

Enhancing Antimicrobial Peptide Activity through Modifications of Charge, Hydrophobicity, and Structure

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Optimization of AMPs
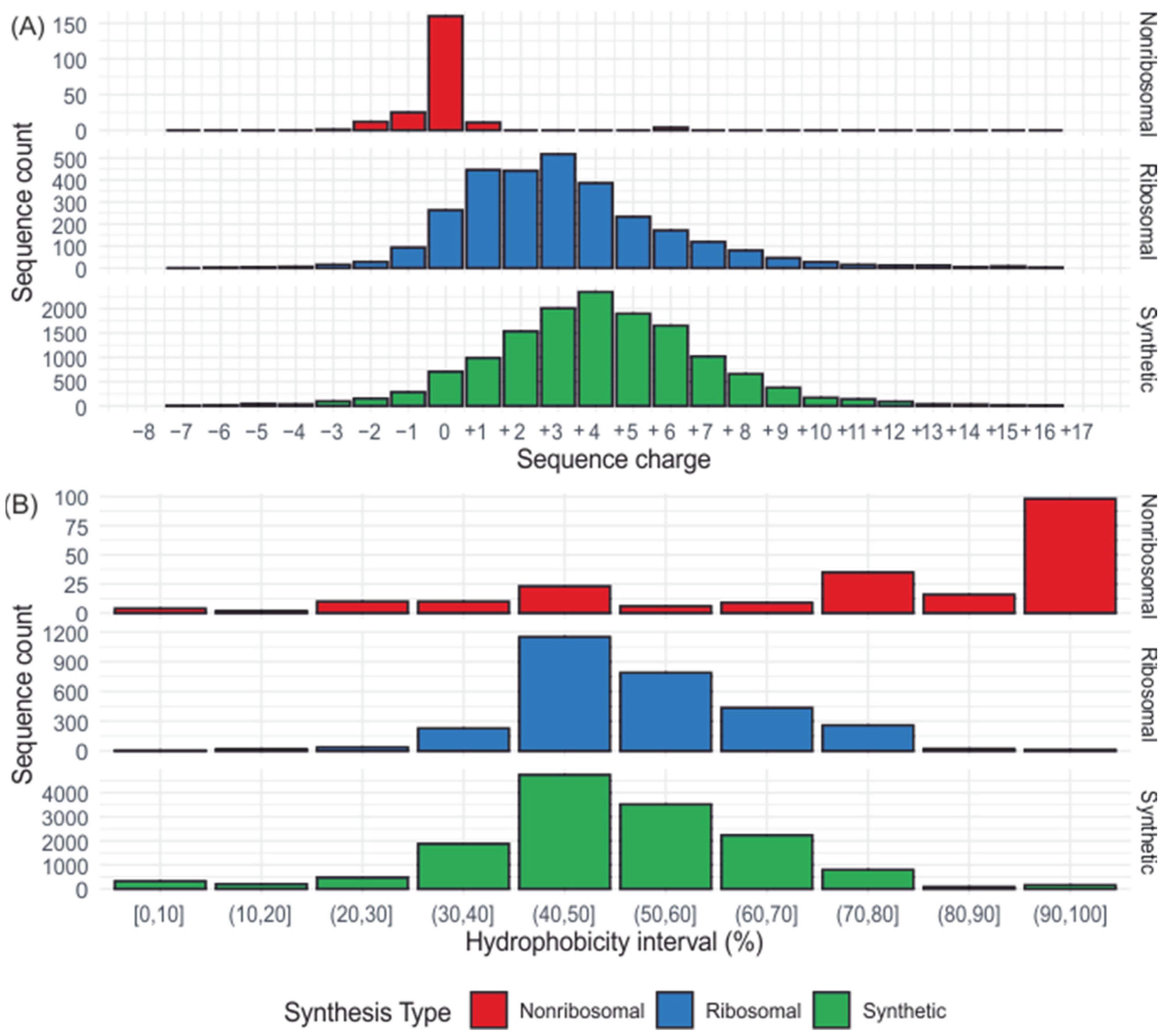
2.1. Charge
2.2. Hydrophobicity
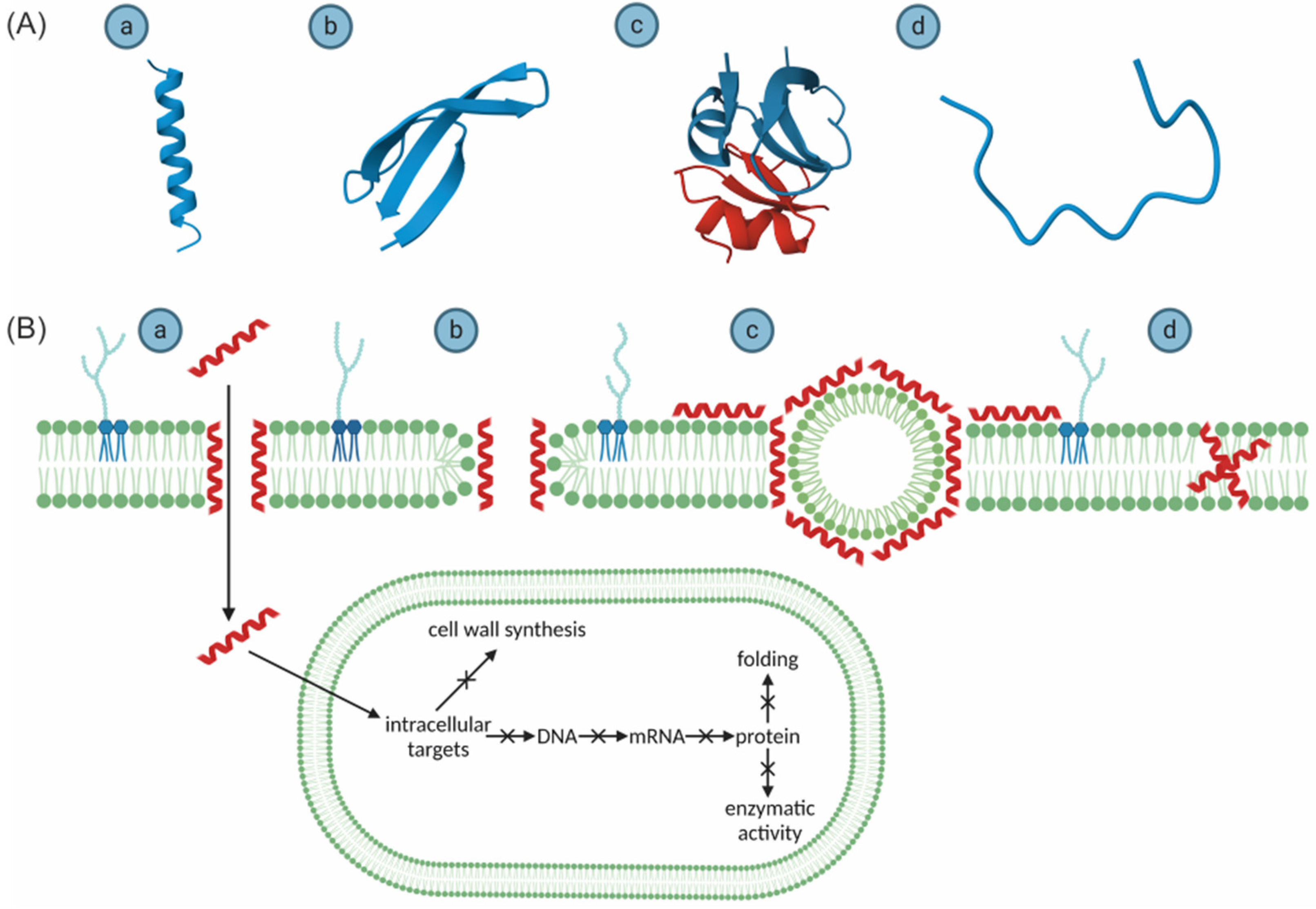
2.3. Structure
2.4. De Novo Design
3. Application of AMPs
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mendelsohn, J.A. “Like All That Lives”: Biology, Medicine and Bacteria in the Age of Pasteur and Koch. Hist. Philos. Life Sci. 2002, 24, 3–36. [Google Scholar] [CrossRef] [PubMed]
- Aminov, R.I. A Brief History of the Antibiotic Era: Lessons Learned and Challenges for the Future. Front. Microbiol. 2010, 1, 134. [Google Scholar] [CrossRef] [PubMed]
- Durand, G.A.; Raoult, D.; Dubourg, G. Antibiotic Discovery: History, Methods and Perspectives. Int. J. Antimicrob. Agents 2019, 53, 371–382. [Google Scholar] [CrossRef]
- Uddin, T.M.; Chakraborty, A.J.; Khusro, A.; Zidan, B.R.M.; Mitra, S.; Emran, T.B.; Dhama, K.; Ripon, M.d.K.H.; Gajdács, M.; Sahibzada, M.U.K.; et al. Antibiotic Resistance in Microbes: History, Mechanisms, Therapeutic Strategies and Future Prospects. J. Infect. Public Health 2021, 14, 1750–1766. [Google Scholar] [CrossRef]
- Blair, J.M.A.; Webber, M.A.; Baylay, A.J.; Ogbolu, D.O.; Piddock, L.J.V. Molecular Mechanisms of Antibiotic Resistance. Nat. Rev. Microbiol. 2015, 13, 42–51. [Google Scholar] [CrossRef] [PubMed]
- MacGowan, A.; Macnaughton, E. Antibiotic Resistance. Medicine 2017, 45, 622–628. [Google Scholar] [CrossRef]
- Laxminarayan, R.; Duse, A.; Wattal, C.; Zaidi, A.K.M.; Wertheim, H.F.L.; Sumpradit, N.; Vlieghe, E.; Hara, G.L.; Gould, I.M.; Goossens, H.; et al. Antibiotic Resistance—The Need for Global Solutions. Lancet Infect. Dis. 2013, 13, 1057–1098. [Google Scholar] [CrossRef]
- Talebi Bezmin Abadi, A.; Rizvanov, A.A.; Haertlé, T.; Blatt, N.L. World Health Organization Report: Current Crisis of Antibiotic Resistance. BioNanoScience 2019, 9, 778–788. [Google Scholar] [CrossRef]
- Kwon, J.H.; Powderly, W.G. The Post-Antibiotic Era Is Here. Science 2021, 373, 471. [Google Scholar] [CrossRef]
- Mancuso, G.; Midiri, A.; Gerace, E.; Biondo, C. Bacterial Antibiotic Resistance: The Most Critical Pathogens. Pathogens 2021, 10, 1310. [Google Scholar] [CrossRef]
- Lucero-Prisno, D.E., III; Shomuyiwa, D.O.; Kouwenhoven, M.B.N.; Dorji, T.; Odey, G.O.; Miranda, A.V.; Ogunkola, I.O.; Adebisi, Y.A.; Huang, J.; Xu, L.; et al. Top 10 Public Health Challenges to Track in 2023: Shifting Focus beyond a Global Pandemic. Public Health Chall. 2023, 2, e86. [Google Scholar] [CrossRef]
- WHO Publishes List of Bacteria for Which New Antibiotics Are Urgently Needed. Available online: https://www.who.int/news/item/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed (accessed on 18 January 2024).
- Chen, C.-Y.; Yang, K.-Y.; Peng, C.-K.; Sheu, C.-C.; Chan, M.-C.; Feng, J.-Y.; Wang, S.-H.; Chen, C.-M.; Zheng, Z.-R.; Liang, S.-J.; et al. Clinical Outcome of Nosocomial Pneumonia Caused by Carbapenem-Resistant Gram-Negative Bacteria in Critically Ill Patients: A Multicenter Retrospective Observational Study. Sci. Rep. 2022, 12, 7501. [Google Scholar] [CrossRef] [PubMed]
- Peleg, A.Y.; Hooper, D.C. Hospital-Acquired Infections Due to Gram-Negative Bacteria. N. Engl. J. Med. 2010, 362, 1804–1813. [Google Scholar] [CrossRef]
- Temkin, E.; Carmeli, Y. Zero or More: Methodological Challenges of Counting and Estimating Deaths Related to Antibiotic-Resistant Infections. Clin. Infect. Dis. 2019, 69, 2029–2034. [Google Scholar] [CrossRef]
- O’Neill, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations; Government of the United Kingdom: London, UK, 2016. [Google Scholar]
- Cassini, A.; Högberg, L.D.; Plachouras, D.; Quattrocchi, A.; Hoxha, A.; Simonsen, G.S.; Colomb-Cotinat, M.; Kretzschmar, M.E.; Devleesschauwer, B.; Cecchini, M.; et al. Attributable Deaths and Disability-Adjusted Life-Years Caused by Infections with Antibiotic-Resistant Bacteria in the EU and the European Economic Area in 2015: A Population-Level Modelling Analysis. Lancet Infect. Dis. 2019, 19, 56–66. [Google Scholar] [CrossRef] [PubMed]
- CDC. Antibiotic Resistance Threats in the United States, 2019; Centres for Disease Control and Prevention, US Department of Health and Human Services: Washington, DC, USA, 2019. [Google Scholar]
