
Combining Photodynamic Therapy and Targeted Drug Delivery Systems: Enhancing Mitochondrial Toxicity for Improved Cancer Outcomes


Abstract
:1. Introduction
2. Photodynamic Therapy
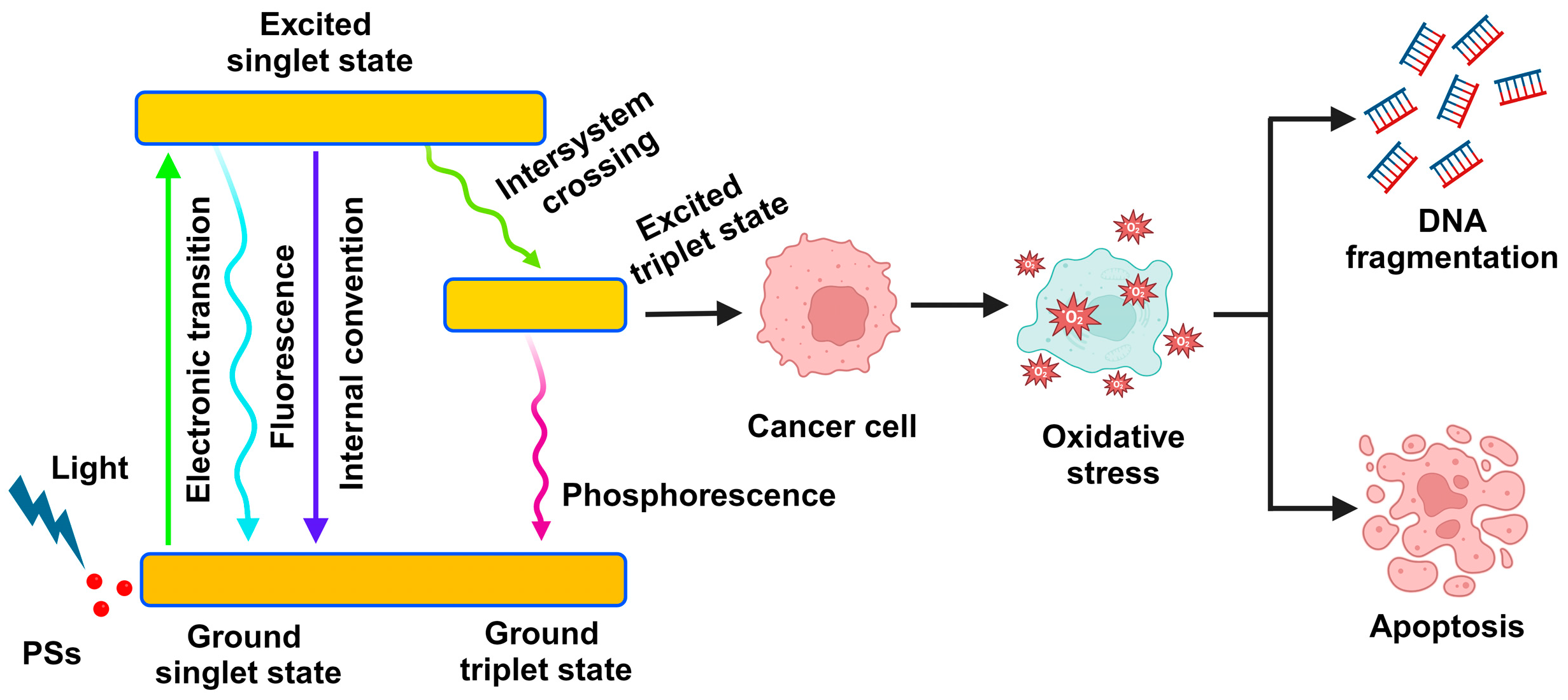
2.1. The Mechanism of Photodynamic Therapy
2.2. Photosensitizers
Types of Photosensitizers
2.3. Light Source
2.4. Drawbacks of PDT
2.5. Challenges in PDT Treatment
3. Targeted Drug Delivery Systems
3.1. Drug Delivery in Cancer Targets Mitochondria
3.2. Drug Targeting Mitochondrial DNA
3.3. Mitochondrial Delivery by Nanoparticles
3.4. Mitochondrial Delivery by Lipid-Based Nanocarriers
3.5. Mitochondrial Delivery by Peptides
3.6. Challenges in Mitochondrial Drug Targeting
4. Drug-Induced Mitochondrial Toxicity
4.1. Mitochondria-Independent Chemotherapeutics
4.2. Effects of PDT on the Mitochondrial Membrane Potential
5. Mitochondria as a Potential Cancer Therapeutic Target
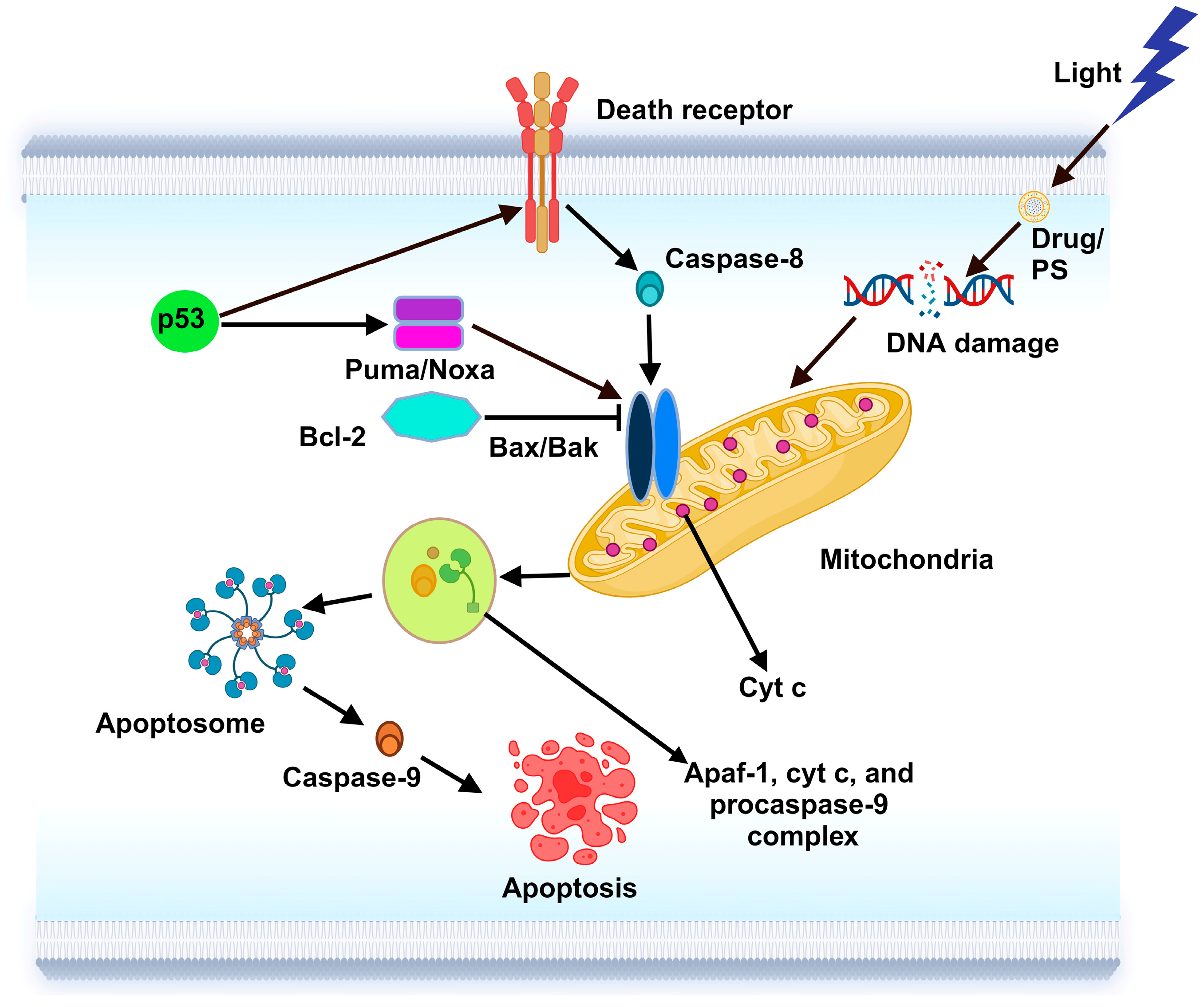
5.1. Apoptosis Regulation
5.2. Metabolic Rewiring
5.3. Therapeutic Strategies
6. Mitochondrial Drug Delivery Mechanisms in Cancer
6.1. Photodynamic Therapy-Induced Mitochondrial Toxicity
6.1.1. Apoptosis
6.1.2. Autophagy
6.2. Nanoparticle-Based Delivery Mechanisms in Cancer
7. Clinical Developments in PDT and Mitochondria Targeting in Cancer
8. Preclinical Studies: Cellular Assays and Animal Models
9. Conclusions and Future Perspectives
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rajan, S.S.; Chandran, R.; Abrahamse, H. Overcoming challenges in cancer treatment: Nano-enabled photodynamic therapy as a viable solution. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2024, 16, 1942. [Google Scholar] [CrossRef] [PubMed]
- Rajan, S.S.; Merlin, J.P.J.; Chandran, R.; Abrahamse, H. Natural Compounds used in targeting cellular organelles for drug delivery. In Interdisciplinary Cancer Research; Springer: Cham, Switzerland, 2024; Volume 287, pp. 1–14. [Google Scholar]
- Missiroli, S.; Perrone, M.; Genovese, I.; Pinton, P.; Giorgi, C. Cancer metabolism and mitochondria: Finding novel mechanisms to fight tumours. EBioMedicine 2020, 59, 102943. [Google Scholar] [CrossRef] [PubMed]
- Merlin, J.P.J.; Mathavarajah, S.; Dellaire, G.; Murphy, K.P.J.; Rupasinghe, H.P.V. A Dietary Antioxidant Formulation Ameliorates DNA Damage Caused by γ-Irradiation in Normal Human Bronchial Epithelial Cells In Vitro. Antioxidants 2022, 11, 1407. [Google Scholar] [CrossRef]
- Merlin, J.P.J.; Dellaire, G.; Murphy, K.; Rupasinghe, H.P.V. Vitamin-containing antioxidant formulation reduces carcinogen-induced dna damage through atr/chk1 signaling in bronchial epithelial cells in vitro. Biomedicines 2021, 9, 1665. [Google Scholar] [CrossRef]
- Suraweera, T.L.; Merlin, J.P.J.; Dellaire, G.; Xu, Z.; Rupasinghe, H.P.V. Genistein and Procyanidin B2 Reduce Carcinogen-Induced Reactive Oxygen Species and DNA Damage through the Activation of Nrf2/ARE Cell Signaling in Bronchial Epithelial Cells In Vitro. Int. J. Mol. Sci. 2023, 24, 3676. [Google Scholar] [CrossRef]
- Merlin, J.P.J.; Prasad, N.R.; Shibli, S.M.A.; Sebeela, M. Ferulic acid loaded Poly- d,l-lactide-co-glycolide nanoparticles: Systematic study of particle size, drug encapsulation efficiency and anticancer effect in non-small cell lung carcinoma cell line in vitro. Biomed. Prev. Nutr. 2012, 2, 69–76. [Google Scholar] [CrossRef]
- Zhang, Z.; Qin, S.; Chen, Y.; Zhou, L.; Yang, M.; Tang, Y.; Zuo, J.; Zhang, J.; Mizokami, A.; Nice, E.C.; et al. Inhibition of NPC1L1 disrupts adaptive responses of drug-tolerant persister cells to chemotherapy. EMBO Mol. Med. 2022, 14, e14903. [Google Scholar] [CrossRef]
- Boulos, J.C.; Yousof Idres, M.R.; Efferth, T. Investigation of cancer drug resistance mechanisms by phosphoproteomics. Pharmacol. Res. 2020, 160, 105091. [Google Scholar] [CrossRef]
- Gong, K.; Guo, G.; Gerber, D.E.; Gao, B.; Peyton, M.; Huang, C.; Minna, J.D.; Hatanpaa, K.J.; Kernstine, K.; Cai, L.; et al. TNF-driven adaptive response mediates resistance to EGFR inhibition in lung cancer. J. Clin. Investig. 2018, 128, 2500–2518. [Google Scholar] [CrossRef]
- Wang, H.; Feng, Z.; Wang, Y.; Zhou, R.; Yang, Z.; Xu, B. Integrating Enzymatic Self-Assembly and Mitochondria Targeting for Selectively Killing Cancer Cells without Acquired Drug Resistance. J. Am. Chem. Soc. 2016, 138, 16046–16055. [Google Scholar] [CrossRef]
- Xiao, Y.; Yu, D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol. Ther. 2021, 221, 107753. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. Targeting lipophilic cations to mitochondria. Biochim. Biophys. Acta 2008, 1777, 1028–1031. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, A.; Zhang, Y.; Khdour, O.M.; Kaye, J.B.; Hecht, S.M. Mitochondrial Nitroreductase Activity Enables Selective Imaging and Therapeutic Targeting. J. Am. Chem. Soc. 2016, 138, 12009–12012. [Google Scholar] [CrossRef]
- Chen, C.; Wang, J.; Li, X.; Liu, X.; Han, X. Recent Advances in Developing Photosensitizers for Photodynamic Cancer Therapy. Comb. Chem. High Throughput Screen. 2017, 20, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Lucky, S.S.; Soo, K.C.; Zhang, Y. Nanoparticles in Photodynamic Therapy. Chem. Rev. 2015, 115, 1990–2042. [Google Scholar] [CrossRef]
- Xiang, H.; Xue, F.; Yi, T.; Tham, H.P.; Liu, J.-G.; Zhao, Y. Cu2- xS Nanocrystals Cross-Linked with Chlorin e6-Functionalized Polyethylenimine for Synergistic Photodynamic and Photothermal Therapy of Cancer. ACS Appl. Mater. Interfaces 2018, 10, 16344–16351. [Google Scholar] [CrossRef]
