
Cannabinoids and Genetic Epilepsy Models: A Review with Focus on CDKL5 Deficiency Disorder

 , and
, and {kind=link}
{kind=link}
Abstract
:1. Introduction
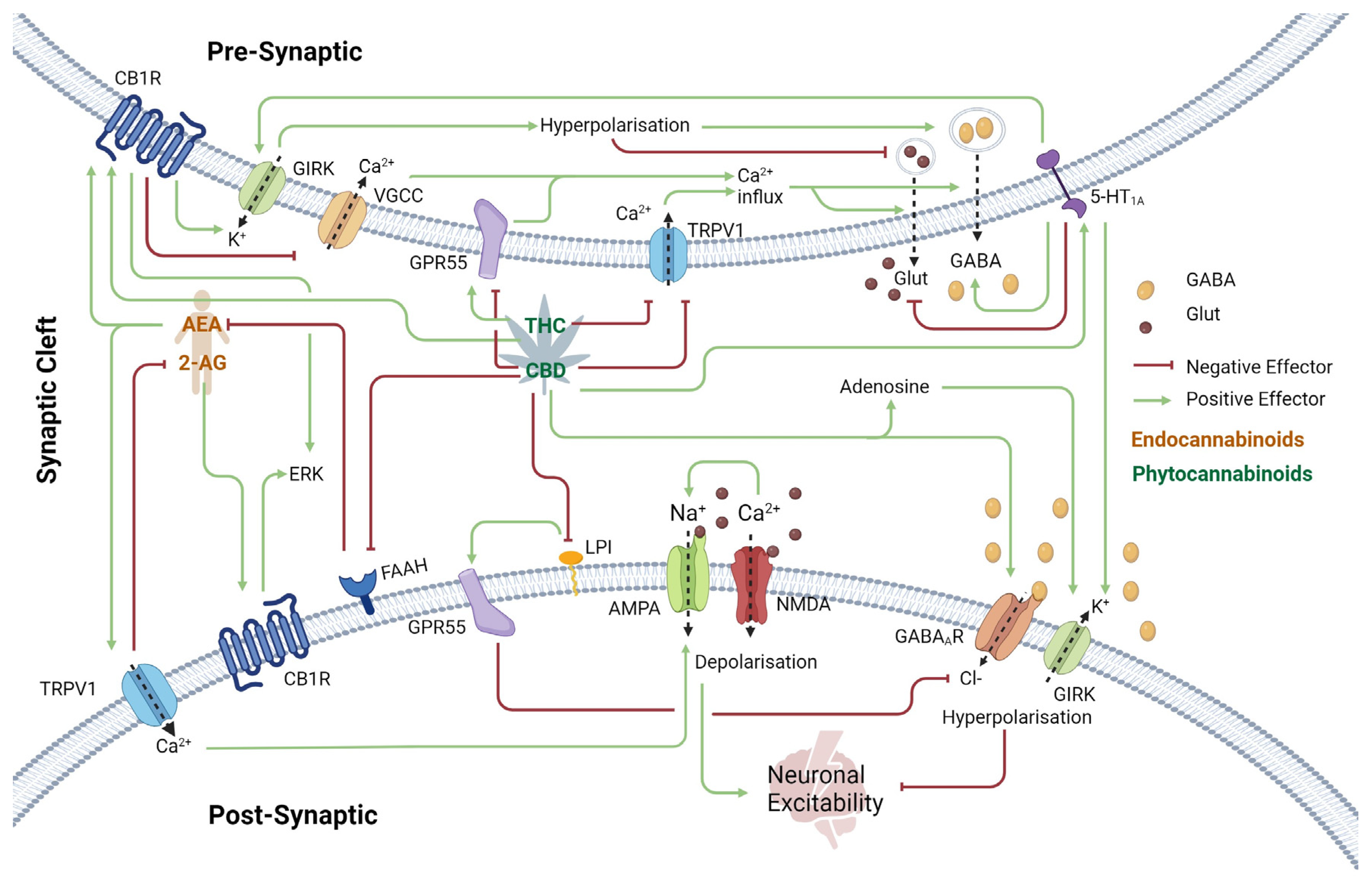
2. The Endocannabinoid System and Its Role in Epilepsy and CDKL5 Deficiency Disorder
2.1. Cannabinoid Receptor 1 (CB1R) in the Central Nervous System
2.2. Transient Receptor Potential Vanilloid 1 (TRPV1) Receptor
2.3. Excitatory and Inhibitory Neurotransmission Receptors
2.4. Other G-Protein-Coupled Receptors
2.5. Neurotransmitter Modulation: Adenosine and Anandamide
3. Human Cannabidiol Trials
Limitations of Cannabinoid Trials
4. Epilepsy Model Systems
4.1. Zebrafish as a Model of CDD and Cannabinoid Use
4.2. Rodent as a Model of CDD and Cannabinoid Use
4.3. CDD-Specific Rodent Models
4.4. Effects of Cannabinoids on Seizure Frequency in Rodents
5. Human iPSC-Derived Neuronal Models
5.1. Two-Dimensional Cell Models
5.2. Three-Dimensional Cell Models
6. Trace Cannabinoids and Their Promising Anti-Epileptic Effects
7. Conclusions
8. Future Directions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Burstein, S. Cannabidiol (CBD) and its analogs: A review of their effects on inflammation. Bioorg. Med. Chem. 2015, 23, 1377–1385. [Google Scholar] [CrossRef]
- Mlost, J.; Bryk, M.; Starowicz, K. Cannabidiol for pain treatment: Focus on pharmacology and mechanism of action. Int. J. Mol. Sci. 2020, 21, 8870. [Google Scholar] [CrossRef] [PubMed]
- Patricio, F.; Morales-Andrade, A.A.; Patricio-Martínez, A.; Limón, I.D. Cannabidiol as a therapeutic target: Evidence of its neuroprotective and neuromodulatory function in Parkinson’s disease. Front. Pharmacol. 2020, 11, 595635. [Google Scholar] [CrossRef] [PubMed]
- Cooray, R.; Gupta, V.; Suphioglu, C. Current aspects of the endocannabinoid system and targeted THC and CBD phytocannabinoids as potential therapeutics for Parkinson’s and Alzheimer’s diseases: A review. Mol. Neurobiol. 2020, 57, 4878–4890. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, T.; Kondo, S.; Sukagawa, A.; Nakane, S.; Shinoda, A.; Itoh, K.; Yamashita, A.; Waku, K. 2-Arachidonoylgylcerol: A possible endogenous cannabinoid receptor ligand in brain. Biochem. Biophys. Res. Commun. 1995, 215, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, V.; Melck, D.; Bisogno, T.; De Petrocellis, L. Endocannabinoids: Endogenous cannabinoid receptor ligands with neuromodulatory action. Trends Neurosci. 1998, 21, 521–528. [Google Scholar] [CrossRef]
- Millán-Guerrero, R.O.; Isais-Millán, S. Cannabis and the exocannabinoid and endocannabinoid systems. Their use and controversies. Gac. Medica Mex. 2019, 155, 471–474. [Google Scholar] [CrossRef]
- Stockings, E.; Zagic, D.; Campbell, G.; Weier, M.; Hall, W.D.; Nielsen, S.; Herkes, G.K.; Farrell, M.; Degenhardt, L. Evidence for cannabis and cannabinoids for epilepsy: A systematic review of controlled and observational evidence. J. Neurol. Neurosurg. Psychiatry 2018, 89, 741–753. [Google Scholar] [CrossRef]
- Weaving, L.S.; Christodoulou, J.; Williamson, S.L.; Friend, K.L.; McKenzie, O.L.; Archer, H.; Evans, J.; Clarke, A.; Pelka, G.J.; Tam, P.P. Mutations of CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and mental retardation. Am. J. Hum. Genet. 2004, 75, 1079–1093. [Google Scholar] [CrossRef]
- Tao, J.; Van Esch, H.; Hagedorn-Greiwe, M.; Hoffmann, K.; Moser, B.; Raynaud, M.; Sperner, J.; Fryns, J.-P.; Schwinger, E.; Gécz, J. Mutations in the X-linked cyclin-dependent kinase–like 5 (CDKL5/STK9) gene are associated with severe neurodevelopmental retardation. Am. J. Hum. Genet. 2004, 75, 1149–1154. [Google Scholar] [CrossRef]
- Fehr, S.; Downs, J.; Ho, G.; de Klerk, N.; Forbes, D.; Christodoulou, J.; Williams, S.; Leonard, H. Functional abilities in children and adults with the CDKL5 disorder. Am. J. Med. Genet. Part A 2016, 170, 2860–2869. [Google Scholar] [CrossRef] [PubMed]
- Fehr, S.; Wong, K.; Chin, R.; Williams, S.; de Klerk, N.; Forbes, D.; Krishnaraj, R.; Christodoulou, J.; Downs, J.; Leonard, H. Seizure variables and their relationship to genotype and functional abilities in the CDKL5 disorder. Neurology 2016, 87, 2206–2213. [Google Scholar] [CrossRef]
- Bahi-Buisson, N.; Nectoux, J.; Rosas-Vargas, H.; Milh, M.; Boddaert, N.; Girard, B.; Cances, C.; Ville, D.; Afenjar, A.; Rio, M. Key clinical features to identify girls with CDKL5 mutations. Brain 2008, 131, 2647–2661. [Google Scholar] [CrossRef] [PubMed]
- Demarest, S.T.; Olson, H.E.; Moss, A.; Pestana-Knight, E.; Zhang, X.; Parikh, S.; Swanson, L.C.; Riley, K.D.; Bazin, G.A.; Angione, K. CDKL5 deficiency disorder: Relationship between genotype, epilepsy, cortical visual impairment, and development. Epilepsia 2019, 60, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Klein, K.; Yendle, S.; Harvey, A.; Antony, J.; Wallace, G.; Bienvenu, T.; Scheffer, I. A distinctive seizure type in patients with CDKL5 mutations: Hypermotor-tonic-spasms sequence. Neurology 2011, 76, 1436–1438. [Google Scholar] [CrossRef]