- Czaplewski, L.; Bax, R.; Clokie, M.; Dawson, M.; Fairhead, H.; Fischetti, V.A.; Foster, S.; Gilmore, B.F.; Hancock, R.E.W.; Harper, D.; et al. Alternatives to Antibiotics-a Pipeline Portfolio Review. Lancet Infect. Dis. 2016, 16, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Magana, M.; Pushpanathan, M.; Santos, A.L.; Leanse, L.; Fernandez, M.; Ioannidis, A.; Giulianotti, M.A.; Apidianakis, Y.; Bradfute, S.; Ferguson, A.L.; et al. The Value of Antimicrobial Peptides in the Age of Resistance. Lancet Infect. Dis. 2020, 20, e216–e230. [Google Scholar] [CrossRef]
- Ryu, M.; Park, J.; Yeom, J.-H.; Joo, M.; Lee, K. Rediscovery of Antimicrobial Peptides as Therapeutic Agents. J. Microbiol. 2021, 59, 113–123. [Google Scholar] [CrossRef]
- Hassan, M.; Flanagan, T.W.; Kharouf, N.; Bertsch, C.; Mancino, D.; Haikel, Y. Antimicrobial Proteins: Structure, Molecular Action, and Therapeutic Potential. Pharmaceutics 2023, 15, 72. [Google Scholar] [CrossRef]
- Pirtskhalava, M.; Amstrong, A.A.; Grigolava, M.; Chubinidze, M.; Alimbarashvili, E.; Vishnepolsky, B.; Gabrielian, A.; Rosenthal, A.; Hurt, D.E.; Tartakovsky, M. DBAASP v3: Database of Antimicrobial/Cytotoxic Activity and Structure of Peptides as a Resource for Development of New Therapeutics. Nucleic Acids Res. 2021, 49, D288–D297. [Google Scholar] [CrossRef]
- Mookherjee, N.; Anderson, M.A.; Haagsman, H.P.; Davidson, D.J. Antimicrobial Host Defence Peptides: Functions and Clinical Potential. Nat. Rev. Drug Discov. 2020, 19, 311–332. [Google Scholar] [CrossRef]
- Ahmed, A.; Siman-Tov, G.; Hall, G.; Bhalla, N.; Narayanan, A. Human Antimicrobial Peptides as Therapeutics for Viral Infections. Viruses 2019, 11, 704. [Google Scholar] [CrossRef]
- Maróti, G.; Kereszt, A.; Kondorosi, É.; Mergaert, P. Natural Roles of Antimicrobial Peptides in Microbes, Plants and Animals. Res. Microbiol. 2011, 162, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.W.; Haney, E.F.; Gill, E.E. The Immunology of Host Defence Peptides: Beyond Antimicrobial Activity. Nat. Rev. Immunol. 2016, 16, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Mahlapuu, M.; Håkansson, J.; Ringstad, L.; Björn, C. Antimicrobial Peptides: An Emerging Category of Therapeutic Agents. Front. Cell. Infect. Microbiol. 2016, 6, 235805. [Google Scholar] [CrossRef]
- de la Fuente-Núñez, C.; Silva, O.N.; Lu, T.K.; Franco, O.L. Antimicrobial Peptides: Role in Human Disease and Potential as Immunotherapies. Pharmacol. Ther. 2017, 178, 132–140. [Google Scholar] [CrossRef]
- Raffatellu, M. Learning from Bacterial Competition in the Host to Develop Antimicrobials. Nat. Med. 2018, 24, 1097–1103. [Google Scholar] [CrossRef] [PubMed]
- Suneja, G.; Nain, S.; Sharma, R. Microbiome: A Source of Novel Bioactive Compounds and Antimicrobial Peptides. In Microbial Diversity in Ecosystem Sustainability and Biotechnological Applications: Volume 1. Microbial Diversity in Normal & Extreme Environments; Satyanarayana, T., Johri, B.N., Das, S.K., Eds.; Springer: Singapore, 2019; pp. 615–630. ISBN 9789811383151. [Google Scholar]
- Travkova, O.G.; Moehwald, H.; Brezesinski, G. The Interaction of Antimicrobial Peptides with Membranes. Adv. Colloid. Interface Sci. 2017, 247, 521–532. [Google Scholar] [CrossRef]
- Kumar, P.; Kizhakkedathu, J.N.; Straus, S.K. Antimicrobial Peptides: Diversity, Mechanism of Action and Strategies to Improve the Activity and Biocompatibility In Vivo. Biomolecules 2018, 8, 4. [Google Scholar] [CrossRef]
- Ahmed, T.A.E.; Hammami, R. Recent Insights into Structure–Function Relationships of Antimicrobial Peptides. J. Food Biochem. 2019, 43, e12546. [Google Scholar] [CrossRef]
- Abraham, D.J.; Leo, A.J. Extension of the Fragment Method to Calculate Amino Acid Zwitterion and Side Chain Partition Coefficients. Proteins Struct. Funct. Bioinform. 1987, 2, 130–152. [Google Scholar] [CrossRef] [PubMed]
- Andersson, D.I.; Hughes, D.; Kubicek-Sutherland, J.Z. Mechanisms and Consequences of Bacterial Resistance to Antimicrobial Peptides. Drug Resist. Updates 2016, 26, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Lázár, V.; Martins, A.; Spohn, R.; Daruka, L.; Grézal, G.; Fekete, G.; Számel, M.; Jangir, P.K.; Kintses, B.; Csörgő, B.; et al. Antibiotic-Resistant Bacteria Show Widespread Collateral Sensitivity to Antimicrobial Peptides. Nat. Microbiol. 2018, 3, 718–731. [Google Scholar] [CrossRef]
- Spohn, R.; Daruka, L.; Lázár, V.; Martins, A.; Vidovics, F.; Grézal, G.; Méhi, O.; Kintses, B.; Számel, M.; Jangir, P.K.; et al. Integrated Evolutionary Analysis Reveals Antimicrobial Peptides with Limited Resistance. Nat. Commun. 2019, 10, 4538. [Google Scholar] [CrossRef]
