google Scholar] [CrossRef]
- Victor, S.P.; Sharma, C.P. Development and evaluation of cyclodextrin complexed hydroxyapatite nanoparticles for preferential albumin adsorption. Colloids Surf. B Biointerfaces 2011, 85, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Ogun, A.S.; Adeyinka, A. Biochemistry, Transferrin. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2018. [Google Scholar]
- Andersson, J.; Ekdahl, K.N.; Lambris, J.D.; Nilsson, B. Binding of C3 fragments on top of adsorbed plasma proteins during complement activation on a model biomaterial surface. Biomaterials 2005, 26, 1477–1485. [Google Scholar] [CrossRef] [PubMed]
- Bahniuk, M.S.; Alshememry, A.K.; Unsworth, L.D. Human plasma protein adsorption to elastin-like polypeptide nanoparticles. Biointerphases 2020, 15, 021007. [Google Scholar] [CrossRef] [PubMed]
- de Nooijer, A.H.; Grondman, I.; Janssen, N.A.; Netea, M.G.; Willems, L.; van de Veerdonk, F.L.; Giamarellos-Bourboulis, E.J.; Toonen, E.J.; Joosten, L.A. Complement activation in the disease course of coronavirus disease 2019 and its effects on clinical outcomes. J. Infect. Dis. 2021, 223, 214–224. [Google Scholar] [CrossRef]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG subclasses and allotypes: From structure to effector functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef]
- Dunkelberger, J.R.; Song, W.-C. Complement and its role in innate and adaptive immune responses. Cell Res. 2010, 20, 34–50. [Google Scholar] [CrossRef]
- Bahniuk, M.S.; Pirayesh, H.; Singh, H.D.; Nychka, J.A.; Unsworth, L.D. Bioactive glass 45S5 powders: Effect of synthesis route and resultant surface chemistry and crystallinity on protein adsorption from human plasma. Biointerphases 2012, 7, 41. [Google Scholar] [CrossRef]
- Harrison, P.M.; Arosio, P. The ferritins: Molecular properties, iron storage function and cellular regulation. Biochim. Biophys. Acta (BBA)-Bioenerg. 1996, 1275, 161–203. [Google Scholar] [CrossRef]
- Stafford, J.L.; Belosevic, M. Transferrin and the innate immune response of fish: Identification of a novel mechanism of macrophage activation. Dev. Comp. Immunol. 2003, 27, 539–554. [Google Scholar] [CrossRef]
- Sano, K.; Asanuma-Date, K.; Arisaka, F.; Hattori, S.; Ogawa, H. Changes in glycosylation of vitronectin modulate multimerization and collagen binding during liver regeneration. Glycobiology 2007, 17, 784–794. [Google Scholar] [CrossRef]
- McNally, A.K.; Jones, J.A.; MacEwan, S.R.; Colton, E.; Anderson, J.M. Vitronectin is a critical protein adhesion substrate for IL-4-induced foreign body giant cell formation. J. Biomed. Mater. Res. Part A Off. J. Soc.Biomater. Jpn. Soc.Biomater. Aust. Soc. Biomater. Korean Soc. Biomater. 2008, 86, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Bale, M.D.; Wohlfahrt, L.A.; Mosher, D.F.; Tomasini, B.; Sutton, R.C. Identification of vitronectin as a major plasma protein adsorbed on polymer surfaces of different copolymer composition. Blood 1989, 74, 2698–2706. [Google Scholar] [CrossRef]