- Takeda, K.; Miyamoto, Y.; Yamamoto, H.; Ishii, A.; Hirose, S.; Yamamoto, H. Clinical features of early myoclonic encephalopathy caused by a CDKL5 mutation. Brain Dev. 2020, 42, 73–76. [Google Scholar] [CrossRef]
- Pintaudi, M.; Baglietto, M.G.; Gaggero, R.; Parodi, E.; Pessagno, A.; Marchi, M.; Russo, S.; Veneselli, E. Clinical and electroencephalographic features in patients with CDKL5 mutations: Two new Italian cases and review of the literature. Epilepsy Behav. 2008, 12, 326–331. [Google Scholar] [CrossRef]
- Fallah, M.S.; Eubanks, J.H. Seizures in mouse models of rare neurodevelopmental disorders. Neurosci. Lett. 2020, 445, 50–68. [Google Scholar] [CrossRef]
- Leonard, H.; Junaid, M.; Wong, K.; Demarest, S.; Downs, J. Exploring quality of life in individuals with a severe developmental and epileptic encephalopathy, CDKL5 Deficiency Disorder. Epilepsy Res. 2021, 169, 106521. [Google Scholar] [CrossRef]
- Gallop, K.; Lloyd, A.J.; Olt, J.; Marshall, J. Impact of developmental and epileptic encephalopathies on caregivers: A literature review. Epilepsy Behav. 2021, 124, 108324. [Google Scholar] [CrossRef]
- Pisano, T.; Numis, A.L.; Heavin, S.B.; Weckhuysen, S.; Angriman, M.; Suls, A.; Podesta, B.; Thibert, R.L.; Shapiro, K.A.; Guerrini, R. Early and effective treatment of KCNQ 2 encephalopathy. Epilepsia 2015, 56, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.; Junaid, M.; Demarest, S.; Saldaris, J.; Benke, T.A.; Marsh, E.D.; Downs, J.; Leonard, H. Factors influencing the attainment of major motor milestones in CDKL5 deficiency disorder. Eur. J. Hum. Genet. 2023, 31, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.R.; Helbig, I.; Kaufman, M.C.; Schust Myers, L.; Conway, L.; Helbig, K.L. Caregiver assessment of quality of life in individuals with genetic developmental and epileptic encephalopathies. Dev. Med. Child Neurol. 2022, 64, 957–964. [Google Scholar] [CrossRef] [PubMed]
- Tangarorang, J.; Leonard, H.; Epstein, A.; Downs, J. A framework for understanding quality of life domains in individuals with the CDKL5 deficiency disorder. Am. J. Med. Genet. Part A 2019, 179, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Kwan, P.; Brodie, M.J. Early identification of refractory epilepsy. New Engl. J. Med. 2000, 342, 314–319. [Google Scholar] [CrossRef]
- Aznar-Laín, G.; Fernández-Mayoralas, D.M.; Caicoya, A.G.; Rocamora, R.; Pérez-Jurado, L.A. CDKL5 deficiency disorder without epilepsy. Pediatr. Neurol. 2023, 144, 84–89. [Google Scholar] [CrossRef]
- Müller, A.; Helbig, I.; Jansen, C.; Bast, T.; Guerrini, R.; Jähn, J.; Muhle, H.; Auvin, S.; Korenke, G.; Philip, S. Retrospective evaluation of low long-term efficacy of antiepileptic drugs and ketogenic diet in 39 patients with CDKL5-related epilepsy. Eur. J. Paediatr. Neurol. 2016, 20, 147–151. [Google Scholar] [CrossRef]
- Wong, K.; Junaid, M.; Alexander, S.; Olson, H.E.; Pestana-Knight, E.M.; Rajaraman, R.R.; Downs, J.; Leonard, H. Caregiver Perspective of Benefits and Side Effects of Anti-Seizure Medications in CDKL5 Deficiency Disorder from an International Database. CNS Drugs 2024, 65, 2186–2199. [Google Scholar] [CrossRef]
- Alexandre, V., Jr.; Capovilla, G.; Fattore, C.; Franco, V.; Gambardella, A.; Guerrini, R.; La Briola, F.; Ladogana, M.; Rosati, E.; Specchio, L.M. Characteristics of a large population of patients with refractory epilepsy attending tertiary referral centers in Italy. Epilepsia 2010, 51, 921–925. [Google Scholar] [CrossRef]
- Canevini, M.P.; De Sarro, G.; Galimberti, C.A.; Gatti, G.; Licchetta, L.; Malerba, A.; Muscas, G.; La Neve, A.; Striano, P.; Perucca, E. Relationship between adverse effects of antiepileptic drugs, number of coprescribed drugs, and drug load in a large cohort of consecutive patients with drug-refractory epilepsy. Epilepsia 2010, 51, 797–804. [Google Scholar] [CrossRef]
- Kanner, A.M.; Balabanov, A.J. The use of monotherapy in patients with epilepsy: An appraisal of the new antiepileptic drugs. Curr. Neurol. Neurosci. Rep. 2005, 5, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Gilliam, F.; Veloso, F.; Bomhof, M.; Gazda, S.; Biton, V.; Ter Bruggen, J.; Neto, W.; Bailey, C.; Pledger, G.; Wu, S.-C. A dose-comparison trial of topiramate as monotherapy in recently diagnosed partial epilepsy. Neurology 2003, 60, 196–202. [Google Scholar] [CrossRef]
- Akyüz, E.; Köklü, B.; Ozenen, C.; Arulsamy, A.; Shaikh, M.F. Elucidating the potential side effects of current anti-seizure drugs for epilepsy. Curr. Neuropharmacol. 2021, 19, 1865. [Google Scholar] [CrossRef] [PubMed]