- Wang, H.; Han, X.; Dong, Z.; Xu, J.; Wang, J.; Liu, Z. Hyaluronidase with pH-responsive Dextran Modification as an Adjuvant Nanomedicine for Enhanced Photodynamic-Immunotherapy of Cancer. Adv. Funct. Mater. 2019, 29, 1902440. [Google Scholar] [CrossRef]
- Hu, D.; Zhong, L.; Wang, M.; Li, H.; Qu, Y.; Liu, Q.; Han, R.; Yuan, L.; Shi, K.; Peng, J.; et al. Perfluorocarbon-Loaded and Redox-Activatable Photosensitizing Agent with Oxygen Supply for Enhancement of Fluorescence/Photoacoustic Imaging Guided Tumor Photodynamic Therapy. Adv. Funct. Mater. 2019, 29, 1806199. [Google Scholar] [CrossRef]
- Zhou, Z.; Song, J.; Nie, L.; Chen, X. Reactive oxygen species generating systems meeting challenges of photodynamic cancer therapy. Chem. Soc. Rev. 2016, 45, 6597–6626. [Google Scholar] [CrossRef]
- Yang, C.; Fu, Y.; Huang, C.; Hu, D.; Zhou, K.; Hao, Y.; Chu, B.; Yang, Y.; Qian, Z. Chlorin e6 and CRISPR-Cas9 dual-loading system with deep penetration for a synergistic tumoral photodynamic-immunotherapy. Biomaterials 2020, 255, 120194. [Google Scholar] [CrossRef]
- Castano, A.P.; Demidova, T.N.; Hamblin, M.R. Mechanisms in photodynamic therapy: Part one—Photosensitizers, photochemistry and cellular localization. Photodiagnosis Photodyn. Ther. 2004, 1, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Merlin, J.P.J.; Abrahamse, H. Optimizing CRISPR/Cas9 precision: Mitigating off-target effects for safe integration with photodynamic and stem cell therapies in cancer treatment. Biomed. Pharmacother. 2024, 180, 117516. [Google Scholar] [CrossRef] [PubMed]
- Merlin, J.P.J.; Crous, A.; Abrahamse, H. Nano-phototherapy: Favorable prospects for cancer treatment. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2024, 16, e1930. [Google Scholar] [CrossRef]
- Kwiatkowski, S.; Knap, B.; Przystupski, D.; Saczko, J.; Kędzierska, E.; Knap-Czop, K.; Kotlińska, J.; Michel, O.; Kotowski, K.; Kulbacka, J. Photodynamic therapy–mechanisms, photosensitizers and combinations. Biomed. Pharmacother. 2018, 106, 1098–1107. [Google Scholar] [CrossRef]
- Zhao, H.; Li, L.; Zheng, C.; Hao, Y.; Niu, M.; Hu, Y.; Chang, J.; Zhang, Z.; Wang, L. An intelligent dual stimuli-responsive photosensitizer delivery system with O2-supplying for efficient photodynamic therapy. Colloids Surf. B Biointerfaces 2018, 167, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Triviño, F.J.; Ayén-Rodríguez, Á.; Llamas-Molina, J.M.; Saenz-Guirado, S.; Ruiz-Villaverde, R. Treatment of superficial basal cell carcinoma with 7.8% 5-aminolaevulinic acid nanoemulsion-based gel (BF-200 ALA) and photodynamic therapy: Results in clinical practice in a tertiary hospital. Dermatol. Ther. 2021, 34, e14558. [Google Scholar] [CrossRef]
- Dave, D.; Desai, U.; Despande, N. Photodynamic Therapy: A View through Light. J. Orofac. Sci. 2012, 2, 82–86. [Google Scholar] [CrossRef]
- Kubrak, T.P.; Kołodziej, P.; Sawicki, J.; Mazur, A.; Koziorowska, K.; Aebisher, D. Some Natural Photosensitizers and Their Medicinal Properties for Use in Photodynamic Therapy. Molecules 2022, 27, 1192. [Google Scholar] [CrossRef]
- Udrea, A.M.; Smarandache, A.; Dinache, A.; Mares, C.; Nistorescu, S.; Avram, S.; Staicu, A. Photosensitizers-Loaded Nanocarriers for Enhancement of Photodynamic Therapy in Melanoma Treatment. Pharmaceutics 2023, 15, 2124. [Google Scholar] [CrossRef]
- Grebinyk, A.; Chepurna, O.; Frohme, M.; Qu, J.; Patil, R.; Vretik, L.O.; Ohulchanskyy, T.Y. Molecular and nanoparticulate agents for photodynamic therapy guided by near infrared imaging. J. Photochem. Photobiol. C Photochem. Rev. 2024, 58, 100652. [Google Scholar] [CrossRef]
- Correia, J.H.; Rodrigues, J.A.; Pimenta, S.; Dong, T.; Yang, Z. Photodynamic Therapy Review: Principles, Photosensitizers, Applications, and Future Directions. Pharmaceutics 2021, 13, 1332. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Zheng, H.; Huang, Z.; Lin, H.; Ke, Z.; Xie, S.; Li, B. Light-Emitting Diode-Based Illumination System for In Vitro Photodynamic Therapy. Int. J. Photoenergy 2012, 2021, 1–6. [Google Scholar] [CrossRef]
- Chilakamarthi, U.; Giribabu, L. Photodynamic Therapy: Past, Present and Future. Chem. Rec. 2017, 17, 775–802. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.M.; Darafsheh, A. Light Sources and Dosimetry Techniques for Photodynamic Therapy. Photochem. Photobiol. 2020, 96, 280–294. [Google Scholar] [CrossRef]
- Borgia, F.; Giuffrida, R.; Caradonna, E.; Vaccaro, M.; Guarneri, F.; Cannavò, S. Early and Late Onset Side Effects of Photodynamic Therapy. Biomedicines 2018, 6, 12. [Google Scholar] [CrossRef]
- Ibbotson, S.H. Adverse effects of topical photodynamic therapy. Photodermatol. Photoimmunol. Photomed. 2011, 27, 116–130. [Google Scholar] [CrossRef]
- Kluger, N.; Jeskanen, L.; Höök-Nikanne, J. Photodynamic therapy-triggered bullous pemphigoid. Int. J. Dermatol. 2017, 56, 13387. [Google Scholar] [CrossRef]
- RaRatour-Bigot, C.; Chemidling, M.; Montlahuc, C.; Abirached, G.; Madjlessi, N.; Bullier, C.; Battistella, M.; Bagot, M.; Lebbe, C.; Basset-Seguin, N. Squamous Cell Carcinoma Following Photodynamic Therapy for Cutaneous Bowen’s Disease in a Series of 105 Patients. Acta Derm. Venereol. 2016, 96, 658–663. [Google Scholar] [CrossRef]
