
Advancements in the Development of Anti-SARS-CoV-2 Therapeutics


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Structural Features and Interacting Receptors of SARS-CoV-2
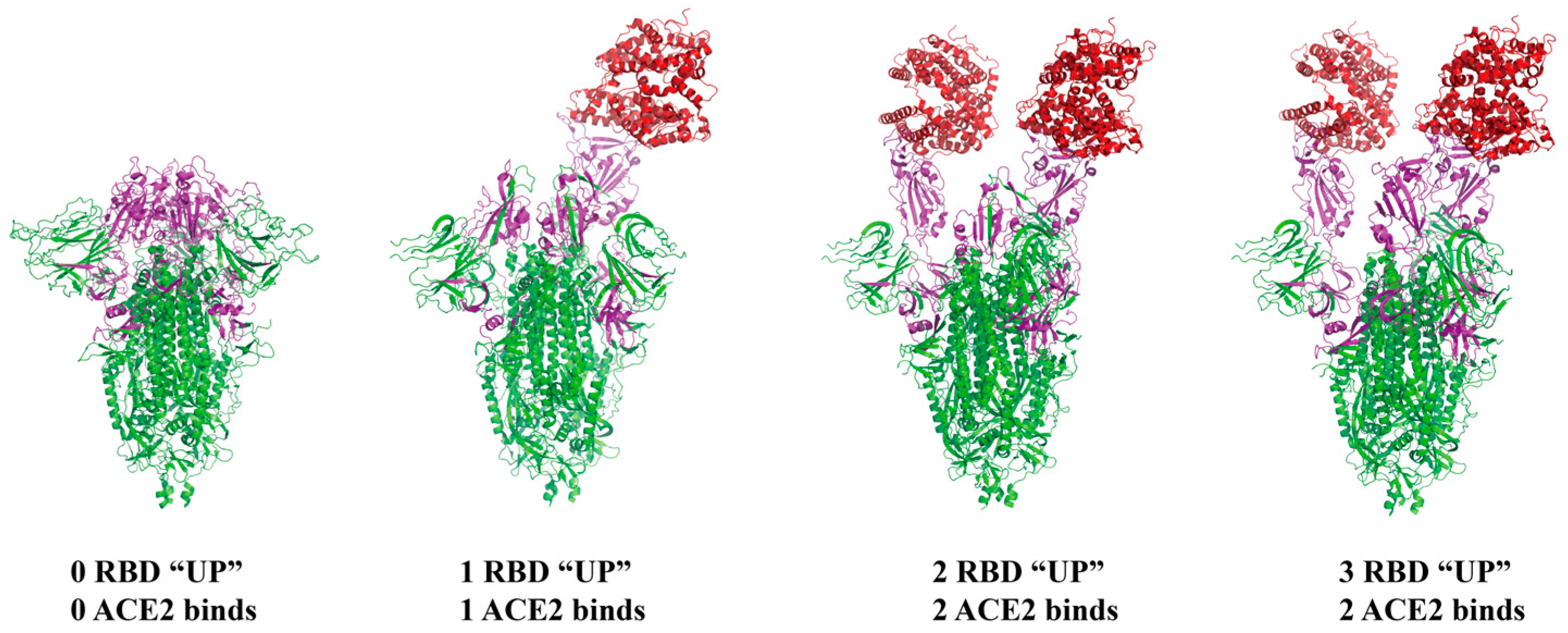
3. Invasion Mechanism of SARS-CoV-2
4. Mutational Characteristics of SARS-CoV-2
4.1. The Alpha (B.1.1.7) Variant
4.2. The Beta (B.1.351) Variant
4.3. The Gamma (P.1) Variant
4.4. The Delta (B.1.617.2) Variant
4.5. The Omicron (B.1.1.529) Variant
5. Anti-SARS-CoV-2 Therapies
5.1. Vaccine Treatment
5.2. Advancements in Research on Drugs Targeting Virus Replication-Related Targets
5.2.1. Inhibitors of RNA-Dependent RNA Polymerase (RdRp)
5.2.2. Inhibitors of 3CL Protease
5.2.3. Inhibitors of Papain-like Protease (PLpro)
5.3. Advances in Research on Drugs Targeting ACE2
5.4. Research Progress on Drugs Targeting Proteases
5.4.1. Furin Inhibitors
5.4.2. TMPRSS2 Inhibitors
5.4.3. Cathepsin Inhibitors
5.4.4. Advancements in Research on Inhibitors Targeting Other Host Enzymes
5.5. Advancements in Research on Drugs Targeting the Spike Protein
5.5.1. Neutralizing Antibodies Targeting S1
5.5.2. Neutralizing Reagents Targeting S2
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients with 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA 2020, 323, 1061–1069. [Google Scholar] [CrossRef]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e286. [Google Scholar] [CrossRef]
- Choi, J.Y.; Smith, D.M. SARS-CoV-2 Variants of Concern. Yonsei Med. J. 2021, 62, 961–968. [Google Scholar] [CrossRef] [PubMed]
- Chenchula, S.; Karunakaran, P.; Sharma, S.; Chavan, M. Current evidence on efficacy of COVID-19 booster dose vaccination against the Omicron variant: A systematic review. J. Med. Virol. 2022, 94, 2969–2976. [Google Scholar] [CrossRef]
- Arya, R.; Kumari, S.; Pandey, B.; Mistry, H.; Bihani, S.C.; Das, A.; Prashar, V.; Gupta, G.D.; Panicker, L.; Kumar, M. Structural insights into SARS-CoV-2 proteins. J. Mol. Biol. 2021, 433, 166725. [Google Scholar] [CrossRef]
- Wang, M.Y.; Zhao, R.; Gao, L.J.; Gao, X.F.; Wang, D.P.; Cao, J.M. SARS-CoV-2: Structure, Biology, and Structure-Based Therapeutics Development. Front. Cell Infect. Microbiol. 2020, 10, 587269. [Google Scholar] [CrossRef] [PubMed]
- Kuriakose, J.; Montezano, A.C.; Touyz, R.M. ACE2/Ang-(1-7)/Mas1 axis and the vascular system: Vasoprotection to COVID-19-associated vascular disease. Clin. Sci. 2021, 135, 387–407. [Google Scholar] [CrossRef]
- Sodhi, C.P.; Nguyen, J.; Yamaguchi, Y.; Werts, A.D.; Lu, P.; Ladd, M.R.; Fulton, W.B.; Kovler, M.L.; Wang, S.; Prindle, T., Jr.; et al. A Dynamic Variation of Pulmonary ACE2 Is Required to Modulate Neutrophilic Inflammation in Response to Pseudomonas aeruginosa Lung Infection in Mice. J. Immunol. 2019, 203, 3000–3012. [Google Scholar] [CrossRef]