- Horbett, T.A. Fibrinogen adsorption to biomaterials. J. Biomed. Mater. Res. Part A 2018, 106, 2777–2788. [Google Scholar] [CrossRef] [PubMed]
- Berclaz, P.-Y.; Trapnell, B.C. Rare Childhood Lung Disorders: α1-Antitrypsin Deficiency, Pulmonary Alveolar Proteinosis, and Pulmonary Alveolar Microlithiasis. In Kendig’s Disorders of the Respiratory Tract in Children; Elsevier: Amsterdam, The Netherlands, 2006; pp. 747–761. [Google Scholar]
- Chidambaranathan-Reghupaty, S.; Fisher, P.B.; Sarkar, D. Hepatocellular carcinoma (HCC): Epidemiology, etiology and molecular classification. Adv. Cancer Res. 2021, 149, 1–61. [Google Scholar]
- Charbgoo, F.; Nejabat, M.; Abnous, K.; Soltani, F.; Taghdisi, S.M.; Alibolandi, M.; Shier, W.T.; Steele, T.W.; Ramezani, M. Gold nanoparticle should understand protein corona for being a clinical nanomaterial. J. Control. Release 2018, 272, 39–53. [Google Scholar] [CrossRef]
- Armstrong, P.B.; Quigley, J.P. α2-macroglobulin: An evolutionarily conserved arm of the innate immune system. Dev. Comp. Immunol. 1999, 23, 375–390. [Google Scholar] [CrossRef]
- Vandooren, J.; Itoh, Y. Alpha-2-macroglobulin in inflammation, immunity and infections. Front. Immunol. 2021, 12, 803244. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.H.; Welsby, I.; Goodnough, L.T. Fibrinogen as a therapeutic target for bleeding: A review of critical levels and replacement therapy. Transfusion 2014, 54, 1389–1405. [Google Scholar] [CrossRef]
- Lowe, G.D.; Rumley, A.; Mackie, I.J. Plasma fibrinogen. Ann. Clin. Biochem. 2004, 41, 430–440. [Google Scholar] [CrossRef]
- Nava-Ortiz, C.A.; Burillo, G.; Concheiro, A.; Bucio, E.; Matthijs, N.; Nelis, H.; Coenye, T.; Alvarez-Lorenzo, C. Cyclodextrin-functionalized biomaterials loaded with miconazole prevent Candida albicans biofilm formation in vitro. Acta Biomater. 2010, 6, 1398–1404. [Google Scholar] [CrossRef]
- Krishnaswamy, S. The transition of prothrombin to thrombin. J. Thromb. Haemost. 2013, 11, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Ashikaga, H.; Chien, K.R. Chapter 28—Blood Coagulation and Atherothrombosis. In Molecular Basis of Cardiovascular Disease, 2nd ed.; Chien, K.R., Ed.; W.B. Saunders: Philadelphia, PA, USA, 2004; pp. 498–518. [Google Scholar]
- Wu, Y. Contact pathway of coagulation and inflammation. Thromb. J. 2015, 13, 17. [Google Scholar] [CrossRef]
- Elechalawar, C.K.; Hossen, M.N.; McNally, L.; Bhattacharya, R.; Mukherjee, P. Analysing the nanoparticle-protein corona for potential molecular target identification. J. Control. Release 2020, 322, 122–136. [Google Scholar] [CrossRef]
- Lundqvist, M.; Augustsson, C.; Lilja, M.; Lundkvist, K.; Dahlbäck, B.; Linse, S.; Cedervall, T. The nanoparticle protein corona formed in human blood or human blood fractions. PLoS ONE 2017, 12, e0175871. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.; Verhamme, I.M.; Sun, M.-F.; Mohammed, B.; Cheng, Q.; Matafonov, A.; Dickeson, S.K.; Joseph, K.; Kaplan, A.P.; Gailani, D. Protease activity in single-chain prekallikrein. Blood J. Am. Soc. Hematol. 2020, 135, 558–567. [Google Scholar] [CrossRef]