- Giri, U.; Sharma, S.D.; Abdulla, M.; Athar, M. Evidence That in Situ Generated Reactive Oxygen Species Act as a Potent Stage I Tumor Promoter in Mouse Skin. Biochem. Biophys. Res. Commun. 1995, 209, 698–705. [Google Scholar] [CrossRef]
- Huis In‘t Veld, R.V.; Heuts, J.; Ma, S.; Cruz, L.J.; Ossendorp, F.A.; Jager, M.J. Current Challenges and Opportunities of Photodynamic Therapy against Cancer. Pharmaceutics 2023, 15, 330. [Google Scholar] [CrossRef]
- Rovini, A.; Savry, A.; Braguer, D.; Carré, M. Microtubule-targeted agents: When mitochondria become essential to chemotherapy. Biochim. Biophys. Acta 2011, 1807, 679–688. [Google Scholar] [CrossRef] [PubMed]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate hypoxic signaling. Curr. Opin. Cell Biol. 2009, 21, 894–899. [Google Scholar] [CrossRef] [PubMed]
- Singh, D. A sojourn on mitochondria targeted drug delivery systems for cancer: Strategies, clinical and future prospects. Mitochondrion 2024, 74, 101826. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A.J.; Hartley, R.C.; Murphy, M.P. Mitochondria-Targeted Small Molecule Therapeutics and Probes. Antioxid. Redox Signal. 2011, 15, 3021–3038. [Google Scholar] [CrossRef] [PubMed]
- Boddapati, S.V.; D’Souza, G.G.M.; Erdogan, S.; Torchilin, V.P.; Weissig, V. Organelle-Targeted Nanocarriers: Specific Delivery of Liposomal Ceramide to Mitochondria Enhances Its Cytotoxicity in Vitro and in Vivo. Nano Lett. 2008, 8, 2559–2563. [Google Scholar] [CrossRef]
- Murphy, M.P.; Smith, R.A.J. Targeting Antioxidants to Mitochondria by Conjugation to Lipophilic Cations. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 629–656. [Google Scholar] [CrossRef]
- Guerra, F.; Arbini, A.A.; Moro, L. Mitochondria and cancer chemoresistance. Biochim. Biophys. Acta 2017, 1858, 686–699. [Google Scholar] [CrossRef]
- Smith, A.L.M.; Whitehall, J.C.; Greaves, L.C. Mitochondrial DNA mutations in ageing and cancer. Mol. Oncol. 2022, 16, 3276–3294. [Google Scholar] [CrossRef]
- Pathania, D.; Millard, M.; Neamati, N. Opportunities in discovery and delivery of anticancer drugs targeting mitochondria and cancer cell metabolism. Adv. Drug Deliv. Rev. 2009, 61, 1250–1275. [Google Scholar] [CrossRef]
- Vellinga, T.T.; Borovski, T.; de Boer, V.C.; Fatrai, S.; van Schelven, S.; Trumpi, K.; Verheem, A.; Snoeren, N.; Emmink, B.L.; Koster, J.; et al. SIRT1/PGC1α-Dependent Increase in Oxidative Phosphorylation Supports Chemotherapy Resistance of Colon Cancer. Clin. Cancer Res. 2015, 21, 2870–2879. [Google Scholar] [CrossRef]
- Dhanasekaran, S.; Venugopal, D.; Al-Dayan, N.; Ravinayagam, V.; Mohammed, A.A. Emerging insights into mitochondria-specific targeting and drug delivering strategies: Recent milestones and therapeutic implications. Saudi J. Biol. Sci. 2020, 27, 3581–3592. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Yang, B.; Huang, Y.; Zhang, Y.; Jiang, Y.; Ma, L.; Shen, Y.-Q. Mitochondrial DNA-targeted therapy: A novel approach to combat cancer. Cell Insight 2023, 2, 100113. [Google Scholar] [CrossRef]
- Merlin, J.P.J.; Li, X. Role of Nanotechnology and Their Perspectives in the Treatment of Kidney Diseases. Front. Genet. 2022, 12, 817974. [Google Scholar] [CrossRef]
- Marin-Hernandez, A.; Gallardo-Perez, J.; Ralph, S.; Rodriguez-Enriquez, S.; Moreno-Sanchez, R. HIF-1α Modulates Energy Metabolism in Cancer Cells by Inducing Over-Expression of Specific Glycolytic Isoforms. Mini-Rev. Med. Chem. 2009, 9, 1084–1101. [Google Scholar] [CrossRef]
- Hu, Q.; Sun, W.; Wang, C.; Gu, Z. Recent advances of cocktail chemotherapy by combination drug delivery systems. Adv. Drug Deliv. Rev. 2016, 98, 19–34. [Google Scholar] [CrossRef]
- Zhang, L.; Gu, F.; Chan, J.; Wang, A.; Langer, R.; Farokhzad, O. Nanoparticles in Medicine: Therapeutic Applications and Developments. Clin. Pharmacol. Ther. 2008, 83, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chan, D.C. Mitochondrial Dynamics in Regulating the Unique Phenotypes of Cancer and Stem Cells. Cell Metab. 2017, 26, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Yousif, L.F.; Stewart, K.M.; Kelley, S.O. Targeting Mitochondria with Organelle-Specific Compounds: Strategies and Applications. ChemBioChem 2009, 10, 1939–1950. [Google Scholar] [CrossRef]
- Cho, H.; Cho, Y.-Y.; Shim, M.S.; Lee, J.Y.; Lee, H.S.; Kang, H.C. Mitochondria-targeted drug delivery in cancers. Biochim. Biophys. Acta 2020, 1866, 165808. [Google Scholar] [CrossRef]
- Meng, J.; Agrahari, V.; Youm, I. Advances in Targeted Drug Delivery Approaches for the Central Nervous System Tumors: The Inspiration of Nanobiotechnology. J. Neuroimmune Pharmacol. 2017, 12, 84–98. [Google Scholar] [CrossRef]
- Hoye, A.T.; Davoren, J.E.; Wipf, P.; Fink, M.P.; Kagan, V.E. Targeting Mitochondria. Acc. Chem. Res. 2008, 41, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Merlin, J.P.J.; Venkadesh, B.; Hussain, R.; Rajan, S.S. Biochemical estimations of multidrug resistance (ferulic acid and paclitaxel) in non-small cells lung carcinoma cells in vitro. Biomed. Aging Pathol. 2013, 3, 47–50. [Google Scholar] [CrossRef]
- Mottis, A.; Herzig, S.; Auwerx, J. Mitocellular communication: Shaping health and disease. Science 2019, 366, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Lin, K.H.; Huang, C.J.; Wei, A.C. MitoTox: A comprehensive mitochondrial toxicity database. BMC Bioinform. 2021, 22, 369. [Google Scholar] [CrossRef] [PubMed]