- Gao, Q.; Chen, R.; Wu, L.; Huang, Q.; Wang, X.X.; Tian, Y.Y.; Zhang, Y.D. Angiotensin-(1-7) reduces α-synuclein aggregation by enhancing autophagic activity in Parkinson’s disease. Neural Regen. Res. 2022, 17, 1138–1145. [Google Scholar] [CrossRef] [PubMed]
- Hamming, I.; Timens, W.; Bulthuis, M.L.; Lely, A.T.; Navis, G.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef]
- Chambers, J.P.; Yu, J.; Valdes, J.J.; Arulanandam, B.P. SARS-CoV-2, Early Entry Events. J. Pathog. 2020, 2020, 9238696. [Google Scholar] [CrossRef] [PubMed]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e278. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef]
- Ravi, V.; Saxena, S.; Panda, P.S. Basic virology of SARS-CoV 2. Indian J. Med. Microbiol. 2022, 40, 182–186. [Google Scholar] [CrossRef]
- Guest, P.C.; Kesharwani, P.; Butler, A.E.; Sahebkar, A. The COVID-19 Pandemic: SARS-CoV-2 Structure, Infection, Transmission, Symptomology, and Variants of Concern. Adv. Exp. Med. Biol. 2023, 1412, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Francisco, R.D.S., Jr.; Benites, L.F.; Lamarca, A.P.; de Almeida, L.G.P.; Hansen, A.W.; Gularte, J.S.; Demoliner, M.; Gerber, A.L.; Guimarães, A.P.d.C.; Antunes, A.K.E.; et al. Pervasive transmission of E484K and emergence of VUI-NP13L with evidence of SARS-CoV-2 co-infection events by two different lineages in Rio Grande do Sul, Brazil. Virus Res. 2021, 296, 198345. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, S.; Wu, B.; Yang, Q.; Chen, A.; Li, Y.; Zhang, Y.; Pan, T.; Zhang, H.; He, X. SARS-CoV-2 Omicron strain exhibits potent capabilities for immune evasion and viral entrance. Signal Transduct. Target. Ther. 2021, 6, 430. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Han, J.; Zhang, Y.; He, J.; Yu, W.; Zhang, X.; Wu, J.; Zhang, S.; Kong, Y.; Guo, Y.; et al. SARS-CoV-2 Omicron Variant: Epidemiological Features, Biological Characteristics, and Clinical Significance. Front. Immunol. 2022, 13, 877101. [Google Scholar] [CrossRef] [PubMed]
- Patone, M.; Thomas, K.; Hatch, R.; Tan, P.S.; Coupland, C.; Liao, W.; Mouncey, P.; Harrison, D.; Rowan, K.; Horby, P.; et al. Mortality and critical care unit admission associated with the SARS-CoV-2 lineage B.1.1.7 in England: An observational cohort study. Lancet Infect. Dis. 2021, 21, 1518–1528. [Google Scholar] [CrossRef]
- Davies, N.G.; Jarvis, C.I.; Edmunds, W.J.; Jewell, N.P.; Diaz-Ordaz, K.; Keogh, R.H. Increased mortality in community-tested cases of SARS-CoV-2 lineage B.1.1.7. Nature 2021, 593, 270–274. [Google Scholar] [CrossRef]