- Szaflarski, J.P.; Bebin, E.M.; Comi, A.M.; Patel, A.D.; Joshi, C.; Checketts, D.; Beal, J.C.; Laux, L.C.; De Boer, L.M.; Wong, M.H. Long-term safety and treatment effects of cannabidiol in children and adults with treatment-resistant epilepsies: Expanded access program results. Epilepsia 2018, 59, 1540–1548. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.D.; Baler, R.D.; Compton, W.M.; Weiss, S.R. Adverse health effects of marijuana use. New Engl. J. Med. 2014, 370, 2219–2227. [Google Scholar] [CrossRef] [PubMed]
- Lagae, L. Cognitive side effects of anti-epileptic drugs: The relevance in childhood epilepsy. Seizure 2006, 15, 235–241. [Google Scholar] [CrossRef]
- Chesney, E.; Oliver, D.; Green, A.; Sovi, S.; Wilson, J.; Englund, A.; Freeman, T.P.; McGuire, P. Adverse effects of cannabidiol: A systematic review and meta-analysis of randomized clinical trials. Neuropsychopharmacology 2020, 45, 1799–1806. [Google Scholar] [CrossRef]
- Tzadok, M.; Uliel-Siboni, S.; Linder, I.; Kramer, U.; Epstein, O.; Menascu, S.; Nissenkorn, A.; Yosef, O.B.; Hyman, E.; Granot, D. CBD-enriched medical cannabis for intractable pediatric epilepsy: The current Israeli experience. Seizure 2016, 35, 41–44. [Google Scholar] [CrossRef]
- Gingrich, J.; Choudhuri, S.; Cournoyer, P.; Downey, J.; Jacobs, K.M. Review of the oral toxicity of cannabidiol (CBD). Food Chem. Toxicol. 2023, 176, 113799. [Google Scholar] [CrossRef]
- Sun, A.Y.; Sullivan, A.; Leffler, J.M.; Hammond, C.J.; Hulvershorn, L.; Miller, L. Review of the Efficacy and Safety of Cannabidiol with a Focus on Children and Adolescents in the Treatment of Psychiatric Symptoms and Disorders. Adolesc. Psychiatry 2023, 13, 143–159. [Google Scholar] [CrossRef]
- O’Shaughnessy, W.B. On the preparations of the Indian hemp, or Gunjah: Cannabis indica their effects on the animal system in health, and their utility in the treatment of tetanus and other convulsive diseases. Prov. Med. J. Retrosp. Med. Sci. 1843, 5, 363. [Google Scholar]
- Vandrey, R.; Raber, J.C.; Raber, M.E.; Douglass, B.; Miller, C.; Bonn-Miller, M.O. Cannabinoid dose and label accuracy in edible medical cannabis products. JAMA 2015, 313, 2491–2493. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.; Kilgore, M.; Babalonis, S. Cannabidiol (CBD) product contamination: Quantitative analysis of Δ9-tetrahydrocannabinol (Δ9-THC) concentrations found in commercially available CBD products. Drug Alcohol Depend. 2022, 237, 109522. [Google Scholar] [CrossRef] [PubMed]
- Pavlovic, R.; Nenna, G.; Calvi, L.; Panseri, S.; Borgonovo, G.; Giupponi, L.; Cannazza, G.; Giorgi, A. Quality traits of “cannabidiol oils”: Cannabinoids content, terpene fingerprint and oxidation stability of European commercially available preparations. Molecules 2018, 23, 1230. [Google Scholar] [CrossRef]
- Calvi, L.; Pentimalli, D.; Panseri, S.; Giupponi, L.; Gelmini, F.; Beretta, G.; Vitali, D.; Bruno, M.; Zilio, E.; Pavlovic, R. Comprehensive quality evaluation of medical Cannabis sativa L. inflorescence and macerated oils based on HS-SPME coupled to GC–MS and LC-HRMS (q-exactive orbitrap®) approach. J. Pharm. Biomed. Anal. 2018, 150, 208–219. [Google Scholar] [CrossRef]
- Krill, C.; Rochfort, S.; Spangenberg, G. A high-throughput method for the comprehensive analysis of terpenes and terpenoids in medicinal cannabis biomass. Metabolites 2020, 10, 276. [Google Scholar] [CrossRef]
- Aizpurua-Olaizola, O.; Soydaner, U.; Ozturk, E.; Schibano, D.; Simsir, Y.; Navarro, P.; Etxebarria, N.; Usobiaga, A. Evolution of the cannabinoid and terpene content during the growth of Cannabis sativa plants from different chemotypes. J. Nat. Prod. 2016, 79, 324–331. [Google Scholar] [CrossRef]
- Russo, E.B. Taming THC: Potential cannabis synergy and phytocannabinoid-terpenoid entourage effects. Br. J. Pharmacol. 2011, 163, 1344–1364. [Google Scholar] [CrossRef]
- Naim-Feil, E.; Elkins, A.C.; Malmberg, M.M.; Ram, D.; Tran, J.; Spangenberg, G.C.; Rochfort, S.J.; Cogan, N.O. The Cannabis Plant as a Complex System: Interrelationships between cannabinoid compositions, morphological, physiological and phenological traits. Plants 2023, 12, 493. [Google Scholar] [CrossRef]
- Geffrey, A.L.; Pollack, S.F.; Bruno, P.L.; Thiele, E.A. Drug–drug interaction between clobazam and cannabidiol in children with refractory epilepsy. Epilepsia 2015, 56, 1246–1251. [Google Scholar] [CrossRef]
- Gaston, T.E.; Bebin, E.M.; Cutter, G.R.; Liu, Y.; Szaflarski, J.P.; UAB CBD Program. Interactions between cannabidiol and commonly used antiepileptic drugs. Epilepsia 2017, 58, 1586–1592. [Google Scholar] [CrossRef] [PubMed]
- Martinez Naya, N.; Kelly, J.; Corna, G.; Golino, M.; Abbate, A.; Toldo, S. Molecular and cellular mechanisms of action of cannabidiol. Molecules 2023, 28, 5980. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.-C.; Mackie, K. Review of the endocannabinoid system. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2021, 6, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Christodoulou, J. BioRender.com/u96b994. Available online: https://app.biorender.com/citation/66f9ef0bc3f734f2723a3f69 (accessed on 4 October 2024).