- Veloso, D.; Colman, R.W. Western blot analyses of prekallikrein and its activation products in human plasma. Thromb. Haemost. 1991, 65, 382–388. [Google Scholar] [CrossRef]
- Raspi, G. Kallikrein and kallikrein-like proteinases: Purification and determination by chromatographic and electrophoretic methods. J. Chromatogr. B Biomed. Sci. Appl. 1996, 684, 265–287. [Google Scholar] [CrossRef] [PubMed]
- Keragala, C.B.; Medcalf, R.L. Plasminogen: An enigmatic zymogen. Blood J. Am. Soc. Hematol. 2021, 137, 2881–2889. [Google Scholar] [CrossRef]
- Woodhouse, K.; Brash, J. Adsorption of plasminogen from plasma to lysine-derivatized polyurethane surfaces. Biomaterials 1992, 13, 1103–1108. [Google Scholar] [CrossRef]
- Rogers, H.J.; Nakashima, M.O.; Kottke-Marchant, K. Hemostasis and thrombosis. In Hematopathology; Elsevier: Amsterdam, The Netherlands, 2018; pp. 57–105.e104. [Google Scholar]
- Gierula, M.; Ahnström, J. Anticoagulant protein S—New insights on interactions and functions. J. Thromb. Haemost. 2020, 18, 2801–2811. [Google Scholar] [CrossRef]
- Harboe, M. A method for determination of hemoglobin in plasma by near-ultraviolet spectrophotometry. Scand. J. Clin. Lab. Investig. 1959, 11, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Beaven, G.H.; Chen, S.H.; D’albis, A.; Gratzer, W.B. A spectroscopic study of the haemin–human-serum-albumin system. Eur. J. Biochem. 1974, 41, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Chipman, D.M.; Grisaro, V.; Sharon, N. The binding of oligosaccharides containing n-acetylglucosamine and n-acetylmuramic acid to lysozyme: The specificity of binding subsites. J. Biol. Chem. 1967, 242, 4388–4394. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MNP Type | Cluster Size via DLS (nm) | Polydispersity Index (PI) | Particle Diameter via TEM (nm) | Zeta Potential (mV) |
|---|---|---|---|---|
| Bare | 301 ± 23 | 0.65 ± 0.22 | 11.36 ± 2.30 | 5.0 ± 0.2 a |
| α-MNP | 94.4 ± 0.7 | 0.14 ± 0.02 | 8.69 ± 1.81 | −24 ± 1.9 b |
| β-MNP | 99.2 ± 0.7 | 0.18 ± 0.03 | 8.50 ± 1.51 | −22 ± 0.6 c |
| γ-MNP | 90.1 ± 0.1 | 0.19 ± 0.01 | 7.69 ± 1.37 | −22 ± 0.5 d |
| MNP Type | Helix | β-Sheet | β-Turn | Random Coil | |
|---|---|---|---|---|---|
| Lysozyme | Blank | 32.7 ± 0.1 | 8.1 ± 0.0 | 18.1 ± 0.0 | 28.5 ± 0.2 |
| α- | 30.6 ± 0.0 | 8.2 ± 0.2 | 18.1 ± 0.0 | 28.9 ± 0.0 | |
| β- | 30.6 ± 0.2 | 8.2 ± 0.2 | 18.1 ± 0.0 | 28.9 ± 0.0 | |
| γ- | 30.4 ± 0.1 | 8.5 ± 0.0 | 17.8 ± 0.1 | 30.2 ± 0.3 | |
| Bare | 32.7 ± 0.3 | 8.1 ± 0.1 | 18.1 ± 0.0 | 28.5 ± 0.1 | |
| Human serum albumin | Blank | 70.9 ± 0.0 | 4.1 ± 0.2 | 12.6 ± 0.1 | 12.2 ± 0.0 |
| α- | 71.2 ± 0.1 | 4.1 ± 0.3 | 12.5 ± 0.2 | 12.1 ± 0.1 | |
| β- | 71.0 ± 0.0 | 4.1 ± 0.2 | 12.5 ± 0.1 | 12.3 ± 0.0 | |