- Robinson, B.H. Lactic acidemia and mitochondrial disease. Mol. Genet. Metab. 2006, 89, 3–13. [Google Scholar] [CrossRef]
- Will, Y.; Shields, J.E.; Wallace, K.B. Drug-induced mitochondrial toxicity in the geriatric population: Challenges and future directions. Biology 2019, 8, 32. [Google Scholar] [CrossRef]
- Dykens, J.A.; Will, Y. The significance of mitochondrial toxicity testing in drug development. Drug Discov. Today 2007, 12, 777–785. [Google Scholar] [CrossRef]
- Merlin, J.P.J.; Venkadesh, B.; Rajan, S.S.; Subramanian, P. Multidrug Resistance for Cancer Treatment: Delivery of Ursolic Acid and Caffeine by Poly (Lactic-Co-Glycolic Acid) Nanoparticles. J. Cancer Sci. Res. 2018, s2, 3. [Google Scholar] [CrossRef]
- Merlin, J.P.J.; Venkadesh, B.; Hussain, R.; Prasad, N.R.; Shibli, S.M.A.; Raj, A.V.; Rajan, S.S. Paclitaxel loaded poly-d,l-lactide-co-glycolide nanoparticles: Enhanced anticancer effect in non-small cell lung carcinoma cell line. Biomed. Prev. Nutr. 2013, 3, 1–9. [Google Scholar] [CrossRef]
- Will, Y.; Dykens, J. Mitochondrial toxicity assessment in industry-a decade of technology development and insight. Expert Opin. Drug Metab. Toxicol. 2014, 10, 1061–1067. [Google Scholar] [CrossRef]
- Kamalian, L.; Chadwick, A.E.; Bayliss, M.; French, N.S.; Monshouwer, M.; Snoeys, J.; Park, B.K. The utility of HepG2 cells to identify direct mitochondrial dysfunction in the absence of cell death. Toxicol. Vitr. 2015, 29, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Hynes, J.; Swiss, R.L.; Will, Y. High-Throughput Analysis of Mitochondrial Oxygen Consumption. Methods Mol. Biol. 2018, 1782, 71–87. [Google Scholar] [PubMed]
- Yang, Y.; An, Y.; Ren, M.; Wang, H.; Bai, J.; Du, W.; Kong, D. The mechanisms of action of mitochondrial targeting agents in cancer: Inhibiting oxidative phosphorylation and inducing apoptosis. Front. Pharmacol. 2023, 14, 1243613. [Google Scholar] [CrossRef]
- Carter, J.L.; Hege, K.; Kalpage, H.A.; Edwards, H.; Hüttemann, M.; Taub, J.W.; Ge, Y. Targeting mitochondrial respiration for the treatment of acute myeloid leukemia. Biochem. Pharmacol. 2020, 182, 114253. [Google Scholar] [CrossRef]
- Tsuji, A.; Akao, T.; Masuya, T.; Murai, M.; Miyoshi, H. IACS-010759, a potent inhibitor of glycolysis-deficient hypoxic tumor cells, inhibits mitochondrial respiratory complex I through a unique mechanism. J. Biol. Chem. 2020, 295, 7481–7491. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Daver, N.; Mahendra, M.; Zhang, J.; Kamiya-Matsuoka, C.; Meric-Bernstam, F.; Kantarjian, H.M.; Ravandi, F.; Collins, M.E.; Francesco, M.E.; et al. Complex I inhibitor of oxidative phosphorylation in advanced solid tumors and acute myeloid leukemia: Phase I trials. Nat. Med. 2023, 29, 115–126. [Google Scholar] [CrossRef]
- Yap, T.A.; Rodon Ahnert, J.; Piha-Paul, S.A.; Fu, S.; Janku, F.; Karp, D.D.; Naing, A.; Ileana Dumbrava, E.E.; Pant, S.; Subbiah, V.; et al. Phase I trial of IACS-010759 (IACS), a potent, selective inhibitor of complex I of the mitochondrial electron transport chain, in patients (pts) with advanced solid tumors. J. Clin. Oncol. 2019, 37, 3014. [Google Scholar] [CrossRef]
- Baran, N.; Lodi, A.; Sweeney, S.R.; Renu, P.; Kuruvilla, V.M.; Cavazos, A.; Herranz, D.; Skwarska, A.; Warmoes, M.; Davis, R.E.; et al. Mitochondrial Complex I Inhibitor Iacs-010759 Reverses the NOTCH1-Driven Metabolic Reprogramming in T-ALL Via Blockade of Oxidative Phosphorylation: Synergy with Chemotherapy and Glutaminase Inhibition. Blood 2018, 132, 4020. [Google Scholar] [CrossRef]
- Martinvalet, D.; Dykxhoorn, D.M.; Ferrini, R.; Lieberman, J. Granzyme A Cleaves a Mitochondrial Complex I Protein to Initiate Caspase-Independent Cell Death. Cell 2008, 133, 681–692. [Google Scholar] [CrossRef]
- Reeves, M.B.; Davies, A.A.; McSharry, B.P.; Wilkinson, G.W.; Sinclair, J.H. Complex I Binding by a Virally Encoded RNA Regulates Mitochondria-Induced Cell Death. Science 2007, 316, 1345–1348. [Google Scholar] [CrossRef]
- Sharma, L.; Lu, J.; Bai, Y. Mitochondrial Respiratory Complex I: Structure, Function and Implication in Human Diseases. Curr. Med. Chem. 2009, 16, 1266–1277. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Xue, J.; Dai, Y.; Liu, H.; Chen, N.; Jia, L.; Huang, J. Inhibition of human hepatocellular carcinoma HepG2 by phthalocyanine photosensitiser PHOTOCYANINE: ROS production, apoptosis, cell cycle arrest. Eur. J. Cancer 2012, 48, 2086–2096. [Google Scholar] [CrossRef] [PubMed]
- Kalpage, H.A.; Wan, J.; Morse, P.T.; Zurek, M.P.; Turner, A.A.; Khobeir, A.; Yazdi, N.; Hakim, L.; Liu, J.; Vaishnav, A.; et al. Cytochrome c phosphorylation: Control of mitochondrial electron transport chain flux and apoptosis. Int. J. Biochem. Cell Biol. 2020, 121, 105704. [Google Scholar] [CrossRef]
- Mahalingam, S.M.; Arumugam, N.; Wong, L.S.; Dhanapal, A.C.T.A.; Djearamane, S. Mechanistic investigation of photodynamic therapy using Visudyne in human KB carcinoma cells. J. King Saud Univ. Sci. 2023, 35, 102871. [Google Scholar] [CrossRef]
- Andrzejewski, S.; Gravel, S.-P.; Pollak, M.; St-Pierre, J. Metformin directly acts on mitochondria to alter cellular bioenergetics. Cancer Metab. 2014, 2, 12. [Google Scholar] [CrossRef]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife 2014, 3, e02242. [Google Scholar] [CrossRef]