- Mackie, K. Distribution of cannabinoid receptors in the central and peripheral nervous system. In Cannabinoids; Springer: Berlin/Heidelberg, Germany, 2005; pp. 299–325. [Google Scholar]
- Guo, J.; Ikeda, S.R. Endocannabinoids modulate N-type calcium channels and G-protein-coupled inwardly rectifying potassium channels via CB1 cannabinoid receptors heterologously expressed in mammalian neurons. Mol. Pharmacol. 2004, 65, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Hill, T.; Cascio, M.G.; Romano, B.; Duncan, M.; Pertwee, R.; Williams, C.; Whalley, B.; Hill, A. Cannabidivarin-rich cannabis extracts are anticonvulsant in mouse and rat via a CB1 receptor-independent mechanism. Br. J. Pharmacol. 2013, 170, 679–692. [Google Scholar] [CrossRef]
- McPartland, J.M.; Duncan, M.; Di Marzo, V.; Pertwee, R.G. Are cannabidiol and Δ9-tetrahydrocannabivarin negative modulators of the endocannabinoid system? A systematic review. Br. J. Pharmacol. 2015, 172, 737–753. [Google Scholar] [CrossRef]
- De Petrocellis, L.; Ligresti, A.; Moriello, A.S.; Allarà, M.; Bisogno, T.; Petrosino, S.; Stott, C.G.; Di Marzo, V. Effects of cannabinoids and cannabinoid-enriched Cannabis extracts on TRP channels and endocannabinoid metabolic enzymes. Br. J. Pharmacol. 2011, 163, 1479–1494. [Google Scholar] [CrossRef]
- Muller, C.; Morales, P.; Reggio, P.H. Cannabinoid ligands targeting TRP channels. Front. Mol. Neurosci. 2019, 11, 487. [Google Scholar] [CrossRef]
- Bisogno, T.; Hanuš, L.; De Petrocellis, L.; Tchilibon, S.; Ponde, D.E.; Brandi, I.; Moriello, A.S.; Davis, J.B.; Mechoulam, R.; Di Marzo, V. Molecular targets for cannabidiol and its synthetic analogues: Effect on vanilloid VR1 receptors and on the cellular uptake and enzymatic hydrolysis of anandamide. Br. J. Pharmacol. 2001, 134, 845–852. [Google Scholar] [CrossRef]
- Musella, A.; De Chiara, V.; Rossi, S.; Prosperetti, C.; Bernardi, G.; Maccarrone, M.; Centonze, D. TRPV1 channels facilitate glutamate transmission in the striatum. Mol. Cell. Neurosci. 2009, 40, 89–97. [Google Scholar] [CrossRef]
- Gray, R.A.; Whalley, B.J. The proposed mechanisms of action of CBD in epilepsy. Epileptic Disord. 2020, 22, S10–S15. [Google Scholar] [CrossRef] [PubMed]
- Vilela, L.R.; Lima, I.V.; Kunsch, É.B.; Pinto, H.P.P.; de Miranda, A.S.; Vieira, É.L.M.; de Oliveira, A.C.P.; Moraes, M.F.D.; Teixeira, A.L.; Moreira, F.A. Anticonvulsant effect of cannabidiol in the pentylenetetrazole model: Pharmacological mechanisms, electroencephalographic profile, and brain cytokine levels. Epilepsy Behav. 2017, 75, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Iannotti, F.A.; Hill, C.L.; Leo, A.; Alhusaini, A.; Soubrane, C.; Mazzarella, E.; Russo, E.; Whalley, B.J.; Di Marzo, V.; Stephens, G.J. Nonpsychotropic plant cannabinoids, cannabidivarin (CBDV) and cannabidiol (CBD), activate and desensitize transient receptor potential vanilloid 1 (TRPV1) channels in vitro: Potential for the treatment of neuronal hyperexcitability. J. ACS Chem. Neurosci. 2014, 5, 1131–1141. [Google Scholar] [CrossRef] [PubMed]
- La Montanara, P.; Hervera, A.; Baltussen, L.L.; Hutson, T.H.; Palmisano, I.; De Virgiliis, F.; Kong, G.; Chadwick, J.; Gao, Y.; Bartus, K. Cyclin-dependent–like kinase 5 is required for pain signaling in human sensory neurons and mouse models. Sci. Transl. Med. 2020, 12, eaax4846. [Google Scholar] [CrossRef]
- Li, X.; Yennawar, M.; Wiest, A.; O’Brien, W.T.; Babrowicz, B.; White, R.S.; Talos, D.M.; Jensen, F.E. Cannabidiol attenuates seizure susceptibility and behavioural deficits in adult CDKL5R59X knock-in mice. Eur. J. Neurosci. 2024, 59, 3337–3352. [Google Scholar] [CrossRef] [PubMed]
- Christodoulou, J. BioRender.com/x98t143. Available online: https://app.biorender.com/citation/66f9ed606fde867d4c3b6570 (accessed on 4 October 2024).
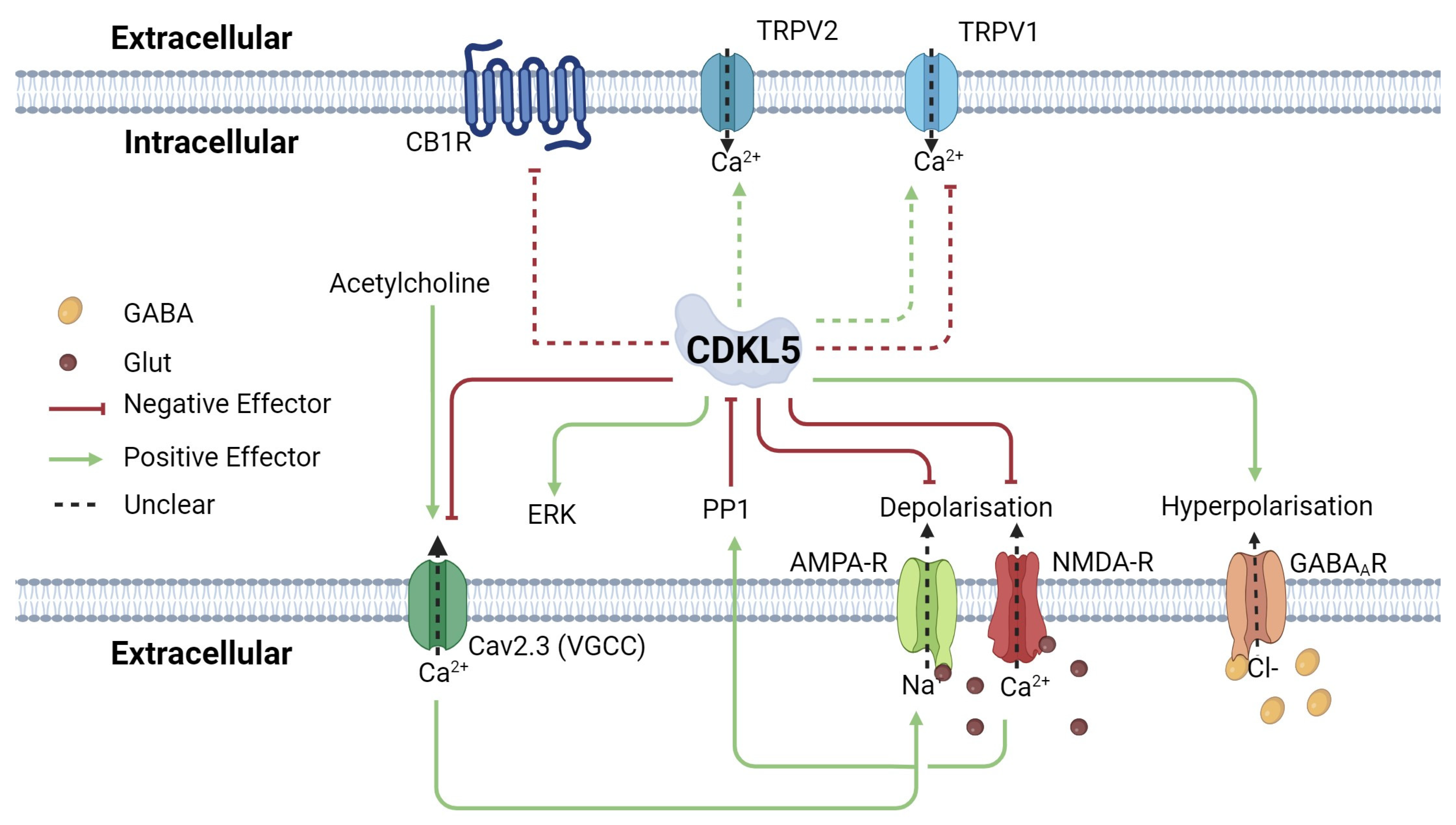
- La Montanara, P.; Rusconi, L.; Locarno, A.; Forti, L.; Barbiero, I.; Tramarin, M.; Chandola, C.; Kilstrup-Nielsen, C.; Landsberger, N. Synaptic synthesis, dephosphorylation, and degradation: A novel paradigm for an activity-dependent neuronal control of CDKL5. J. Biol. Chem. 2015, 290, 4512–4527. [Google Scholar] [CrossRef]
- Rusconi, L.; Kilstrup-Nielsen, C.; Landsberger, N. Extrasynaptic N-methyl-D-aspartate (NMDA) receptor stimulation induces cytoplasmic translocation of the CDKL5 kinase and its proteasomal degradation. J. Biol. Chem. 2011, 286, 36550–36558. [Google Scholar] [CrossRef]
- Okuda, K.; Kobayashi, S.; Fukaya, M.; Watanabe, A.; Murakami, T.; Hagiwara, M.; Sato, T.; Ueno, H.; Ogonuki, N.; Komano-Inoue, S. CDKL5 controls postsynaptic localization of GluN2B-containing NMDA receptors in the hippocampus and regulates seizure susceptibility. Neurobiol. Dis. 2017, 106, 158–170. [Google Scholar] [CrossRef]
- Simões de Oliveira, L.; O’Leary, H.E.; Nawaz, S.; Loureiro, R.; Davenport, E.C.; Baxter, P.; Louros, S.R.; Dando, O.; Perkins, E.; Peltier, J. Enhanced hippocampal LTP but normal NMDA receptor and AMPA receptor function in a rat model of CDKL5 deficiency disorder. J Mol. Autism 2024, 15, 28. [Google Scholar] [CrossRef]