| γ- | 70.3 ± 0.0 | 4.3 ± 0.1 | 12.8 ± 0.0 | 12.8 ± 0.3 | |
| Bare | 71.4 ± 0.1 | 4.1 ± 0.0 | 12.5 ± 0.2 | 12.1 ± 0.1 | |
| α-Lactalbumin | Blank | 32.3 ± 0.2 | 20.8 ± 0.1 | 18.0 ± 0.0 | 28.9 ± 0.1 |
| α- | 32.6 ± 0.0 | 20.3 ± 0.0 | 17.9 ± 0.1 | 29.3 ± 0.2 | |
| β- | 32.6 ± 0.1 | 20.3 ± 0.2 | 17.9 ± 0.1 | 29.3 ± 0.1 | |
| γ- | 31.5 ± 0.2 | 21.1 ± 0.3 | 17.9 ± 0.1 | 29.4 ± 0.2 | |
| Bare | 32.3 ± 0.2 | 20.8 ± 0.1 | 18.0 ± 0.0 | 28.9 ± 0.0 |
| MNP Type | |||||||
|---|---|---|---|---|---|---|---|
| Plasma Proteins | Fragment Size (kDa) | Bare | α-CD | β-CD | γ-CD | ||
| Albumin | 66 | 9 | 9 | 8 | 9 | 0 | |
| 1 to 3 | |||||||
| Immune response-related | C3 | 4 to 5 | |||||
| 6 to 7 | |||||||
| Whole C3 | 187 | 4 | 4 | 3 | 3 | 8 to 9 | |
| α chain | 115 | 6 | 5 | 3 | 3 | 10 to 11 | |
| β chain | 70 | 9 | 9 | 8 | 8 | 12 | |
| Activation fragment | 42 | 6 | 6 | 6 | 6 | ||
| IgG | |||||||
| Heavy chain | 55 | 6 | 6 | 6 | 6 | ||
| Light chain | 27 | 9 | 9 | 8 | 8 | ||
| Transferrin | 77 | 9 | 9 | 8 | 8 | ||
| Vitronectin | 54 | 8 | 9 | 9 | 9 | ||
| α1 antitrypsin | 54 | 6 | 7 | 7 | 7 | ||
| α2 macroglobulin | 163 | 1 | 1 | 0 | 0 | ||
| Coagulation-related | Fibrinogen | ||||||
| α chain | 68 | 5 | 5 | 4 | 4 | ||
| β chain | 56 | 4 | 4 | 4 | 4 | ||
| γ chain | 48 | 4 | 5 | 4 | 4 | ||
| Cleavage fragments | <48 | 1 | 4 | 4 | 4 | ||
| Prothrombin | 72 | 2 | 2 | 2 | 2 | ||
| Antithrombin | 53 | 8 | 7 | 7 | 7 | ||
| Factor I | 88 | 0 | 0 | 0 | 0 | ||
| Factor XII | 80 | 6 | 7 | 7 | 7 | ||
| Factor XI | 70 | 9 | 9 | 10 | 10 | ||
| Prekallikrein | 85 | 5 | 5 | 4 | 4 | ||
| 50 | 7 | 6 | 6 | 6 | |||
| Plasminogen | 91 | 9 | 9 | 7 | 6 | ||
| Kininogen | 120 | 0 | 0 | 0 | 0 | ||
| Fibronectin | 440 | 0 | 0 | 0 | 0 | ||
| Protein C | 62 | 0 | 0 | 0 | 0 | ||
| Protein S | 69 | 0 | 0 | 0 | 0 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghaffari Sharaf, M.; Li, S.; Rowe, E.M.; Devine, D.V.; Unsworth, L.D. Characterization and Hemocompatibility of α, β, and γ Cyclodextrin-Modified Magnetic Nano-Adsorbents. Int. J. Mol. Sci. 2024, 25, 10710. https://doi.org/10.3390/ijms251910710
Ghaffari Sharaf M, Li S, Rowe EM, Devine DV, Unsworth LD. Characterization and Hemocompatibility of α, β, and γ Cyclodextrin-Modified Magnetic Nano-Adsorbents. International Journal of Molecular Sciences. 2024; 25(19):10710. https://doi.org/10.3390/ijms251910710
Chicago/Turabian StyleGhaffari Sharaf, Mehdi, Shuhui Li, Elyn M. Rowe, Dana V. Devine, and Larry D. Unsworth. 2024. "Characterization and Hemocompatibility of α, β, and γ Cyclodextrin-Modified Magnetic Nano-Adsorbents" International Journal of Molecular Sciences 25, no. 19: 10710. https://doi.org/10.3390/ijms251910710
APA StyleGhaffari Sharaf, M., Li, S., Rowe, E. M., Devine, D. V., & Unsworth, L. D. (2024). Characterization and Hemocompatibility of α, β, and γ Cyclodextrin-Modified Magnetic Nano-Adsorbents. International Journal of Molecular Sciences, 25(19), 10710. https://doi.org/10.3390/ijms251910710