- Le, C.-F.; Fang, C.-M.; Sekaran, S.D. Intracellular Targeting Mechanisms by Antimicrobial Peptides. Antimicrob. Agents Chemother. 2017, 61, e02340-16. [Google Scholar] [CrossRef]
- Scocchi, M.; Mardirossian, M.; Runti, G.; Benincasa, M. Non-Membrane Permeabilizing Modes of Action of Antimicrobial Peptides on Bacteria. Curr. Top. Med. Chem. 2016, 16, 76–88. [Google Scholar] [CrossRef]
- Costa, F.; Teixeira, C.; Gomes, P.; Martins, M.C.L. Clinical Application of AMPs. In Antimicrobial Peptides: Basics for Clinical Application; Matsuzaki, K., Ed.; Advances in Experimental Medicine and Biology; Springer: Singapore, 2019; pp. 281–298. ISBN 9789811335884. [Google Scholar]
- Mahlapuu, M.; Björn, C.; Ekblom, J. Antimicrobial Peptides as Therapeutic Agents: Opportunities and Challenges. Crit. Rev. Biotechnol. 2020, 40, 978–992. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Dou, X.; Song, J.; Lyu, Y.; Zhu, X.; Xu, L.; Li, W.; Shan, A. Antimicrobial Peptides: Promising Alternatives in the Post Feeding Antibiotic Era. Med. Res. Rev. 2019, 39, 831–859. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Yang, N.; Wang, X.; Hao, Y.; Mao, R.; Li, Z.; Wang, Z.; Teng, D.; Wang, J. An Enhanced Variant Designed from DLP4 Cationic Peptide Against Staphylococcus Aureus CVCC 546. Front. Microbiol. 2020, 11, 1057. [Google Scholar] [CrossRef]
- Hwang, P.M.; Vogel, H.J. Structure–Function Relationships of Antimicrobial Peptides. Biochem. Cell Biol. 1998, 76, 235–246. [Google Scholar] [CrossRef]
- Li, S.; Wang, Y.; Xue, Z.; Jia, Y.; Li, R.; He, C.; Chen, H. The structure-mechanism relationship and mode of actions of antimicrobial peptides: A review. Trends Food Sci. Technol. 2021, 109, 103–115. [Google Scholar] [CrossRef]
- Tan, P.; Fu, H.; Ma, X. Design, Optimization, and Nanotechnology of Antimicrobial Peptides: From Exploration to Applications. Nano Today 2021, 39, 101229. [Google Scholar] [CrossRef]
- Wang, G.; Li, X.; Wang, Z. APD3: The Antimicrobial Peptide Database as a Tool for Research and Education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef] [PubMed]
- Burdukiewicz, M.; Sidorczuk, K.; Rafacz, D.; Pietluch, F.; Chilimoniuk, J.; Rödiger, S.; Gagat, P. Proteomic Screening for Prediction and Design of Antimicrobial Peptides with AmpGram. Int. J. Mol. Sci. 2020, 21, 4310. [Google Scholar] [CrossRef]
- Sidorczuk, K.; Gagat, P.; Pietluch, F.; Kała, J.; Rafacz, D.; Bąkała, L.; Słowik, J.; Kolenda, R.; Rödiger, S.; Fingerhut, L.C. Benchmarks in Antimicrobial Peptide Prediction Are Biased Due to the Selection of Negative Data. Brief. Bioinform. 2022, 23, bbac343. [Google Scholar] [CrossRef]
- Santos-Júnior, C.D.; Pan, S.; Zhao, X.-M.; Coelho, L.P. Macrel: Antimicrobial Peptide Screening in Genomes and Metagenomes. PeerJ 2020, 8, e10555. [Google Scholar] [CrossRef] [PubMed]
- Fingerhut, L.C.H.W.; Miller, D.J.; Strugnell, J.M.; Daly, N.L.; Cooke, I.R. Ampir: An R Package for Fast Genome-Wide Prediction of Antimicrobial Peptides. Bioinformatics 2021, 36, 5262–5263. [Google Scholar] [CrossRef]
- Veltri, D.; Kamath, U.; Shehu, A. Deep Learning Improves Antimicrobial Peptide Recognition. Bioinformatics 2018, 34, 2740–2747. [Google Scholar] [CrossRef]
- Lawrence, T.J.; Carper, D.L.; Spangler, M.K.; Carrell, A.A.; Rush, T.A.; Minter, S.J.; Weston, D.J.; Labbé, J.L. amPEPpy 1.0: A Portable and Accurate Antimicrobial Peptide Prediction Tool. Bioinformatics 2021, 37, 2058–2060. [Google Scholar] [CrossRef]
- Gautier, R.; Douguet, D.; Antonny, B.; Drin, G. HELIQUEST: A Web Server to Screen Sequences with Specific α-Helical Properties. Bioinformatics 2008, 24, 2101–2102. [Google Scholar] [CrossRef]
- Zhou, X.; Zheng, W.; Li, Y.; Pearce, R.; Zhang, C.; Bell, E.W.; Zhang, G.; Zhang, Y. I-TASSER-MTD: A Deep-Learning-Based Platform for Multi-Domain Protein Structure and Function Prediction. Nat. Protoc. 2022, 17, 2326–2353. [Google Scholar] [CrossRef] [PubMed]
- Lamiable, A.; Thévenet, P.; Rey, J.; Vavrusa, M.; Derreumaux, P.; Tufféry, P. PEP-FOLD3: Faster de Novo Structure Prediction for Linear Peptides in Solution and in Complex. Nucleic Acids Res. 2016, 44, W449–W454. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T. Protein Secondary Structure Prediction Based on Position-Specific Scoring Matrices 1 1Edited by G. Von Heijne. J. Mol. Biol. 1999, 292, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Ramos-Llorens, M.; Bello-Madruga, R.; Valle, J.; Andreu, D.; Torrent, M. PyAMPA: A High-Throughput Prediction and Optimization Tool for Antimicrobial Peptides. mSystems 2024, 9, e01358-23. [Google Scholar] [CrossRef]