- Ortega, M.A.; García-Montero, C.; Fraile-Martinez, O.; Colet, P.; Baizhaxynova, A.; Mukhtarova, K.; Alvarez-Mon, M.; Kanatova, K.; Asúnsolo, A.; Sarría-Santamera, A. Recapping the Features of SARS-CoV-2 and Its Main Variants: Status and Future Paths. J. Pers. Med. 2022, 12, 995. [Google Scholar] [CrossRef]
- Cegolon, L.; Magnano, G.; Negro, C.; Larese Filon, F. SARS-CoV-2 Reinfections in Health-Care Workers, 1 March 2020-31 January 2023. Viruses 2023, 15, 1551. [Google Scholar] [CrossRef]
- Yue, C.; Song, W.; Wang, L.; Jian, F.; Chen, X.; Gao, F.; Shen, Z.; Wang, Y.; Wang, X.; Cao, Y. ACE2 binding and antibody evasion in enhanced transmissibility of XBB.1.5. Lancet Infect. Dis. 2023, 23, 278–280. [Google Scholar] [CrossRef]
- Uraki, R.; Ito, M.; Furusawa, Y.; Yamayoshi, S.; Iwatsuki-Horimoto, K.; Adachi, E.; Saito, M.; Koga, M.; Tsutsumi, T.; Yamamoto, S.; et al. Humoral immune evasion of the omicron subvariants BQ.1.1 and XBB. Lancet Infect. Dis. 2023, 23, 30–32. [Google Scholar] [CrossRef]
- Volz, E.; Mishra, S.; Chand, M.; Barrett, J.C.; Johnson, R.; Geidelberg, L.; Hinsley, W.R.; Laydon, D.J.; Dabrera, G.; O’Toole, Á.; et al. Assessing transmissibility of SARS-CoV-2 lineage B.1.1.7 in England. Nature 2021, 593, 266–269. [Google Scholar] [CrossRef]
- Galloway, S.E.; Paul, P.; MacCannell, D.R.; Johansson, M.A.; Brooks, J.T.; MacNeil, A.; Slayton, R.B.; Tong, S.; Silk, B.J.; Armstrong, G.L.; et al. Emergence of SARS-CoV-2 B.1.1.7 Lineage—United States, December 29, 2020-January 12, 2021. MMWR Morb. Mortal. Wkly. Rep. 2021, 70, 95–99. [Google Scholar] [CrossRef]
- McCarthy, K.R.; Rennick, L.J.; Nambulli, S.; Robinson-McCarthy, L.R.; Bain, W.G.; Haidar, G.; Duprex, W.P. Recurrent deletions in the SARS-CoV-2 spike glycoprotein drive antibody escape. Science 2021, 371, 1139–1142. [Google Scholar] [CrossRef]
- Kemp, S.A.; Collier, D.A.; Datir, R.P.; Ferreira, I.; Gayed, S.; Jahun, A.; Hosmillo, M.; Rees-Spear, C.; Mlcochova, P.; Lumb, I.U.; et al. SARS-CoV-2 evolution during treatment of chronic infection. Nature 2021, 592, 277–282. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, Q.; Wei, P.; Chen, Z.; Aviszus, K.; Yang, J.; Downing, W.; Jiang, C.; Liang, B.; Reynoso, L.; et al. The basis of a more contagious 501Y.V1 variant of SARS-CoV-2. Cell Res. 2021, 31, 720–722. [Google Scholar] [CrossRef]
- Daniloski, Z.; Jordan, T.X.; Ilmain, J.K.; Guo, X.; Bhabha, G.; tenOever, B.R.; Sanjana, N.E. The Spike D614G mutation increases SARS-CoV-2 infection of multiple human cell types. eLife 2021, 10, e65365. [Google Scholar] [CrossRef]