- Xu, Z.; Han, X.; Ou, D.; Liu, T.; Li, Z.; Jiang, G.; Liu, J.; Zhang, J. Targeting PI3K/AKT/mTOR-mediated autophagy for tumor therapy. Appl. Microbiol. Biotechnol. 2020, 104, 575–587. [Google Scholar] [CrossRef]
- Caino, M.C.; Ghosh, J.C.; Chae, Y.C.; Vaira, V.; Rivadeneira, D.B.; Faversani, A.; Rampini, P.; Kossenkov, A.V.; Aird, K.M.; Zhang, R.; et al. PI3K therapy reprograms mitochondrial trafficking to fuel tumor cell invasion. Proc. Natl. Acad. Sci. USA 2015, 112, 8638–8643. [Google Scholar] [CrossRef]
- Xia, M.; Yan, X.; Zhou, L.; Xu, L.; Zhang, L.; Yi, H.; Su, J. p62 Suppressed VK3-induced Oxidative Damage through Keap1/Nrf2 Pathway in Human Ovarian Cancer Cells. J. Cancer 2020, 11, 1299–1307. [Google Scholar] [CrossRef]
- Marchi, S.; Giorgi, C.; Galluzzi, L.; Pinton, P. Ca2+ Fluxes and Cancer. Mol. Cell. 2020, 78, 1055–1069. [Google Scholar] [CrossRef]
- Mahalingam, D.; Wilding, G.; Denmeade, S.; Sarantopoulas, J.; Cosgrove, D.; Cetnar, J.; Azad, N.; Bruce, J.; Kurman, M.; Allgood, V.E.; et al. Mipsagargin, a novel thapsigargin-based PSMA-activated prodrug: Results of a first-in-man phase I clinical trial in patients with refractory, advanced or metastatic solid tumours. Br. J. Cancer 2016, 114, 986–994. [Google Scholar] [CrossRef] [PubMed]
- Molina, J.R.; Sun, Y.; Protopopova, M.; Gera, S.; Bandi, M.; Bristow, C.; McAfoos, T.; Morlacchi, P.; Ackroyd, J.; Agip, A.-N.A.; et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat. Med. 2018, 24, 1036–1046. [Google Scholar] [CrossRef] [PubMed]
- Rohlenova, K.; Sachaphibulkij, K.; Stursa, J.; Bezawork-Geleta, A.; Blecha, J.; Endaya, B.; Werner, L.; Cerny, J.; Zobalova, R.; Goodwin, J.; et al. Selective Disruption of Respiratory Supercomplexes as a New Strategy to Suppress Her2 high Breast Cancer. Antioxid. Redox Signal. 2017, 26, 84–103. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.-F.; Jameson, V.J.A.; Tilly, D.; Cerny, J.; Mahdavian, E.; Marín-Hernández, A.; Hernández-Esquivel, L.; Rodríguez-Enríquez, S.; Stursa, J.; Witting, P.K.; et al. Mitochondrial Targeting of Vitamin E Succinate Enhances Its Pro-apoptotic and Anti-cancer Activity via Mitochondrial Complex II. J. Biol. Chem. 2011, 286, 3717–3728. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Shestov, A.A.; Worth, A.J.; Nath, K.; Nelson, D.S.; Leeper, D.B.; Glickson, J.D.; Blair, I.A. Inhibition of Mitochondrial Complex II by the Anticancer Agent Lonidamine. J. Biol. Chem. 2016, 291, 42–57. [Google Scholar] [CrossRef]
- Brummer, C.; Faerber, S.; Bruss, C.; Blank, C.; Lacroix, R.; Haferkamp, S.; Herr, W.; Kreutz, M.; Renner, K. Metabolic targeting synergizes with MAPK inhibition and delays drug resistance in melanoma. Cancer Lett. 2019, 442, 453–463. [Google Scholar] [CrossRef]
- Lucantoni, F.; Düssmann, H.; Llorente-Folch, I.; Prehn, J.H.M. BCL2 and BCL(X)L selective inhibitors decrease mitochondrial ATP production in breast cancer cells and are synthetically lethal when combined with 2-deoxy-D-glucose. Oncotarget 2018, 9, 26046–26063. [Google Scholar] [CrossRef]
- Chae, Y.C.; Caino, M.C.; Lisanti, S.; Ghosh, J.C.; Dohi, T.; Danial, N.N.; Villanueva, J.; Ferrero, S.; Vaira, V.; Santambrogio, L.; et al. Control of Tumor Bioenergetics and Survival Stress Signaling by Mitochondrial HSP90s. Cancer Cell 2012, 22, 331–344. [Google Scholar] [CrossRef]
- Stuart, S.D.; Schauble, A.; Gupta, S.; Kennedy, A.D.; Keppler, B.R.; Bingham, P.M.; Zachar, Z. A strategically designed small molecule attacks alpha-ketoglutarate dehydrogenase in tumor cells through a redox process. Cancer Metab. 2014, 2, 4. [Google Scholar] [CrossRef]
- Pardee, T.S.; Anderson, R.G.; Pladna, K.M.; Isom, S.; Ghiraldeli, L.P.; Miller, L.D.; Chou, J.W.; Jin, G.; Zhang, W.; Ellis, L.R.; et al. A Phase I Study of CPI-613 in Combination with High-Dose Cytarabine and Mitoxantrone for Relapsed or Refractory Acute Myeloid Leukemia. Clin. Cancer Res. 2018, 24, 2060–2073. [Google Scholar] [CrossRef]
- Egawa, Y.; Saigo, C.; Kito, Y.; Moriki, T.; Takeuchi, T. Therapeutic potential of CPI-613 for targeting tumorous mitochondrial energy metabolism and inhibiting autophagy in clear cell sarcoma. PLoS ONE 2018, 13, e0198940. [Google Scholar] [CrossRef] [PubMed]
- Rani, R.; Kumar, V. Recent Update on Human Lactate Dehydrogenase Enzyme 5 (h LDH5) Inhibitors: A Promising Approach for Cancer Chemotherapy. J. Med. Chem. 2016, 59, 487–496. [Google Scholar] [CrossRef]
- Liu, P.-F.; Tsai, K.-L.; Hsu, C.-J.; Tsai, W.-L.; Cheng, J.-S.; Chang, H.-W.; Shiau, C.-W.; Goan, Y.-G.; Tseng, H.-H.; Wu, C.-H.; et al. Drug Repurposing Screening Identifies Tioconazole as an ATG4 Inhibitor that Suppresses Autophagy and Sensitizes Cancer Cells to Chemotherapy. Theranostics 2018, 8, 830–845. [Google Scholar] [CrossRef]
- Donohue, E.; Thomas, A.; Maurer, N.; Manisali, I.; Zeisser-Labouebe, M.; Zisman, N.; Anderson, H.J.; Ng, S.S.W.; Webb, M.; Bally, M.; et al. The Autophagy Inhibitor Verteporfin Moderately Enhances the Antitumor Activity of Gemcitabine in a Pancreatic Ductal Adenocarcinoma Model. J. Cancer 2013, 4, 585–596. [Google Scholar] [CrossRef]
- Kon, N.; Murakoshi, M.; Isobe, A.; Kagechika, K.; Miyoshi, N.; Nagayama, T. DS16570511 is a small-molecule inhibitor of the mitochondrial calcium uniporter. Cell Death Discov. 2017, 3, 17045. [Google Scholar] [CrossRef] [PubMed]