- Bessalle, R.; Haas, H.; Goria, A.; Shalit, I.; Fridkin, M. Augmentation of the Antibacterial Activity of Magainin by Positive-Charge Chain Extension. Antimicrob. Agents Chemother. 1992, 36, 313–317. [Google Scholar] [CrossRef]
- Higgs, R.; Lynn, D.J.; Cahalane, S.; Alaña, I.; Hewage, C.M.; James, T.; Lloyd, A.T.; O’Farrelly, C. Modification of Chicken Avian β-Defensin-8 at Positively Selected Amino Acid Sites Enhances Specific Antimicrobial Activity. Immunogenetics 2007, 59, 573–580. [Google Scholar] [CrossRef]
- Jiang, Z.; Vasil, A.I.; Hale, J.D.; Hancock, R.E.W.; Vasil, M.L.; Hodges, R.S. Effects of Net Charge and the Number of Positively Charged Residues on the Biological Activity of Amphipathic A-helical Cationic Antimicrobial Peptides. Biopolymers 2008, 90, 369–383. [Google Scholar] [CrossRef]
- Yount, N.Y.; Bayer, A.S.; Xiong, Y.Q.; Yeaman, M.R. Advances in Antimicrobial Peptide Immunobiology. Biopolymers 2006, 84, 435–458. [Google Scholar] [CrossRef]
- Frederiksen, N.; Hansen, P.R.; Zabicka, D.; Tomczak, M.; Urbas, M.; Domraceva, I.; Björkling, F.; Franzyk, H. Alternating Cationic-Hydrophobic Peptide/Peptoid Hybrids: Influence of Hydrophobicity on Antibacterial Activity and Cell Selectivity. ChemMedChem 2020, 15, 2544–2561. [Google Scholar] [CrossRef]
- Shang, D.; Zhang, Q.; Dong, W.; Liang, H.; Bi, X. The Effects of LPS on the Activity of Trp-Containing Antimicrobial Peptides against Gram-Negative Bacteria and Endotoxin Neutralization. Acta Biomater. 2016, 33, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Yount, N.; Yeaman, M. Immunocontinuum: Perspectives in Antimicrobial Peptide Mechanisms of Action and Resistance. Protein Pept. Lett. 2005, 12, 49–67. [Google Scholar] [CrossRef] [PubMed]
- Dennison, S.; Harris, F.; Phoenix, D. Are Oblique Orientated α-Helices Used by Antimicrobial Peptides for Membrane Invasion? Protein Pept. Lett. 2005, 12, 27–29. [Google Scholar] [CrossRef] [PubMed]
- Glukhov, E.; Stark, M.; Burrows, L.L.; Deber, C.M. Basis for Selectivity of Cationic Antimicrobial Peptides for Bacterial Versus Mammalian Membranes. J. Biol. Chem. 2005, 280, 33960–33967. [Google Scholar] [CrossRef]
- Haney, E.F.; Lau, F.; Vogel, H.J. Solution Structures and Model Membrane Interactions of Lactoferrampin, an Antimicrobial Peptide Derived from Bovine Lactoferrin. Biochim. Biophys. Acta (BBA) Biomembr. 2007, 1768, 2355–2364. [Google Scholar] [CrossRef]
- Sparks, K.A.; Gleason, N.J.; Gist, R.; Langston, R.; Greathouse, D.V.; Koeppe, R.E.I. Comparisons of Interfacial Phe, Tyr, and Trp Residues as Determinants of Orientation and Dynamics for GWALP Transmembrane Peptides. Biochemistry 2014, 53, 3637–3645. [Google Scholar] [CrossRef]
- Shahmiri, M.; Cornell, B.; Mechler, A. Phenylalanine Residues Act as Membrane Anchors in the Antimicrobial Action of Aurein 1.2. Biointerphases 2017, 12, 05G605. [Google Scholar] [CrossRef]
- Lander, A.J.; Mercado, L.D.; Li, X.; Taily, I.M.; Findlay, B.L.; Jin, Y.; Luk, L.Y.P. Roles of Inter- and Intramolecular Tryptophan Interactions in Membrane-Active Proteins Revealed by Racemic Protein Crystallography. Commun. Chem. 2023, 6, 154. [Google Scholar] [CrossRef]
- Schmidtchen, A.; Pasupuleti, M.; Mörgelin, M.; Davoudi, M.; Alenfall, J.; Chalupka, A.; Malmsten, M. Boosting Antimicrobial Peptides by Hydrophobic Oligopeptide End Tags. J. Biol. Chem. 2009, 284, 17584–17594. [Google Scholar] [CrossRef]
- Schmidtchen, A.; Ringstad, L.; Kasetty, G.; Mizuno, H.; Rutland, M.W.; Malmsten, M. Membrane Selectivity by W-Tagging of Antimicrobial Peptides. Biochim. Biophys. Acta (BBA) Biomembr. 2011, 1808, 1081–1091. [Google Scholar] [CrossRef]
- Malmsten, M.; Kasetty, G.; Pasupuleti, M.; Alenfall, J.; Schmidtchen, A. Highly Selective End-Tagged Antimicrobial Peptides Derived from PRELP. PLoS ONE 2011, 6, e16400. [Google Scholar] [CrossRef] [PubMed]