- Weissman, D.; Alameh, M.G.; de Silva, T.; Collini, P.; Hornsby, H.; Brown, R.; LaBranche, C.C.; Edwards, R.J.; Sutherland, L.; Santra, S.; et al. D614G Spike Mutation Increases SARS CoV-2 Susceptibility to Neutralization. Cell Host Microbe 2021, 29, 23–31.e24. [Google Scholar] [CrossRef]
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science 2021, 372, eabg3055. [Google Scholar] [CrossRef]
- Thakur, N.; Gallo, G.; Newman, J.; Peacock, T.P.; Biasetti, L.; Hall, C.N.; Wright, E.; Barclay, W.; Bailey, D. SARS-CoV-2 variants of concern alpha, beta, gamma and delta have extended ACE2 receptor host ranges. J. Gen. Virol. 2022, 103, 001735. [Google Scholar] [CrossRef]
- Barton, M.I.; MacGowan, S.A.; Kutuzov, M.A.; Dushek, O.; Barton, G.J.; van der Merwe, P.A. Effects of common mutations in the SARS-CoV-2 Spike RBD and its ligand, the human ACE2 receptor on binding affinity and kinetics. eLife 2021, 10, e70658. [Google Scholar] [CrossRef]
- Wibmer, C.K.; Ayres, F.; Hermanus, T.; Madzivhandila, M.; Kgagudi, P.; Oosthuysen, B.; Lambson, B.E.; de Oliveira, T.; Vermeulen, M.; van der Berg, K.; et al. SARS-CoV-2 501Y.V2 escapes neutralization by South African COVID-19 donor plasma. Nat. Med. 2021, 27, 622–625. [Google Scholar] [CrossRef]
- Faria, N.R.; Mellan, T.A.; Whittaker, C.; Claro, I.M.; Candido, D.D.S.; Mishra, S.; Crispim, M.A.E.; Sales, F.C.S.; Hawryluk, I.; McCrone, J.T.; et al. Genomics and epidemiology of the P.1 SARS-CoV-2 lineage in Manaus, Brazil. Science 2021, 372, 815–821. [Google Scholar] [CrossRef] [PubMed]
- Rahman, F.I.; Ether, S.A.; Islam, M.R. The “Delta Plus” COVID-19 variant has evolved to become the next potential variant of concern: Mutation history and measures of prevention. J. Basic Clin. Physiol. Pharmacol. 2022, 33, 109–112. [Google Scholar] [CrossRef]
- Liu, C.; Ginn, H.M.; Dejnirattisai, W.; Supasa, P.; Wang, B.; Tuekprakhon, A.; Nutalai, R.; Zhou, D.; Mentzer, A.J.; Zhao, Y.; et al. Reduced neutralization of SARS-CoV-2 B.1.617 by vaccine and convalescent serum. Cell 2021, 184, 4220–4236.e4213. [Google Scholar] [CrossRef]
- Ao, Z.; Ouyang, M.J.; Olukitibi, T.A.; Yao, X. SARS-CoV-2 Delta spike protein enhances the viral fusogenicity and inflammatory cytokine production. iScience 2022, 25, 104759. [Google Scholar] [CrossRef]
- Garcia-Beltran, W.F.; Lam, E.C.; St Denis, K.; Nitido, A.D.; Garcia, Z.H.; Hauser, B.M.; Feldman, J.; Pavlovic, M.N.; Gregory, D.J.; Poznansky, M.C.; et al. Multiple SARS-CoV-2 variants escape neutralization by vaccine-induced humoral immunity. Cell 2021, 184, 2372–2383.e2379. [Google Scholar] [CrossRef] [PubMed]
- Motozono, C.; Toyoda, M.; Zahradnik, J.; Saito, A.; Nasser, H.; Tan, T.S.; Ngare, I.; Kimura, I.; Uriu, K.; Kosugi, Y.; et al. SARS-CoV-2 spike L452R variant evades cellular immunity and increases infectivity. Cell Host Microbe 2021, 29, 1124–1136.e1111. [Google Scholar] [CrossRef]