- Hall, B.J.; Ripley, B.; Ghosh, A. NR2B signaling regulates the development of synaptic AMPA receptor current. J. Neurosci. 2007, 27, 13446–13456. [Google Scholar] [CrossRef]
- Yennawar, M.; White, R.S.; Jensen, F.E. AMPA receptor dysregulation and therapeutic interventions in a mouse model of CDKL5 deficiency disorder. J. Neurosci. 2019, 39, 4814–4828. [Google Scholar] [CrossRef] [PubMed]
- Mulcahey, P.J.; Tang, S.; Takano, H.; White, A.; Portillo, D.R.D.; Kane, O.M.; Marsh, E.D.; Zhou, Z.; Coulter, D.A. Aged heterozygous Cdkl5 mutant mice exhibit spontaneous epileptic spasms. Exp. Neurol. 2020, 332, 113388. [Google Scholar] [CrossRef] [PubMed]
- Bakas, T.; Van Nieuwenhuijzen, P.; Devenish, S.; McGregor, I.; Arnold, J.; Chebib, M. The direct actions of cannabidiol and 2-arachidonoyl glycerol at GABAA receptors. J. Pharmacol. Res. 2017, 119, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Van Bergen, N.J.; Massey, S.; Quigley, A.; Rollo, B.; Harris, A.R.; Kapsa, R.M.; Christodoulou, J. CDKL5 deficiency disorder: Molecular insights and mechanisms of pathogenicity to fast-track therapeutic development. Biochem. Soc. Trans. 2022, 50, 1207–1224. [Google Scholar] [CrossRef] [PubMed]
- Sylantyev, S.; Jensen, T.P.; Ross, R.A.; Rusakov, D.A. Cannabinoid-and lysophosphatidylinositol-sensitive receptor GPR55 boosts neurotransmitter release at central synapses. Proc. Natl. Acad. Sci. USA 2013, 110, 5193–5198. [Google Scholar] [CrossRef]
- Lauckner, J.E.; Jensen, J.B.; Chen, H.-Y.; Lu, H.-C.; Hille, B.; Mackie, K. GPR55 is a cannabinoid receptor that increases intracellular calcium and inhibits M current. Proc. Natl. Acad. Sci. USA 2008, 105, 2699–2704. [Google Scholar] [CrossRef]
- Rosenberg, E.C.; Chamberland, S.; Bazelot, M.; Nebet, E.R.; Wang, X.; McKenzie, S.; Jain, S.; Greenhill, S.; Wilson, M.; Marley, N. Cannabidiol modulates excitatory-inhibitory ratio to counter hippocampal hyperactivity. Neuron 2023, 111, 1282–1300.e8. [Google Scholar] [CrossRef]
- Ryberg, E.; Larsson, N.; Sjögren, S.; Hjorth, S.; Hermansson, N.O.; Leonova, J.; Elebring, T.; Nilsson, K.; Drmota, T.; Greasley, P. The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. Pharmacol. 2007, 152, 1092–1101. [Google Scholar] [CrossRef]
- De Rosa, R.; Valastro, S.; Cambria, C.; Barbiero, I.; Puricelli, C.; Tramarin, M.; Randi, S.; Bianchi, M.; Antonucci, F.; Kilstrup-Nielsen, C. Loss of CDKL5 causes synaptic GABAergic defects that can be restored with the neuroactive steroid pregnenolone-methyl-ether. Int. J. Mol. Sci. 2022, 24, 68. [Google Scholar] [CrossRef]
- Burman, R.J.; Rosch, R.E.; Wilmshurst, J.M.; Sen, A.; Ramantani, G.; Akerman, C.J.; Raimondo, J.V. Why won’t it stop? The dynamics of benzodiazepine resistance in status epilepticus. Nat. Rev. Neurol. 2022, 18, 428–441. [Google Scholar] [CrossRef]
- Chuang, S.-H.; Westenbroek, R.E.; Stella, N.; Catterall, W.A. Combined antiseizure efficacy of cannabidiol and clonazepam in a conditional mouse model of dravet syndrome. J. Exp. Neurol. 2021, 2, 81. [Google Scholar] [PubMed]
- Hess, E.J.; Moody, K.A.; Geffrey, A.L.; Pollack, S.F.; Skirvin, L.A.; Bruno, P.L.; Paolini, J.L.; Thiele, E.A. Cannabidiol as a new treatment for drug-resistant epilepsy in tuberous sclerosis complex. Epilepsia 2016, 57, 1617–1624. [Google Scholar] [CrossRef] [PubMed]
- Devinsky, O.; Marsh, E.; Friedman, D.; Thiele, E.; Laux, L.; Sullivan, J.; Miller, I.; Flamini, R.; Wilfong, A.; Filloux, F. Cannabidiol in patients with treatment-resistant epilepsy: An open-label interventional trial. J. Lancet Neurol. 2016, 15, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.A.; Reitman, M.L. Adenosine-related mechanisms in non-adenosine receptor drugs. Cells 2020, 9, 956. [Google Scholar] [CrossRef]
- Weltha, L.; Reemmer, J.; Boison, D. The role of adenosine in epilepsy. Brain Res. Bull. 2019, 151, 46–54. [Google Scholar] [CrossRef]
- Rosenberg, E.C.; Patra, P.H.; Whalley, B.J. Therapeutic effects of cannabinoids in animal models of seizures, epilepsy, epileptogenesis, and epilepsy-related neuroprotection. Epilepsy Behav. 2017, 70, 319–327. [Google Scholar] [CrossRef]
- Szaflarski, J.; Bebin, E.; DeWolfe, J.; Dure, L.; Gaston, T.; Harsanyi, K.; Houston, J.; McGrath, T.; Perry, L.; Singh, R. Seizure response to cannabidiol in a state-sponsored open-label program (S14. 006). Neurology 2016, 86, S14.006. [Google Scholar] [CrossRef]
- Olson, H.E.; Daniels, C.I.; Haviland, I.; Swanson, L.C.; Greene, C.A.; Denny, A.M.M.; Demarest, S.T.; Pestana-Knight, E.; Zhang, X.; Moosa, A.N. Current neurologic treatment and emerging therapies in CDKL5 deficiency disorder. J. Neurodev. Disord. 2021, 13, 40. [Google Scholar] [CrossRef]
- Anderson, L.L.; Low, I.K.; McGregor, I.S.; Arnold, J.C. Interactions between cannabidiol and Δ9-tetrahydrocannabinol in modulating seizure susceptibility and survival in a mouse model of Dravet syndrome. Br. J. Pharmacol. 2020, 177, 4261–4274. [Google Scholar] [CrossRef]