- Molhoek, E.M.; Van Dijk, A.; Veldhuizen, E.J.A.; Dijk-Knijnenburg, H.; Mars-Groenendijk, R.H.; Boele, L.C.L.; Kaman-van Zanten, W.E.; Haagsman, H.P.; Bikker, F.J. Chicken Cathelicidin-2-Derived Peptides with Enhanced Immunomodulatory and Antibacterial Activities against Biological Warfare Agents. Int. J. Antimicrob. Agents 2010, 36, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, A.; Molhoek, E.M.; Veldhuizen, E.J.A.; Bokhoven, J.L.M.T.; Wagendorp, E.; Bikker, F.; Haagsman, H.P. Identification of Chicken Cathelicidin-2 Core Elements Involved in Antibacterial and Immunomodulatory Activities. Mol. Immunol. 2009, 46, 2465–2473. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Shen, T.; Liu, Y.; Zhou, J.; Shi, S.; Wang, Y.; Zhao, Z.; Yan, Z.; Liao, C.; Wang, C. Enhancing the Antibacterial Activity of Antimicrobial Peptide PMAP-37(F34-R) by Cholesterol Modification. BMC Vet. Res. 2020, 16, 419. [Google Scholar] [CrossRef]
- Zhou, J.; Chen, L.; Liu, Y.; Shen, T.; Zhang, C.; Liu, Z.; Feng, X.; Wang, C. Antimicrobial Peptide PMAP-37 Analogs: Increasing the Positive Charge to Enhance the Antibacterial Activity of PMAP-37. J. Pept. Sci. 2019, 25, e3220. [Google Scholar] [CrossRef]
- Liu, T.; Zhu, N.; Zhong, C.; Zhu, Y.; Gou, S.; Chang, L.; Bao, H.; Liu, H.; Zhang, Y.; Ni, J. Effect of N-Methylated and Fatty Acid Conjugation on Analogs of Antimicrobial Peptide Anoplin. Eur. J. Pharm. Sci. 2020, 152, 105453. [Google Scholar] [CrossRef] [PubMed]
- Zhong, C.; Zhu, N.; Zhu, Y.; Liu, T.; Gou, S.; Xie, J.; Yao, J.; Ni, J. Antimicrobial Peptides Conjugated with Fatty Acids on the Side Chain of D-Amino Acid Promises Antimicrobial Potency against Multidrug-Resistant Bacteria. Eur. J. Pharm. Sci. 2020, 141, 105123. [Google Scholar] [CrossRef]
- Liu, H.; Yang, N.; Teng, D.; Mao, R.; Hao, Y.; Ma, X.; Wang, X.; Wang, J. Fatty Acid Modified-Antimicrobial Peptide Analogues with Potent Antimicrobial Activity and Topical Therapeutic Efficacy against Staphylococcus Hyicus. Appl. Microbiol. Biotechnol. 2021, 105, 5845–5859. [Google Scholar] [CrossRef]
- Chatterjee, J.; Rechenmacher, F.; Kessler, H. N-Methylation of Peptides and Proteins: An Important Element for Modulating Biological Functions. Angew. Chem. Int. Ed. 2013, 52, 254–269. [Google Scholar] [CrossRef]
- Smirnova, M.P.; Kolodkin, N.I.; Kolobov, A.A.; Afonin, V.G.; Afonina, I.V.; Stefanenko, L.I.; Shpen’, V.M.; Shamova, O.V.; Kolobov, A.A. Indolicidin Analogs with Broad-Spectrum Antimicrobial Activity and Low Hemolytic Activity. Peptides 2020, 132, 170356. [Google Scholar] [CrossRef]
- Yeaman, M.R.; Yount, N.Y. Mechanisms of Antimicrobial Peptide Action and Resistance. Pharmacol. Rev. 2003, 55, 27–55. [Google Scholar] [CrossRef]
- Dathe, M.; Wieprecht, T. Structural Features of Helical Antimicrobial Peptides: Their Potential to Modulate Activity on Model Membranes and Biological Cells. Biochim. Biophys. Acta (BBA) Biomembr. 1999, 1462, 71–87. [Google Scholar] [CrossRef]
- Dempsey, C.E. The Actions of Melittin on Membranes. Biochim. Biophys. Acta (BBA) Rev. Biomembr. 1990, 1031, 143–161. [Google Scholar] [CrossRef]
- Asthana, N.; Yadav, S.P.; Ghosh, J.K. Dissection of Antibacterial and Toxic Activity of Melittin. J. Biol. Chem. 2004, 279, 55042–55050. [Google Scholar] [CrossRef]
- Chen, Y.; Mant, C.T.; Farmer, S.W.; Hancock, R.E.W.; Vasil, M.L.; Hodges, R.S. Rational Design of α-Helical Antimicrobial Peptides with Enhanced Activities and Specificity/Therapeutic Index. J. Biol. Chem. 2005, 280, 12316–12329. [Google Scholar] [CrossRef] [PubMed]
- Hawrani, A.; Howe, R.A.; Walsh, T.R.; Dempsey, C.E. Origin of Low Mammalian Cell Toxicity in a Class of Highly Active Antimicrobial Amphipathic Helical Peptides. J. Biol. Chem. 2008, 283, 18636–18645. [Google Scholar] [CrossRef]
- Zhang, S.-K.; Song, J.; Gong, F.; Li, S.-B.; Chang, H.-Y.; Xie, H.-M.; Gao, H.-W.; Tan, Y.-X.; Ji, S.-P. Design of an α-Helical Antimicrobial Peptide with Improved Cell-Selective and Potent Anti-Biofilm Activity. Sci. Rep. 2016, 6, 27394. [Google Scholar] [CrossRef]
- Srivastava, S.; Kumar, A.; Tripathi, A.K.; Tandon, A.; Ghosh, J.K. Modulation of Anti-Endotoxin Property of Temporin L by Minor Amino Acid Substitution in Identified Phenylalanine Zipper Sequence. Biochem. J. 2016, 473, 4045–4062. [Google Scholar] [CrossRef]
- Song, J.; Wang, J.; Zhan, N.; Sun, T.; Yu, W.; Zhang, L.; Shan, A.; Zhang, A. Therapeutic Potential of Trp-Rich Engineered Amphiphiles by Single Hydrophobic Amino Acid End-Tagging. ACS Appl. Mater. Interfaces 2019, 11, 43820–43834. [Google Scholar] [CrossRef]
- Li, Y.; Bionda, N.; Yongye, A.; Geer, P.; Stawikowski, M.; Cudic, P.; Martinez, K.; Houghten, R.A. Dissociation of Antimicrobial and Hemolytic Activities of Gramicidin S through N–Methylation Modification. ChemMedChem 2013, 8, 1865–1872. [Google Scholar] [CrossRef]