- Cao, Y.; Jian, F.; Wang, J.; Yu, Y.; Song, W.; Yisimayi, A.; Wang, J.; An, R.; Chen, X.; Zhang, N.; et al. Imprinted SARS-CoV-2 humoral immunity induces convergent Omicron RBD evolution. Nature 2023, 614, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Jeworowski, L.M.; Mühlemann, B.; Walper, F.; Schmidt, M.L.; Jansen, J.; Krumbholz, A.; Simon-Lorière, E.; Jones, T.C.; Corman, V.M.; Drosten, C. Humoral immune escape by current SARS-CoV-2 variants BA.2.86 and JN.1, December 2023. Eurosurveillance 2024, 29, 2300740. [Google Scholar] [CrossRef]
- Yang, S.; Yu, Y.; Xu, Y.; Jian, F.; Song, W.; Yisimayi, A.; Wang, P.; Wang, J.; Liu, J.; Yu, L.; et al. Fast evolution of SARS-CoV-2 BA.2·86 to JN.1 under heavy immune pressure. Lancet Infect. Dis. 2024, 24, e70–e72. [Google Scholar] [CrossRef]
- Kaku, Y.; Okumura, K.; Padilla-Blanco, M.; Kosugi, Y.; Uriu, K.; Hinay, A.A., Jr.; Chen, L.; Plianchaisuk, A.; Kobiyama, K.; Ishii, K.J.; et al. Virological characteristics of the SARS-CoV-2 JN.1 variant. Lancet Infect. Dis. 2024, 24, e82. [Google Scholar] [CrossRef]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef] [PubMed]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Andrews, N.; Stowe, J.; Kirsebom, F.; Toffa, S.; Rickeard, T.; Gallagher, E.; Gower, C.; Kall, M.; Groves, N.; O’Connell, A.M.; et al. Covid-19 Vaccine Effectiveness against the Omicron (B.1.1.529) Variant. N. Engl. J. Med. 2022, 386, 1532–1546. [Google Scholar] [CrossRef] [PubMed]
- Planas, D.; Saunders, N.; Maes, P.; Guivel-Benhassine, F.; Planchais, C.; Buchrieser, J.; Bolland, W.-H.; Porrot, F.; Staropoli, I.; Lemoine, F.; et al. Considerable escape of SARS-CoV-2 Omicron to antibody neutralization. Nature 2022, 602, 671–675. [Google Scholar] [CrossRef]
- Lu, L.; Mok, B.W.Y.; Chen, L.L.; Chan, J.M.C.; Tsang, O.T.Y.; Lam, B.H.S.; Chuang, V.W.M.; Chu, A.W.H.; Chan, W.M.; Ip, J.D.; et al. Neutralization of Severe Acute Respiratory Syndrome Coronavirus 2 Omicron Variant by Sera From BNT162b2 or CoronaVac Vaccine Recipients. Clin. Infect. Dis. 2022, 75, e822–e826. [Google Scholar] [CrossRef]
- Saciuk, Y.; Kertes, J.; Shamir Stein, N.; Ekka Zohar, A. Effectiveness of a Third Dose of BNT162b2 mRNA Vaccine. J. Infect. Dis. 2022, 225, 30–33. [Google Scholar] [CrossRef]
- Garcia-Beltran, W.F.; St Denis, K.J.; Hoelzemer, A.; Lam, E.C.; Nitido, A.D.; Sheehan, M.L.; Berrios, C.; Ofoman, O.; Chang, C.C.; Hauser, B.M.; et al. mRNA-based COVID-19 vaccine boosters induce neutralizing immunity against SARS-CoV-2 Omicron variant. Cell 2022, 185, 457–466.e454. [Google Scholar] [CrossRef]