- Pamplona, F.A.; Da Silva, L.R.; Coan, A.C. Potential clinical benefits of CBD-rich cannabis extracts over purified CBD in treatment-resistant epilepsy: Observational data meta-analysis. Front. Neurol. 2018, 9, 392084. [Google Scholar] [CrossRef]
- Arzimanoglou, A.; Brandl, U.; Cross, J.H.; Gil-Nagel, A.; Lagae, L.; Landmark, C.J.; Specchio, N.; Nabbout, R.; Thiele, E.A.; Gubbay, O. Epilepsy and cannabidiol: A guide to treatment. Epileptic Disord. 2020, 22, 1–14. [Google Scholar] [PubMed]
- Dale, T.; Downs, J.; Wong, K.; Leonard, H. The perceived effects of cannabis products in the management of seizures in CDKL5 Deficiency Disorder. Epilepsy Behav. 2021, 122, 108152. [Google Scholar] [CrossRef] [PubMed]
- Lattanzi, S.; Trinka, E.; Striano, P.; Rocchi, C.; Salvemini, S.; Silvestrini, M.; Brigo, F. Highly purified cannabidiol for epilepsy treatment: A systematic review of epileptic conditions beyond dravet syndrome and Lennox–Gastaut syndrome. CNS Drugs 2021, 35, 265–281. [Google Scholar] [CrossRef] [PubMed]
- Caraballo, R.; Reyes, G.; Demirdjian, G.; Huaman, M.; Gutierrez, R. Long-term use of cannabidiol-enriched medical cannabis in a prospective cohort of children with drug-resistant developmental and epileptic encephalopathy. J. Seizure 2022, 95, 56–63. [Google Scholar] [CrossRef]
- Devinsky, O.; Verducci, C.; Thiele, E.A.; Laux, L.C.; Patel, A.D.; Filloux, F.; Szaflarski, J.P.; Wilfong, A.; Clark, G.D.; Park, Y.D.; et al. Open-label use of highly purified CBD (Epidiolex®) in patients with CDKL5 deficiency disorder and Aicardi, Dup15q, and Doose syndromes. Epilepsy 2018, 86, 131–137. [Google Scholar] [CrossRef]
- Sands, T.T.; Rahdari, S.; Oldham, M.S.; Caminha Nunes, E.; Tilton, N.; Cilio, M.R. Long-term safety, tolerability, and efficacy of cannabidiol in children with refractory epilepsy: Results from an expanded access program in the US. J. CNS Drugs 2019, 33, 47–60. [Google Scholar] [CrossRef]
- Avoli, M.; Louvel, J.; Pumain, R.; Köhling, R. Cellular and molecular mechanisms of epilepsy in the human brain. Prog. Neurobiol. 2005, 77, 166–200. [Google Scholar] [CrossRef]
- Sumadewi, K.T.; Harkitasari, S.; Tjandra, D.C. Biomolecular mechanisms of epileptic seizures and epilepsy: A review. Acta Epileptol. 2023, 5, 28. [Google Scholar] [CrossRef]
- Negraes, P.D.; Trujillo, C.A.; Yu, N.-K.; Wu, W.; Yao, H.; Liang, N.; Lautz, J.D.; Kwok, E.; McClatchy, D.; Diedrich, J. Altered network and rescue of human neurons derived from individuals with early-onset genetic epilepsy. Mol. Psychiatry 2021, 26, 7047–7068. [Google Scholar] [CrossRef]
- Sun, X.; Wang, T. Research progress on the pathogenesis of CDKL5 pathogenic variants and related encephalopathy. Eur. J. Pediatr. 2023, 182, 3049–3056. [Google Scholar] [CrossRef]
- Shen, D.; Chen, J.; Liu, D.; Shen, M.; Wang, X.; Wu, Y.; Ke, S.; Macdonald, R.L.; Zhang, Q. The GABRG2 F343L allele causes spontaneous seizures in a novel transgenic zebrafish model that can be treated with suberanilohydroxamic acid (SAHA). J. Ann. Transl. Med. 2020, 8, 1560. [Google Scholar] [CrossRef] [PubMed]
- Serrano, R.J.; Lee, C.; Douek, A.M.; Kaslin, J.; Bryson-Richardson, R.J.; Sztal, T.E. Novel preclinical model for CDKL5 deficiency disorder. J Dis. Models Mech. 2022, 15, dmm049094. [Google Scholar] [CrossRef] [PubMed]
- Varela, T.; Varela, D.; Martins, G.; Conceição, N.; Cancela, M.L. Cdkl5 mutant zebrafish shows skeletal and neuronal alterations mimicking human CDKL5 deficiency disorder. Sci. Rep. 2022, 12, 9325. [Google Scholar] [CrossRef] [PubMed]
- Sztal, T.E.; Ruparelia, A.A.; Williams, C.; Bryson-Richardson, R.J. Using touch-evoked response and locomotion assays to assess muscle performance and function in zebrafish. JoVE 2016, 13, e54431. [Google Scholar]
- Thornton, C.; Dickson, K.E.; Carty, D.R.; Ashpole, N.M.; Willett, K.L. Cannabis constituents reduce seizure behavior in chemically-induced and scn1a-mutant zebrafish. Epilepsy Behav. 2020, 110, 107152. [Google Scholar] [CrossRef]
- Samarut, É.; Nixon, J.; Kundap, U.P.; Drapeau, P.; Ellis, L.D. Single and synergistic effects of cannabidiol and Δ-9-tetrahydrocannabinol on zebrafish models of neuro-hyperactivity. Front. Pharmacol. 2019, 10, 226. [Google Scholar] [CrossRef]
- Jones, N.A.; Glyn, S.E.; Akiyama, S.; Hill, T.D.; Hill, A.J.; Weston, S.E.; Burnett, M.D.; Yamasaki, Y.; Stephens, G.J.; Whalley, B.J. Cannabidiol exerts anti-convulsant effects in animal models of temporal lobe and partial seizures. Seizure 2012, 21, 344–352. [Google Scholar] [CrossRef]
- Jones, N.A.; Hill, A.J.; Smith, I.; Bevan, S.A.; Williams, C.M.; Whalley, B.J.; Stephens, G.J. Cannabidiol displays antiepileptiform and antiseizure properties in vitro and in vivo. J. Pharmacol. Exp. Ther. 2010, 332, 569–577. [Google Scholar] [CrossRef]
- Marshall, G.F.; Gonzalez-Sulser, A.; Abbott, C.M. Modelling epilepsy in the mouse: Challenges and solutions. Dis. Models Mech. 2021, 14, dmm047449. [Google Scholar] [CrossRef]