- Woods, J.J.; Nemani, N.; Shanmughapriya, S.; Kumar, A.; Zhang, M.; Nathan, S.R.; Thomas, M.; Carvalho, E.; Ramachandran, K.; Srikantan, S.; et al. A Selective and Cell-Permeable Mitochondrial Calcium Uniporter (MCU) Inhibitor Preserves Mitochondrial Bioenergetics after Hypoxia/Reoxygenation Injury. ACS Cent. Sci. 2019, 5, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Le, A.; Lane, A.N.; Hamaker, M.; Bose, S.; Gouw, A.; Barbi, J.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Zhang, H.; et al. Glucose-Independent Glutamine Metabolism via TCA Cycling for Proliferation and Survival in B Cells. Cell Metab. 2012, 15, 110–121. [Google Scholar] [CrossRef]
- Xue, C.; Wang, C.; Liu, Q.; Meng, Q.; Sun, H.; Huo, X.; Ma, X.; Liu, Z.; Ma, X.; Peng, J.; et al. Targeting P-glycoprotein expression and cancer cell energy metabolism: Combination of metformin and 2-deoxyglucose reverses the multidrug resistance of K562/Dox cells to doxorubicin. Tumor Biol. 2016, 37, 8587–8597. [Google Scholar] [CrossRef]
- Wang, H.; Wang, L.; Zhang, Y.; Wang, J.; Deng, Y.; Lin, D. Inhibition of glycolytic enzyme hexokinase II (HK2) suppresses lung tumor growth. Cancer Cell. Int. 2016, 16, 9. [Google Scholar] [CrossRef]
- Zhang, D.; Li, J.; Wang, F.; Hu, J.; Wang, S.; Sun, Y. 2-Deoxy-D-glucose targeting of glucose metabolism in cancer cells as a potential therapy. Cancer Lett. 2014, 355, 176–183. [Google Scholar] [CrossRef]
- Kurtoglu, M.; Maher, J.C.; Lampidis, T.J. Differential Toxic Mechanisms of 2-Deoxy-D-Glucose versus 2-Fluorodeoxy-D -Glucose in Hypoxic and Normoxic Tumor Cells. Antioxid. Redox Signal. 2007, 9, 1383–1390. [Google Scholar] [CrossRef]
- Alistar, A.; Morris, B.B.; Desnoyer, R.; Klepin, H.D.; Hosseinzadeh, K.; Clark, C.; Cameron, A.; Leyendecker, J.; D’Agostino, R.; Topaloglu, U.; et al. Safety and tolerability of the first-in-class agent CPI-613 in combination with modified FOLFIRINOX in patients with metastatic pancreatic cancer: A single-centre, open-label, dose-escalation, phase 1 trial. Lancet Oncol. 2017, 18, 770–778. [Google Scholar] [CrossRef] [PubMed]
- Jha, M.K.; Suk, K. Pyruvate Dehydrogenase Kinase as a Potential Therapeutic Target for Malignant Gliomas. Brain Tumor Res. Treat. 2013, 1, 57. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, S.-L.; Hu, X.; Tam, K.Y. Targeting Tumor Metabolism for Cancer Treatment: Is Pyruvate Dehydrogenase Kinases (PDKs) a Viable Anticancer Target? Int. J. Biol. Sci. 2015, 11, 1390–1400. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Z.H.; Cai, Q. Mitochondrial transport in neurons: Impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 2012, 13, 77–93. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.-H.; Ding, Z.-B.; Zhou, J.; Hui, B.; Shi, G.-M.; Ke, A.-W.; Wang, X.-Y.; Dai, Z.; Peng, Y.-F.; Gu, C.-Y.; et al. Targeting autophagy enhances sorafenib lethality for hepatocellular carcinoma via ER stress-related apoptosis. Autophagy 2011, 7, 1159–1172. [Google Scholar] [CrossRef]
- Biswas, S.; Dodwadkar, N.S.; Deshpande, P.P.; Torchilin, V.P. Liposomes loaded with paclitaxel and modified with novel triphenylphosphonium-PEG-PE conjugate possess low toxicity, target mitochondria and demonstrate enhanced antitumor effects in vitro and in vivo. J. Control. Release 2012, 159, 393–402. [Google Scholar] [CrossRef]
- Oliveira, C.S.; Turchiello, R.; Kowaltowski, A.J.; Indig, G.L.; Baptista, M.S. Major determinants of photoinduced cell death: Subcellular localization versus photosensitization efficiency. Free Radic. Biol. Med. 2011, 51, 824–833. [Google Scholar] [CrossRef]
- Yaqoob, M.D.; Xu, L.; Li, C.; Leong, M.M.L.; Xu, D.D. Targeting mitochondria for cancer photodynamic therapy. Photodiagnosis Photodyn. Ther. 2022, 38, 102830. [Google Scholar] [CrossRef]
- Kurokawa, H.; Ito, H.; Inoue, M.; Tabata, K.; Sato, Y.; Yamagata, K.; Kizaka-Kondoh, S.; Kadonosono, T.; Yano, S.; Inoue, M.; et al. High-resolution imaging of intracellular oxygen concentration by phosphorescence lifetime. Sci. Rep. 2015, 5, 10657. [Google Scholar] [CrossRef]
- Celli, J.P.; Solban, N.; Liang, A.; Pereira, S.P.; Hasan, T. Verteporfin-based photodynamic therapy overcomes gemcitabine insensitivity in a panel of pancreatic cancer cell lines. Lasers Surg. Med. 2011, 43, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Zielonka, J.; Ouari, O.; Lopez, M.; McAllister, D.; Boyle, K.; Barrios, C.S.; Weber, J.J.; Johnson, B.D.; Hardy, M.; et al. Mitochondria-Targeted Analogues of Metformin Exhibit Enhanced Antiproliferative and Radiosensitizing Effects in Pancreatic Cancer Cells. Cancer Res. 2016, 76, 3904–3915. [Google Scholar] [CrossRef]
- Millard, M.; Gallagher, J.D.; Olenyuk, B.Z.; Neamati, N. A Selective Mitochondrial-Targeted Chlorambucil with Remarkable Cytotoxicity in Breast and Pancreatic Cancers. J. Med. Chem. 2013, 56, 9170–9179. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.; Shao, W.; Zhu, C.; Wen, G.; Yue, X.; Wang, R.; Quan, J.; Du, J.; Bu, X. Mitochondria-Targeted Approach: Remarkably Enhanced Cellular Bioactivities of TPP2a as Selective Inhibitor and Probe toward TrxR. ACS Chem. Biol. 2016, 11, 425–434. [Google Scholar] [CrossRef]
- Mahalingam, S.M.; Ordaz, J.D.; Low, P.S. Targeting of a Photosensitizer to the Mitochondrion Enhances the Potency of Photodynamic Therapy. ACS Omega 2018, 3, 6066–6074. [Google Scholar] [CrossRef]
- Peng, F.; Liao, M.; Qin, R.; Zhu, S.; Peng, C.; Fu, L.; Chen, Y.; Han, B. Regulated cell death (RCD) in cancer: Key pathways and targeted therapies. Signal Transduct. Target Ther. 2022, 7, 286. [Google Scholar] [CrossRef]