- Gordon, C.J.; Tchesnokov, E.P.; Woolner, E.; Perry, J.K.; Feng, J.Y.; Porter, D.P.; Götte, M. Remdesivir is a direct-acting antiviral that inhibits RNA-dependent RNA polymerase from severe acute respiratory syndrome coronavirus 2 with high potency. J. Biol. Chem. 2020, 295, 6785–6797. [Google Scholar] [CrossRef]
- Gbinigie, O.; Ogburn, E.; Allen, J.; Dorward, J.; Dobson, M.; Madden, T.A.; Yu, L.M.; Lowe, D.M.; Rahman, N.; Petrou, S.; et al. Platform adaptive trial of novel antivirals for early treatment of COVID-19 In the community (PANORAMIC): Protocol for a randomised, controlled, open-label, adaptive platform trial of community novel antiviral treatment of COVID-19 in people at increased risk of more severe disease. BMJ Open 2023, 13, e069176. [Google Scholar] [CrossRef]
- Gordon, C.J.; Tchesnokov, E.P.; Feng, J.Y.; Porter, D.P.; Götte, M. The antiviral compound remdesivir potently inhibits RNA-dependent RNA polymerase from Middle East respiratory syndrome coronavirus. J. Biol. Chem. 2020, 295, 4773–4779. [Google Scholar] [CrossRef] [PubMed]
- Kabinger, F.; Stiller, C.; Schmitzová, J.; Dienemann, C.; Kokic, G.; Hillen, H.S.; Höbartner, C.; Cramer, P. Mechanism of molnupiravir-induced SARS-CoV-2 mutagenesis. Nat. Struct. Mol. Biol. 2021, 28, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Butler, C.C.; Hobbs, F.D.R.; Gbinigie, O.A.; Rahman, N.M.; Hayward, G.; Richards, D.B.; Dorward, J.; Lowe, D.M.; Standing, J.F.; Breuer, J.; et al. Molnupiravir plus usual care versus usual care alone as early treatment for adults with COVID-19 at increased risk of adverse outcomes (PANORAMIC): An open-label, platform-adaptive randomised controlled trial. Lancet 2023, 401, 281–293. [Google Scholar] [CrossRef]
- Khoo, S.H.; FitzGerald, R.; Saunders, G.; Middleton, C.; Ahmad, S.; Edwards, C.J.; Hadjiyiannakis, D.; Walker, L.; Lyon, R.; Shaw, V.; et al. Molnupiravir versus placebo in unvaccinated and vaccinated patients with early SARS-CoV-2 infection in the UK (AGILE CST-2): A randomised, placebo-controlled, double-blind, phase 2 trial. Lancet. Infect. Dis. 2023, 23, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Abdelnabi, R.; Foo, C.S.; Kaptein, S.J.F.; Zhang, X.; Do, T.N.D.; Langendries, L.; Vangeel, L.; Breuer, J.; Pang, J.; Williams, R.; et al. The combined treatment of Molnupiravir and Favipiravir results in a potentiation of antiviral efficacy in a SARS-CoV-2 hamster infection model. EBioMedicine 2021, 72, 103595. [Google Scholar] [CrossRef]