- Hodge, R.D.; Bakken, T.E.; Miller, J.A.; Smith, K.A.; Barkan, E.R.; Graybuck, L.T.; Close, J.L.; Long, B.; Johansen, N.; Penn, O. Conserved cell types with divergent features in human versus mouse cortex. Nature 2019, 573, 61–68. [Google Scholar] [CrossRef]
- Whalley, B.J.; Lin, H.; Bell, L.; Hill, T.; Patel, A.; Gray, R.A.; Elizabeth Roberts, C.; Devinsky, O.; Bazelot, M.; Williams, C.M. Species-specific susceptibility to cannabis-induced convulsions. Br. J. Pharmacol. 2019, 176, 1506–1523. [Google Scholar] [CrossRef] [PubMed]
- Amendola, E.; Zhan, Y.; Mattucci, C.; Castroflorio, E.; Calcagno, E.; Fuchs, C.; Lonetti, G.; Silingardi, D.; Vyssotski, A.L.; Farley, D. Mapping pathological phenotypes in a mouse model of CDKL5 disorder. PLoS ONE 2014, 9, e91613. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.-T.J.; Allen, M.; Goffin, D.; Zhu, X.; Fairless, A.H.; Brodkin, E.S.; Siegel, S.J.; Marsh, E.D.; Blendy, J.A.; Zhou, Z. Loss of CDKL5 disrupts kinome profile and event-related potentials leading to autistic-like phenotypes in mice. Proc. Natl. Acad. Sci. USA 2012, 109, 21516–21521. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Wang, I.-T.J.; Yue, C.; Takano, H.; Terzic, B.; Pance, K.; Lee, J.Y.; Cui, Y.; Coulter, D.A.; Zhou, Z. Loss of CDKL5 in glutamatergic neurons disrupts hippocampal microcircuitry and leads to memory impairment in mice. J. Neurosci. 2017, 37, 7420–7437. [Google Scholar] [CrossRef] [PubMed]
- Di Nardo, A.; Rühmkorf, A.; Award, P.; Brennecke, A.; Fagiolini, M.; Sahin, M. Phenotypic characterization of Cdkl5-knockdown neurons establishes elongated cilia as a functional assay for CDKL5 Deficiency Disorder. Neurosci. Res. 2022, 176, 73–78. [Google Scholar] [CrossRef]
- Fuchs, C.; Gennaccaro, L.; Trazzi, S.; Bastianini, S.; Bettini, S.; Martire, V.L.; Ren, E.; Medici, G.; Zoccoli, G.; Rimondini, R. Heterozygous CDKL5 knockout female mice are a valuable animal model for CDKL5 disorder. Neural Plast. 2018, 2018, 9726950. [Google Scholar] [CrossRef]
- Jhang, C.-L.; Huang, T.-N.; Hsueh, Y.-P.; Liao, W. Mice lacking cyclin-dependent kinase-like 5 manifest autistic and ADHD-like behaviors. Hum. Mol. Genet. 2017, 26, 3922–3934. [Google Scholar] [CrossRef]
- Tassinari, M.; Uguagliati, B.; Trazzi, S.; Cerchier, C.B.; Cavina, O.V.; Mottolese, N.; Loi, M.; Candini, G.; Medici, G.; Ciani, E. Early-onset brain alterations during postnatal development in a mouse model of CDKL5 deficiency disorder. Neurobiol. Dis. 2023, 182, 106146. [Google Scholar] [CrossRef]
- Hill, A.; Mercier, M.; Hill, T.; Glyn, S.; Jones, N.; Yamasaki, Y.; Futamura, T.; Duncan, M.; Stott, C.; Stephens, G. Cannabidivarin is anticonvulsant in mouse and rat. Br. J. Pharmacol. 2012, 167, 1629–1642. [Google Scholar] [CrossRef]
- Patra, P.H.; Barker-Haliski, M.; White, H.S.; Whalley, B.J.; Glyn, S.; Sandhu, H.; Jones, N.; Bazelot, M.; Williams, C.M.; McNeish, A.J. Cannabidiol reduces seizures and associated behavioral comorbidities in a range of animal seizure and epilepsy models. Epilepsia 2019, 60, 303–314. [Google Scholar] [CrossRef]
- Shapiro, L.; Escayg, A.; Wong, J.C. Cannabidiol increases seizure resistance and improves behavior in an scn8a mouse model. Front. Pharmacol. 2022, 13, 815950. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.S.; Stella, N.; Catterall, W.A.; Westenbroek, R.E. Cannabidiol attenuates seizures and social deficits in a mouse model of Dravet syndrome. Proc. Natl. Acad. Sci. USA 2017, 114, 11229–11234. [Google Scholar] [CrossRef] [PubMed]
- Lazarini-Lopes, W.; Do Val-da Silva, R.A.; da Silva-Júnior, R.M.; Silva-Cardoso, G.K.; Leite-Panissi, C.R.; Leite, J.P.; Garcia-Cairasco, N. Chronic cannabidiol (CBD) administration induces anticonvulsant and antiepileptogenic effects in a genetic model of epilepsy. Epilepsy Behav. 2021, 119, 107962. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Rollo, B.; Javaid, M.S.; Huang, Z.; He, W.; Xu, H.; Kwan, P.; Zhang, C. An integrated in vitro human iPSCs-derived neuron and in vivo animal approach for preclinical screening of anti-seizure compounds. J. Adv. Res. 2023, 64, 249–262. [Google Scholar] [CrossRef]
- Sun, Y.; Dolmetsch, R.E. Investigating the therapeutic mechanism of cannabidiol in a human induced pluripotent stem cell (iPSC)-based model of Dravet syndrome. In Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2018. [Google Scholar]
- Wu, X.; Sosunov, A.A.; Lado, W.; Teoh, J.J.; Ham, A.; Li, H.; Al-Dalahmah, O.; Gill, B.J.; Arancio, O.; Schevon, C.A. Synaptic hyperexcitability of cytomegalic pyramidal neurons contributes to epileptogenesis in tuberous sclerosis complex. Cell Rep. 2022, 40, 111085. [Google Scholar] [CrossRef]
- Paşca, A.M.; Sloan, S.A.; Clarke, L.E.; Tian, Y.; Makinson, C.D.; Huber, N.; Kim, C.H.; Park, J.-Y.; O’Rourke, N.A.; Nguyen, K.D. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat. Methods 2015, 12, 671–678. [Google Scholar] [CrossRef]