- Tait, S.W.G.; de Vries, E.; Maas, C.; Keller, A.M.; D’Santos, C.S.; Borst, J. Apoptosis induction by Bid requires unconventional ubiquitination and degradation of its N-terminal fragment. J. Cell Biol. 2007, 179, 1453–1466. [Google Scholar] [CrossRef] [PubMed]
- Del Re, D.P.; Amgalan, D.; Linkermann, A.; Liu, Q.; Kitsis, R.N. Fundamental Mechanisms of Regulated Cell Death and Implications for Heart Disease. Physiol. Rev. 2019, 99, 1765–1817. [Google Scholar] [CrossRef]
- Chipuk, J.E.; Moldoveanu, T.; Llambi, F.; Parsons, M.J.; Green, D.R. The BCL-2 Family Reunion. Mol. Cell 2010, 37, 299–310. [Google Scholar] [CrossRef]
- Chipuk, J.E.; McStay, G.P.; Bharti, A.; Kuwana, T.; Clarke, C.J.; Siskind, L.J.; Obeid, L.M.; Green, D.R. Sphingolipid Metabolism Cooperates with BAK and BAX to Promote the Mitochondrial Pathway of Apoptosis. Cell 2012, 148, 988–1000. [Google Scholar] [CrossRef]
- Zou, H.; Henzel, W.J.; Liu, X.; Lutschg, A.; Wang, X. Apaf-1, a Human Protein Homologous to C. elegans CED-4, Participates in Cytochrome c–Dependent Activation of Caspase-3. Cell 1997, 90, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Merlin, J.P.J.; Rupasinghe, H.P.V.; Dellaire, G.; Murphy, K. Role of Dietary Antioxidants in p53-Mediated Cancer Chemoprevention and Tumor Suppression. Oxid. Med. Cell. Longev. 2021, 2021, 9924328. [Google Scholar] [CrossRef]
- Edlich, F.; Banerjee, S.; Suzuki, M.; Cleland, M.M.; Arnoult, D.; Wang, C.; Neutzner, A.; Tjandra, N.; Youle, R.J. Bcl-xL Retrotranslocates Bax from the Mitochondria into the Cytosol. Cell 2011, 145, 104–116. [Google Scholar] [CrossRef]
- Wani, A.K.; Akhtar, N.; Mir, T.U.G.; Singh, R.; Jha, P.K.; Mallik, S.K.; Sinha, S.; Tripathi, S.K.; Jain, A.; Jha, A.; et al. Targeting Apoptotic Pathway of Cancer Cells with Phytochemicals and Plant-Based Nanomaterials. Biomolecules 2023, 13, 194. [Google Scholar] [CrossRef]
- Pandya, S.R.; Singh, H.; Desimone, M.F.; Singh, J.; George, N.; Jasani, S. Circumventing challenges in mitochondrial targeting for cancer treatment: Leveraging nanoplatforms for effective solutions. Mater. Adv. 2024, 5, 409–431. [Google Scholar] [CrossRef]
- Oleinick, N.L.; Morris, R.L.; Belichenko, I. The role of apoptosis in response to photodynamic therapy: What, where, why, and how. Photochem. Photobiol. Sci. 2002, 1, 1–21. [Google Scholar] [CrossRef]
- Usuda, J.; Azizuddin, K.; Chiu, S.; Oleinick, N.L. Association Between the Photodynamic Loss of Bcl-2 and the Sensitivity to Apoptosis Caused by Phthalocyanine Photodynamic Therapy. Photochem Photobiol. 2003, 78, 1–8. [Google Scholar] [CrossRef]
- Shimizu, S.; Kanaseki, T.; Mizushima, N.; Mizuta, T.; Arakawa-Kobayashi, S.; Thompson, C.B.; Tsujimoto, Y. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat. Cell Biol. 2004, 6, 1221–1228. [Google Scholar] [CrossRef] [PubMed]
- Pattingre, S.; Levine, B. Bcl-2 Inhibition of Autophagy: A New Route to Cancer? Cancer Res. 2006, 66, 2885–2888. [Google Scholar] [CrossRef]
- Dewaele, M.; Martinet, W.; Rubio, N.; Verfaillie, T.; de Witte, P.A.; Piette, J.; Agostinis, P. Autophagy pathways activated in response to PDT contribute to cell resistance against ROS damage. J. Cell. Mol. Med. 2011, 15, 1402–1414. [Google Scholar] [CrossRef]
- Wang, H.; Fang, Z.-Z.; Zheng, Y.; Zhou, K.; Hu, C.; Krausz, K.W.; Sun, D.; Idle, J.R.; Gonzalez, F.J. Metabolic profiling of praziquantel enantiomers. Biochem. Pharmacol. 2014, 90, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wu, D.; Wang, X.; Cederbaum, A.I. Cytochrome P4502E1, oxidative stress, JNK, and autophagy in acute alcohol-induced fatty liver. Free Radic. Biol. Med. 2012, 53, 1170–1180. [Google Scholar] [CrossRef] [PubMed]
- Aniogo, E.C.; George, B.P.; Abrahamse, H. Molecular Effectors of Photodynamic Therapy-Mediated Resistance to Cancer Cells. Int. J. Mol. Sci. 2021, 22, 13182. [Google Scholar] [CrossRef] [PubMed]
- Edis, Z.; Wang, J.; Waqas, M.K.; Ijaz, M.; Ijaz, M. Nanocarriers-Mediated Drug Delivery Systems for Anticancer Agents: An Overview and Perspectives. Int. J. Nanomed. 2021, 16, 1313–1330. [Google Scholar] [CrossRef] [PubMed]
- Sultana, A.; Zare, M.; Thomas, V.; Kumar, T.S.S.; Ramakrishna, S. Nano-based drug delivery systems: Conventional drug delivery routes, recent developments and future prospects. Med. Drug Discov. 2022, 15, 100134. [Google Scholar] [CrossRef]
- Awad, N.S.; Salkho, N.M.; Abuwatfa, W.H.; Paul, V.; AlSawaftah, N.M.; Husseini, G.A. Tumor vasculature vs tumor cell targeting: Understanding the latest trends in using functional nanoparticles for cancer treatment. Open Nano 2023, 11, 100136. [Google Scholar] [CrossRef]
- Singh, S.; Singh, S.; Lillard Jr, J.W.; Singh, R. Drug delivery approaches for breast cancer. Int. J. Nanomed. 2017, 12, 6205–6218. [Google Scholar] [CrossRef]
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A.; Kessel, D.; et al. Photodynamic therapy of cancer: An update. CA Cancer J. Clin. 2011, 61, 250–281. [Google Scholar] [CrossRef]
- Celli, J.P.; Spring, B.Q.; Rizvi, I.; Evans, C.L.; Samkoe, K.S.; Verma, S.; Pogue, B.W.; Hasan, T. Imaging and Photodynamic Therapy: Mechanisms, Monitoring, and Optimization. Chem. Rev. 2010, 110, 2795–2838. [Google Scholar] [CrossRef]