- He, R.; Di Bonaventura, I.; Visini, R.; Gan, B.-H.; Fu, Y.; Probst, D.; Lüscher, A.; Köhler, T.; Van Delden, C.; Stocker, A.; et al. Design, Crystal Structure and Atomic Force Microscopy Study of Thioether Ligated D,L-Cyclic Antimicrobial Peptides against Multidrug Resistant Pseudomonas Aeruginosa. Chem. Sci. 2017, 8, 7464–7475. [Google Scholar] [CrossRef] [PubMed]
- Mani, R.; Tang, M.; Wu, X.; Buffy, J.J.; Waring, A.J.; Sherman, M.A.; Hong, M. Membrane-Bound Dimer Structure of a β-Hairpin Antimicrobial Peptide from Rotational-Echo Double-Resonance Solid-State NMR. Biochemistry 2006, 45, 8341–8349. [Google Scholar] [CrossRef]
- Harwig, S.S.L.; Waring, A.; Yang, H.J.; Cho, Y.; Tan, L.; Lehrer, R.I. Intramolecular Disulfide Bonds Enhance the Antimicrobial and Lytic Activities of Protegrins at Physiological Sodium Chloride Concentrations. Eur. J. Biochem. 1996, 240, 352–357. [Google Scholar] [CrossRef]
- Kokryakov, V.N.; Harwig, S.S.L.; Panyutich, E.A.; Shevchenko, A.A.; Aleshina, G.M.; Shamova, O.V.; Korneva, H.A.; Lehrer, R.I. Protegrins: Leukocyte Antimicrobial Peptides That Combine Features of Corticostatic Defensins and Tachyplesins. FEBS Lett. 1993, 327, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Tam, J.P.; Wu, C.; Yang, J. Membranolytic Selectivity of Cystine-stabilized Cyclic Protegrins. Eur. J. Biochem. 2000, 267, 3289–3300. [Google Scholar] [CrossRef]
- Wiradharma, N.; Khoe, U.; Hauser, C.A.E.; Seow, S.V.; Zhang, S.; Yang, Y.-Y. Synthetic Cationic Amphiphilic α-Helical Peptides as Antimicrobial Agents. Biomaterials 2011, 32, 2204–2212. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Chen, C.; Zhang, S.; Zhao, X.; Xu, H.; Zhao, X.; Lu, J.R. Designed Antimicrobial and Antitumor Peptides with High Selectivity. Biomacromolecules 2011, 12, 3839–3843. [Google Scholar] [CrossRef]
- Chen, C.; Hu, J.; Zeng, P.; Chen, Y.; Xu, H.; Lu, J.R. High Cell Selectivity and Low-Level Antibacterial Resistance of Designed Amphiphilic Peptide G(IIKK)3I-NH2. ACS Appl. Mater. Interfaces 2014, 6, 16529–16536. [Google Scholar] [CrossRef]
- Ahmad, A.; Asthana, N.; Azmi, S.; Srivastava, R.M.; Pandey, B.K.; Yadav, V.; Ghosh, J.K. Structure–Function Study of Cathelicidin-Derived Bovine Antimicrobial Peptide BMAP-28: Design of Its Cell-Selective Analogs by Amino Acid Substitutions in the Heptad Repeat Sequences. Biochim. Biophys. Acta (BBA) Biomembr. 2009, 1788, 2411–2420. [Google Scholar] [CrossRef]
- Ahmad, A.; Azmi, S.; Srivastava, R.M.; Srivastava, S.; Pandey, B.K.; Saxena, R.; Bajpai, V.K.; Ghosh, J.K. Design of Nontoxic Analogues of Cathelicidin-Derived Bovine Antimicrobial Peptide BMAP-27: The Role of Leucine as Well as Phenylalanine Zipper Sequences in Determining Its Toxicity. Biochemistry 2009, 48, 10905–10917. [Google Scholar] [CrossRef]
- Kumar, A.; Tripathi, A.K.; Kathuria, M.; Shree, S.; Tripathi, J.K.; Purshottam, R.K.; Ramachandran, R.; Mitra, K.; Ghosh, J.K. Single Amino Acid Substitutions at Specific Positions of the Heptad Repeat Sequence of Piscidin-1 Yielded Novel Analogs That Show Low Cytotoxicity and In Vitro and In Vivo Antiendotoxin Activity. Antimicrob. Agents Chemother. 2016, 60, 3687–3699. [Google Scholar] [CrossRef] [PubMed]
- Cohen, C.; Parry, D.A.D. α-Helical Coiled Coils and Bundles: How to Design an A-helical Protein. Proteins 1990, 7, 1–15. [Google Scholar] [CrossRef]
- Jelesarov, I.; Dürr, E.; Thomas, R.M.; Bosshard, H.R. Salt Effects on Hydrophobic Interaction and Charge Screening in the Folding of a Negatively Charged Peptide to a Coiled Coil (Leucine Zipper). Biochemistry 1998, 37, 7539–7550. [Google Scholar] [CrossRef]
- Dou, X.; Zhu, X.; Wang, J.; Dong, N.; Shan, A. Novel Design of Heptad Amphiphiles To Enhance Cell Selectivity, Salt Resistance, Antibiofilm Properties and Their Membrane-Disruptive Mechanism. J. Med. Chem. 2017, 60, 2257–2270. [Google Scholar] [CrossRef] [PubMed]
- Edwards, I.A.; Elliott, A.G.; Kavanagh, A.M.; Zuegg, J.; Blaskovich, M.A.T.; Cooper, M.A. Contribution of Amphipathicity and Hydrophobicity to the Antimicrobial Activity and Cytotoxicity of β-Hairpin Peptides. ACS Infect. Dis. 2016, 2, 442–450. [Google Scholar] [CrossRef]
- Shao, C.; Zhu, Y.; Jian, Q.; Lai, Z.; Tan, P.; Li, G.; Shan, A. Cross-Strand Interaction, Central Bending, and Sequence Pattern Act as Biomodulators of Simplified β-Hairpin Antimicrobial Amphiphiles. Small 2021, 17, 2003899. [Google Scholar] [CrossRef]