- Qian, X.; Nguyen, H.N.; Song, M.M.; Hadiono, C.; Ogden, S.C.; Hammack, C.; Yao, B.; Hamersky, G.R.; Jacob, F.; Zhong, C. Brain-region-specific organoids using mini-bioreactors for modeling ZIKV exposure. J. Cell Rep. 2016, 165, 1238–1254. [Google Scholar] [CrossRef]
- Santos, A.C.; Nader, G.; El Soufi El Sabbagh, D.; Urban, K.; Attisano, L.; Carlen, P.L. Treating Hyperexcitability in Human Cerebral Organoids Resulting from Oxygen-Glucose Deprivation. Cells 2023, 12, 1949. [Google Scholar] [CrossRef]
- Hill, A.J.; Weston, S.E.; Jones, N.A.; Smith, I.; Bevan, S.A.; Williamson, E.M.; Stephens, G.J.; Williams, C.M.; Whalley, B.J. Δ9-Tetrahydrocannabivarin suppresses in vitro epileptiform and in vivo seizure activity in adult rats. Epilepsia 2010, 51, 1522–1532. [Google Scholar] [CrossRef]
- Amada, N.; Yamasaki, Y.; Williams, C.M.; Whalley, B.J. Cannabidivarin (CBDV) suppresses pentylenetetrazole (PTZ)-induced increases in epilepsy-related gene expression. PeerJ 2013, 1, e214. [Google Scholar] [CrossRef]
- Benson, M.J.; Anderson, L.L.; Low, I.K.; Luo, J.L.; Kevin, R.C.; Zhou, C.; McGregor, I.S.; Arnold, J.C. Evaluation of the possible anticonvulsant effect of Δ9-tetrahydrocannabinolic acid in murine seizure models. ACS Chem. Neurosci. 2022, 7, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.L.; Ametovski, A.; Lin Luo, J.; Everett-Morgan, D.; McGregor, I.S.; Banister, S.D.; Arnold, J.C. Cannabichromene, related phytocannabinoids, and 5-fluoro-cannabichromene have anticonvulsant properties in a mouse model of Dravet Syndrome. ACS Chem. Neurosci. 2021, 12, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.L.; Heblinski, M.; Absalom, N.L.; Hawkins, N.A.; Bowen, M.T.; Benson, M.J.; Zhang, F.; Bahceci, D.; Doohan, P.T.; Chebib, M. Cannabigerolic acid, a major biosynthetic precursor molecule in cannabis, exhibits divergent effects on seizures in mouse models of epilepsy. Br. J. Pharmacol. 2021, 178, 4826–4841. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.L.; Udoh, M.; Everett-Morgan, D.; Heblinski, M.; McGregor, I.S.; Banister, S.D.; Arnold, J.C. Olivetolic acid, a cannabinoid precursor in Cannabis sativa, but not CBGA methyl ester exhibits a modest anticonvulsant effect in a mouse model of Dravet syndrome. J. Cannabis Res. 2022, 4, 1–9. [Google Scholar] [CrossRef]
- Thomas, A.; Stevenson, L.A.; Wease, K.N.; Price, M.R.; Baillie, G.; Ross, R.A.; Pertwee, R.G. Evidence that the plant cannabinoid Δ9-tetrahydrocannabivarin is a cannabinoid CB1 and CB2 receptor antagonist. Br. J. Pharmacol. 2005, 146, 917. [Google Scholar] [CrossRef]
- Cascio, M.G.; Gauson, L.A.; Stevenson, L.A.; Ross, R.A.; Pertwee, R.G. Evidence that the plant cannabinoid cannabigerol is a highly potent α2-adrenoceptor agonist and moderately potent 5HT1A receptor antagonist. Br. J. Pharmacol. 2010, 159, 129–141. [Google Scholar] [CrossRef]
- Suzuki, S.; Wakano, C.; Monteilh-Zoller, M.K.; Cullen, A.J.; Fleig, A.; Penner, R. Cannabigerolic Acid (CBGA) inhibits the TRPM7 ion channel through its kinase domain. J. Funct. 2024, 5, 69. [Google Scholar] [CrossRef]
- De Petrocellis, L.; Vellani, V.; Schiano-Moriello, A.; Marini, P.; Magherini, P.C.; Orlando, P.; Di Marzo, V. Plant-derived cannabinoids modulate the activity of transient receptor potential channels of ankyrin type-1 and melastatin type-8. J. Pharmacol. Exp. Ther. 2008, 325, 1007–1015. [Google Scholar] [CrossRef]
- Khalil, A.; Shekh-Ahmad, T.; Kovac, S.; Wykes, R.C.; Horgen, F.D.; Fleig, A.; Walker, M.C. Drugs acting at TRPM7 channels inhibit seizure-like activity. Epilepsia Open 2023, 8, 1169–1174. [Google Scholar] [CrossRef]
- Moriyama, H.; Nomura, S.; Imoto, H.; Inoue, T.; Fujiyama, Y.; Haji, K.; Maruta, Y.; Ishihara, H.; Suzuki, M. Suppressive effects of transient receptor potential melastatin 8 agonist on epileptiform discharges and epileptic seizures. Front. Pharmacol. 2021, 12, 766782. [Google Scholar] [CrossRef]
- Hill, A.J.; Jones, N.A.; Smith, I.; Hill, C.L.; Williams, C.M.; Stephens, G.J.; Whalley, B.J. Voltage-gated sodium (NaV) channel blockade by plant cannabinoids does not confer anticonvulsant effects per se. J. Neurosci. Lett. 2014, 566, 269–274. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Massey, S.; Quigley, A.; Rochfort, S.; Christodoulou, J.; Van Bergen, N.J. Cannabinoids and Genetic Epilepsy Models: A Review with Focus on CDKL5 Deficiency Disorder. Int. J. Mol. Sci. 2024, 25, 10768. https://doi.org/10.3390/ijms251910768
Massey S, Quigley A, Rochfort S, Christodoulou J, Van Bergen NJ. Cannabinoids and Genetic Epilepsy Models: A Review with Focus on CDKL5 Deficiency Disorder. International Journal of Molecular Sciences. 2024; 25(19):10768. https://doi.org/10.3390/ijms251910768
Chicago/Turabian StyleMassey, Sean, Anita Quigley, Simone Rochfort, John Christodoulou, and Nicole J. Van Bergen. 2024. "Cannabinoids and Genetic Epilepsy Models: A Review with Focus on CDKL5 Deficiency Disorder" International Journal of Molecular Sciences 25, no. 19: 10768. https://doi.org/10.3390/ijms251910768