- van Straten, D.; Mashayekhi, V.; de Bruijn, H.; Oliveira, S.; Robinson, D. Oncologic Photodynamic Therapy: Basic Principles, Current Clinical Status and Future Directions. Cancers 2017, 9, 19. [Google Scholar] [CrossRef]
- Li, J.; Wang, S.; Fontana, F.; Tapeinos, C.; Shahbazi, M.A.; Han, H.; Santos, H.A. Nanoparticles-based phototherapy systems for cancer treatment: Current status and clinical potential. Bioact. Mater. 2022, 23, 471–507. [Google Scholar] [CrossRef] [PubMed]
- Weissig, V. Mitochondria-Specific Nanocarriers for Improving the Proapoptotic Activity of Small Molecules. Methods Enzymol. 2012, 508, 131–155. [Google Scholar]
- Modica-Napolitano, J.S.; Singh, K. Mitochondria as targets for detection and treatment of cancer. Expert Rev. Mol. Med. 2002, 4, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, M.; Vousden, K.H. Regulation of p53 stability. Oncogene 1999, 18, 7637–7643. [Google Scholar] [CrossRef] [PubMed]
- Fan, T.; Zhang, M.; Yang, J.; Zhu, Z.; Cao, W.; Dong, C. Therapeutic cancer vaccines: Advancements, challenges and prospects. Signal Transduct. Target Ther. 2023, 8, 450. [Google Scholar] [CrossRef]
- Alpert, N.M.; Pelletier-Galarneau, M.; Kim, S.J.W.; Petibon, Y.; Sun, T.; Ramos-Torres, K.M.; Normandin, M.D.; El Fakhri, G. In-vivo Imaging of Mitochondrial Depolarization of Myocardium with Positron Emission Tomography and a Proton Gradient Uncoupler. Front. Physiol. 2020, 11, 00491. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Drug | Type of Cancer | Model | Mitochondria Function | Mechanism of Action | References |
|---|---|---|---|---|---|
| Metformin | Breast, prostate, melanoma, ovary, and lung | Clinical trials | Bioenergetic | Inhibition complex I | [86,87] |
| Vitamin K3 | Ovary | Clinical trials | Signaling | Increasing generation of ROS | [90] |
| Mipsagargin (G-202) | Glioblastoma and liver | Clinical trials | Signaling | Mitochondrial Ca2+ transfer modulation | [92] |
| BAY 87-2243 | Lung and prostate | Clinical trials | Bioenergetic | Inhibition complex I | [93] |
| IACS-010,759 | Acute myeloid leukemia and chronic lymphocytic leukemia | Clinical trials | Bioenergetic | Inhibition complex I | [94] |
| MitoVES | Breast | Preclinical | Bioenergetic | Inhibition complex I and II | [95] |
| Lonidamine | Lung | Clinical trials | Bioenergetic | Inhibition complex II | [96] |
| Venetoclax and WEHI-539 | Breast | Preclinical | Bioenergetic | Reducing bioenergetic | [97] |
| VLX600 | Colon | Clinical trials | Bioenergetic | ETC inhibitor | [98] |
| Gamitrinib | Lung and prostate | Preclinical | Bioenergetic | Inhibition HSP90 and TRAP-1 activity | [99] |
| CPI-613 | Pancreas and hematological cancers | Clinical trials | Bioenergetic | PDH and α-KGDH inhibitor | [100,101] |
| Enasidenib and Ivosidenib | Acute myeloid leukemia | Clinical trials | Bioenergetic | Mutant IDH inhibitors | [102] |
| Dichloroacetate | Lung and liver | Clinical trials | Bioenergetic | PDKs inhibitor | |
| Gossypol | Breast, brain, and prostate | Clinical trials | Bioenergetic | LDHA inhibitor and NADH competitor | [103] |
| Tioconazole | Colon | Clinical trials | Signaling | Blocking autophagy targeting ATG4B | [104] |
| Verteporfin | Pancreas | Clinical trials | Signaling | Blocking autophagosome formation | [105] |
| Mitoxantrone and pixantrone | B-cell non-Hodgkin’s lymphoma | Clinical trials | Signaling | MCU complex inhibition | [106] |
| GLS inhibitors | Breast and Burkitt lymphoma | Clinical trials | Biosynthesis | Reducing glutamine catabolism | [107,108] |
| Experimental Condition | Therapeutic Agent | Treatment Method | Reference |
|---|---|---|---|
| Ovarian cancer | Paclitaxel-NPs | Chemotherapy | [45] |
| Ovarian cancer | Cisplatin-based peptide delivery | Chemotherapy | [46] |
| Several cancers | PEGylated-NPs | Drug solubility | [47] |
| Cancer therapy | ROS-sensitive NPs | Intracellular release | [50] |
| Mitochondrial disorders | Mitochondrial transcription factors | Gene therapy | [55] |
| Neurodegenerative diseases | Antioxidant-NPs | Oxidative stress reduction | [56] |
| Gene therapy | Oligonucleotide-NPs | Nucleic acid delivery | [57] |
| Neurodegenerative diseases | Curcumin-NPs | Antioxidant therapy | [61] |
| Cardioprotection | Mitochondria-targeted Peptides | Cellular uptake | [62] |
| Mitochondrial diseases | Mitochondrial proteins | Mitochondrial dynamics | [118] |
| Breast cancer | Doxorubicin-NPs | Chemotherapy | [153] |
| Solid tumors | Antibody-NP complexes | Targeted therapy | [154] |
| Cancer treatment | Mitochondria-targeted aptamers | Targeted therapy | [155] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merlin, J.P.J.; Crous, A.; Abrahamse, H. Combining Photodynamic Therapy and Targeted Drug Delivery Systems: Enhancing Mitochondrial Toxicity for Improved Cancer Outcomes. Int. J. Mol. Sci. 2024, 25, 10796. https://doi.org/10.3390/ijms251910796
Merlin JPJ, Crous A, Abrahamse H. Combining Photodynamic Therapy and Targeted Drug Delivery Systems: Enhancing Mitochondrial Toxicity for Improved Cancer Outcomes. International Journal of Molecular Sciences. 2024; 25(19):10796. https://doi.org/10.3390/ijms251910796
Chicago/Turabian StyleMerlin, J. P. Jose, Anine Crous, and Heidi Abrahamse. 2024. "Combining Photodynamic Therapy and Targeted Drug Delivery Systems: Enhancing Mitochondrial Toxicity for Improved Cancer Outcomes" International Journal of Molecular Sciences 25, no. 19: 10796. https://doi.org/10.3390/ijms251910796