- Cardoso, M.H.; Orozco, R.Q.; Rezende, S.B.; Rodrigues, G.; Oshiro, K.G.N.; Cândido, E.S.; Franco, O.L. Computer-Aided Design of Antimicrobial Peptides: Are We Generating Effective Drug Candidates? Front. Microbiol. 2020, 10, 3097. [Google Scholar] [CrossRef]
- Agüero-Chapin, G.; Galpert-Cañizares, D.; Domínguez-Pérez, D.; Marrero-Ponce, Y.; Pérez-Machado, G.; Teijeira, M.; Antunes, A. Emerging Computational Approaches for Antimicrobial Peptide Discovery. Antibiotics 2022, 11, 936. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, Y.; Zhu, Y.; Huang, P.; Gao, Q.; Li, X.; Chen, Z.; Liu, Y.; Jiang, J.; Gao, Y.; et al. Machine Learning and Genetic Algorithm-Guided Directed Evolution for the Development of Antimicrobial Peptides. J. Adv. Res. 2024, in press. [CrossRef]
- Yoshida, M.; Hinkley, T.; Tsuda, S.; Abul-Haija, Y.M.; McBurney, R.T.; Kulikov, V.; Mathieson, J.S.; Reyes, S.G.; Castro, M.D.; Cronin, L. Using Evolutionary Algorithms and Machine Learning to Explore Sequence Space for the Discovery of Antimicrobial Peptides. Chem 2018, 4, 533–543. [Google Scholar] [CrossRef]
- Maccari, G.; Luca, M.D.; Nifosí, R.; Cardarelli, F.; Signore, G.; Boccardi, C.; Bifone, A. Antimicrobial Peptides Design by Evolutionary Multiobjective Optimization. PLoS Comput. Biol. 2013, 9, e1003212. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Li, F.; Leier, A.; Xiang, D.; Shen, H.-H.; Marquez Lago, T.T.; Li, J.; Yu, D.-J.; Song, J. Comprehensive Assessment of Machine Learning-Based Methods for Predicting Antimicrobial Peptides. Brief. Bioinform. 2021, 22, bbab083. [Google Scholar] [CrossRef] [PubMed]
- Suda, S.; Field, D.; Barron, N. Antimicrobial Peptide Production and Purification. In Protein Chromatography: Methods and Protocols; Walls, D., Loughran, S.T., Eds.; Springer: New York, NY, USA, 2017; pp. 401–410. ISBN 978-1-4939-6412-3. [Google Scholar]
- Gaglione, R.; Pane, K.; Dell’Olmo, E.; Cafaro, V.; Pizzo, E.; Olivieri, G.; Notomista, E.; Arciello, A. Cost-Effective Production of Recombinant Peptides in Escherichia coli. New Biotechnol. 2019, 51, 39–48. [Google Scholar] [CrossRef]
- Tiwari, P.; Srivastava, Y.; Sharma, A.; Vinayagam, R. Antimicrobial Peptides: The Production of Novel Peptide-Based Therapeutics in Plant Systems. Life 2023, 13, 1875. [Google Scholar] [CrossRef] [PubMed]
- Papo, N.; Oren, Z.; Pag, U.; Sahl, H.-G.; Shai, Y. The Consequence of Sequence Alteration of an Amphipathic α-Helical Antimicrobial Peptide and Its Diastereomers. J. Biol. Chem. 2002, 277, 33913–33921. [Google Scholar] [CrossRef]
- Li, Y.; Liu, T.; Liu, Y.; Tan, Z.; Ju, Y.; Yang, Y.; Dong, W. Antimicrobial Activity, Membrane Interaction and Stability of the D-Amino Acid Substituted Analogs of Antimicrobial Peptide W3R6. J. Photochem. Photobiol. B Biol. 2019, 200, 111645. [Google Scholar] [CrossRef]
- Zai, Y.; Ying, Y.; Ye, Z.; Zhou, M.; Ma, C.; Shi, Z.; Chen, X.; Xi, X.; Chen, T.; Wang, L. Broad-Spectrum Antimicrobial Activity and Improved Stability of a D-Amino Acid Enantiomer of DMPC-10A, the Designed Derivative of Dermaseptin Truncates. Antibiotics 2020, 9, 627. [Google Scholar] [CrossRef]
- Gentilucci, L.; De Marco, R.; Cerisoli, L. Chemical Modifications Designed to Improve Peptide Stability: Incorporation of Non-Natural Amino Acids, Pseudo-Peptide Bonds, and Cyclization. Curr. Pharm. Des. 2010, 16, 3185–3203. [Google Scholar] [CrossRef]
- Rozek, A.; Powers, J.-P.S.; Friedrich, C.L.; Hancock, R.E.W. Structure-Based Design of an Indolicidin Peptide Analogue with Increased Protease Stability. Biochemistry 2003, 42, 14130–14138. [Google Scholar] [CrossRef] [PubMed]
- Schafmeister, C.E.; Po, J.; Verdine, G.L. An All-Hydrocarbon Cross-Linking System for Enhancing the Helicity and Metabolic Stability of Peptides. J. Am. Chem. Soc. 2000, 122, 5891–5892. [Google Scholar] [CrossRef]
- Pham, T.K.; Kim, D.-H.; Lee, B.-J.; Kim, Y.-W. Truncated and Constrained Helical Analogs of Antimicrobial Esculentin-2EM. Bioorganic Med. Chem. Lett. 2013, 23, 6717–6720. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Hu, Y.; Pu, Q.; He, T.; Zhang, Q.; Wu, W.; Xia, X.; Zhang, J. Novel Stapling by Lysine Tethering Provides Stable and Low Hemolytic Cationic Antimicrobial Peptides. J. Med. Chem. 2020, 63, 4081–4089. [Google Scholar] [CrossRef] [PubMed]
- Park, I.Y.; Cho, J.H.; Kim, K.S.; Kim, Y.-B.; Kim, M.S.; Kim, S.C. Helix Stability Confers Salt Resistance upon Helical Antimicrobial Peptides *. J. Biol. Chem. 2004, 279, 13896–13901. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.-Y.; Tu, C.-H.; Yip, B.-S.; Chen, H.-L.; Cheng, H.-T.; Huang, K.-C.; Lo, H.-J.; Cheng, J.-W. Easy Strategy To Increase Salt Resistance of Antimicrobial Peptides. Antimicrob. Agents Chemother. 2011, 55, 4918–4921. [Google Scholar] [CrossRef] [PubMed]