- Zhang, J.-L.; Li, Y.-H.; Wang, L.-L.; Liu, H.-Q.; Lu, S.-Y.; Liu, Y.; Li, K.; Liu, B.; Li, S.-Y.; Shao, F.-M.; et al. Azvudine is a thymus-homing anti-SARS-CoV-2 drug effective in treating COVID-19 patients. Signal Transduct. Target. Ther. 2021, 6, 414. [Google Scholar] [CrossRef]
- Yang, H.; Wang, Z.; Jiang, C.; Zhang, Y.; Zhang, Y.; Xu, M.; Zhang, Y.; Wang, Y.; Liu, X.; An, Z.; et al. Oral azvudine for mild-to-moderate COVID-19 in high risk, nonhospitalized adults: Results of a real-world study. J. Med. Virol. 2023, 95, e28947. [Google Scholar] [CrossRef]
- Meng, W.; Guo, S.; Cao, S.; Shuda, M.; Robinson-McCarthy, L.R.; McCarthy, K.R.; Shuda, Y.; Paniz Mondolfi, A.E.; Bryce, C.; Grimes, Z.; et al. Development and characterization of a new monoclonal antibody against SARS-CoV-2 NSP12 (RdRp). J. Med. Virol. 2023, 95, e28246. [Google Scholar] [CrossRef]
- He, S.-t.; Qin, H.; Guan, L.; Liu, K.; Hong, B.; Zhang, X.; Lou, F.; Li, M.; Lin, W.; Chen, Y.; et al. Bovine lactoferrin inhibits SARS-CoV-2 and SARS-CoV-1 by targeting the RdRp complex and alleviates viral infection in the hamster model. J. Med. Virol. 2023, 95, e28281. [Google Scholar] [CrossRef]
- Minetti, C.A.; Remeta, D.P.; Hashimoto, K.; Bonala, R.; Chennamshetti, R.; Yin, X.; Garcia-Diaz, M.; Grollman, A.P.; Johnson, F.; Sidorenko, V.S. Characterization of Aurintricarboxylic Acid (ATA) Interactions with Plasma Transporter Protein and SARS-CoV-2 Viral Targets: Correlation of Functional Activity and Binding Energetics. Life 2022, 12, 872. [Google Scholar] [CrossRef]
- Prajapati, K.S.; Singh, A.K.; Kushwaha, P.P.; Shuaib, M.; Maurya, S.K.; Gupta, S.; Senapati, S.; Singh, S.P.; Waseem, M.; Kumar, S. Withaniasomnifera phytochemicals possess SARS-CoV-2 RdRp and human TMPRSS2 protein binding potential. Vegetos 2023, 36, 701–720. [Google Scholar] [CrossRef] [PubMed]
- David, A.B.; Diamant, E.; Dor, E.; Barnea, A.; Natan, N.; Levin, L.; Chapman, S.; Mimran, L.C.; Epstein, E.; Zichel, R.; et al. Identification of SARS-CoV-2 Receptor Binding Inhibitors by In Vitro Screening of Drug Libraries. Molecules 2021, 26, 3213. [Google Scholar] [CrossRef] [PubMed]
- Hammond, J.; Leister-Tebbe, H.; Gardner, A.; Abreu, P.; Bao, W.; Wisemandle, W.; Baniecki, M.; Hendrick, V.M.; Damle, B.; Simón-Campos, A.; et al. Oral Nirmatrelvir for High-Risk, Nonhospitalized Adults with Covid-19. N. Engl. J. Med. 2022, 386, 1397–1408. [Google Scholar] [CrossRef]
- Liu, J.; Pan, X.; Zhang, S.; Li, M.; Ma, K.; Fan, C.; Lv, Y.; Guan, X.; Yang, Y.; Ye, X.; et al. Efficacy and safety of Paxlovid in severe adult patients with SARS-Cov-2 infection: A multicenter randomized controlled study. Lancet Reg. Health. West. Pac. 2023, 33, 100694. [Google Scholar] [CrossRef]