- Hayes, H.C.; Luk, L.Y.P.; Tsai, Y.-H. Approaches for Peptide and Protein Cyclisation. Org. Biomol. Chem. 2021, 19, 3983–4001. [Google Scholar] [CrossRef]
- Ye, Z.; Zhu, X.; Acosta, S.; Kumar, D.; Sang, T.; Aparicio, C. Self-Assembly Dynamics and Antimicrobial Activity of All L- and D-Amino Acid Enantiomers of a Designer Peptide. Nanoscale 2018, 11, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, X.; Wang, Q.; Meng, D. Unnatural Amino Acids: Promising Implications for the Development of New Antimicrobial Peptides. Crit. Rev. Microbiol. 2023, 49, 231–255. [Google Scholar] [CrossRef]
- Zhu, Y.; Hao, W.; Wang, X.; Ouyang, J.; Deng, X.; Yu, H.; Wang, Y. Antimicrobial Peptides, Conventional Antibiotics, and Their Synergistic Utility for the Treatment of Drug-Resistant Infections. Med. Res. Rev. 2022, 42, 1377–1422. [Google Scholar] [CrossRef]
- Kim, E.Y.; Rajasekaran, G.; Shin, S.Y. LL-37-Derived Short Antimicrobial Peptide KR-12-A5 and Its d-Amino Acid Substituted Analogs with Cell Selectivity, Anti-Biofilm Activity, Synergistic Effect with Conventional Antibiotics, and Anti-Inflammatory Activity. Eur. J. Med. Chem. 2017, 136, 428–441. [Google Scholar] [CrossRef]
- Reffuveille, F.; de la Fuente-Núñez, C.; Mansour, S.; Hancock, R.E.W. A Broad-Spectrum Antibiofilm Peptide Enhances Antibiotic Action against Bacterial Biofilms. Antimicrob. Agents Chemother. 2014, 58, 5363–5371. [Google Scholar] [CrossRef]
- Song, M.; Liu, Y.; Huang, X.; Ding, S.; Wang, Y.; Shen, J.; Zhu, K. A Broad-Spectrum Antibiotic Adjuvant Reverses Multidrug-Resistant Gram-Negative Pathogens. Nat. Microbiol. 2020, 5, 1040–1050. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.-Y.; Yan, Z.-B.; Meng, Y.-M.; Hong, X.-Y.; Shao, G.; Ma, J.-J.; Cheng, X.-R.; Liu, J.; Kang, J.; Fu, C.-Y. Antimicrobial Peptides: Mechanism of Action, Activity and Clinical Potential. Mil. Med. Res. 2021, 8, 48. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.K.; Kumari, T.; Harioudh, M.K.; Yadav, P.K.; Kathuria, M.; Shukla, P.K.; Mitra, K.; Ghosh, J.K. Identification of GXXXXG Motif in Chrysophsin-1 and Its Implication in the Design of Analogs with Cell-Selective Antimicrobial and Anti-Endotoxin Activities. Sci. Rep. 2017, 7, 3384. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.K.; Kumari, T.; Tandon, A.; Sayeed, M.; Afshan, T.; Kathuria, M.; Shukla, P.K.; Mitra, K.; Ghosh, J.K. Selective Phenylalanine to Proline Substitution for Improved Antimicrobial and Anticancer Activities of Peptides Designed on Phenylalanine Heptad Repeat. Acta Biomater. 2017, 57, 170–186. [Google Scholar] [CrossRef]
- Porto, W.F.; Fensterseifer, I.C.M.; Ribeiro, S.M.; Franco, O.L. Joker: An Algorithm to Insert Patterns into Sequences for Designing Antimicrobial Peptides. Biochim. Biophys. Acta (BBA) Gen. Subj. 2018, 1862, 2043–2052. [Google Scholar] [CrossRef]
- Porto, W.F.; Irazazabal, L.; Alves, E.S.F.; Ribeiro, S.M.; Matos, C.O.; Pires, Á.S.; Fensterseifer, I.C.M.; Miranda, V.J.; Haney, E.F.; Humblot, V.; et al. In Silico Optimization of a Guava Antimicrobial Peptide Enables Combinatorial Exploration for Peptide Design. Nat. Commun. 2018, 9, 1490. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gagat, P.; Ostrówka, M.; Duda-Madej, A.; Mackiewicz, P. Enhancing Antimicrobial Peptide Activity through Modifications of Charge, Hydrophobicity, and Structure. Int. J. Mol. Sci. 2024, 25, 10821. https://doi.org/10.3390/ijms251910821
Gagat P, Ostrówka M, Duda-Madej A, Mackiewicz P. Enhancing Antimicrobial Peptide Activity through Modifications of Charge, Hydrophobicity, and Structure. International Journal of Molecular Sciences. 2024; 25(19):10821. https://doi.org/10.3390/ijms251910821
Chicago/Turabian StyleGagat, Przemysław, Michał Ostrówka, Anna Duda-Madej, and Paweł Mackiewicz. 2024. "Enhancing Antimicrobial Peptide Activity through Modifications of Charge, Hydrophobicity, and Structure" International Journal of Molecular Sciences 25, no. 19: 10821. https://doi.org/10.3390/ijms251910821
APA StyleGagat, P., Ostrówka, M., Duda-Madej, A., & Mackiewicz, P. (2024). Enhancing Antimicrobial Peptide Activity through Modifications of Charge, Hydrophobicity, and Structure. International Journal of Molecular Sciences, 25(19), 10821. https://doi.org/10.3390/ijms251910821