- Unoh, Y.; Uehara, S.; Nakahara, K.; Nobori, H.; Yamatsu, Y.; Yamamoto, S.; Maruyama, Y.; Taoda, Y.; Kasamatsu, K.; Suto, T.; et al. Discovery of S-217622, a Noncovalent Oral SARS-CoV-2 3CL Protease Inhibitor Clinical Candidate for Treating COVID-19. J. Med. Chem. 2022, 65, 6499–6512. [Google Scholar] [CrossRef]
- Mukae, H.; Yotsuyanagi, H.; Ohmagari, N.; Doi, Y.; Sakaguchi, H.; Sonoyama, T.; Ichihashi, G.; Sanaki, T.; Baba, K.; Tsuge, Y.; et al. Efficacy and Safety of Ensitrelvir in Patients with Mild-to-Moderate Coronavirus Disease 2019: The Phase 2b Part of a Randomized, Placebo-Controlled, Phase 2/3 Study. Clin. Infect. Dis. 2023, 76, 1403–1411. [Google Scholar] [CrossRef]
- Balakrishnan, A.; Reyes, A.; Shen, R.; Bisht, N.; Sweeney, J.; Levene, R.; McAllister, N.; Cressey, T.; Manalo, N.; Rhodin, M.H.; et al. Molecular Basis for Antiviral Action of EDP-235: A Potent and Selective SARS-CoV-2 3CLpro Inhibitor for the Treatment of Covid 19. FASEB J. 2022, 36. [Google Scholar] [CrossRef]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 2020, 587, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Gao, X.; Tang, K.; Cai, J.P.; Hu, M.; Luo, P.; Wen, L.; Ye, Z.W.; Luo, C.; Tsang, J.O.; et al. Targeting papain-like protease for broad-spectrum coronavirus inhibition. Protein Cell 2022, 13, 940–953. [Google Scholar] [CrossRef]
- Napolitano, V.; Dabrowska, A.; Schorpp, K.; Mourão, A.; Barreto-Duran, E.; Benedyk, M.; Botwina, P.; Brandner, S.; Bostock, M.; Chykunova, Y.; et al. Acriflavine, a clinically approved drug, inhibits SARS-CoV-2 and other betacoronaviruses. Cell Chem. Biol. 2022, 29, 774–784.e778. [Google Scholar] [CrossRef]
- Song, S.; Kwon, O.S.; Chung, Y.B. Pharmacokinetics and metabolism of acriflavine in rats following intravenous or intramuscular administration of AG60, a mixture of acriflavine and guanosine, a potential antitumour agent. Xenobiotica 2005, 35, 755–773. [Google Scholar] [CrossRef] [PubMed]
- Ratia, K.; Pegan, S.; Takayama, J.; Sleeman, K.; Coughlin, M.; Baliji, S.; Chaudhuri, R.; Fu, W.; Prabhakar, B.S.; Johnson, M.E.; et al. A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc. Natl. Acad. Sci. USA 2008, 105, 16119–16124. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Huang, B.; Tang, J.; Liu, S.; Liu, M.; Ye, Y.; Liu, Z.; Xiong, Y.; Zhu, W.; Cao, D.; et al. The complex structure of GRL0617 and SARS-CoV-2 PLpro reveals a hot spot for antiviral drug discovery. Nat. Commun. 2021, 12, 488. [Google Scholar] [CrossRef] [PubMed]
- Savarino, A.; Di Trani, L.; Donatelli, I.; Cauda, R.; Cassone, A. New insights into the antiviral effects of chloroquine. Lancet Infect. Dis. 2006, 6, 67–69. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J.; Jain, S.; Meena, J.; Yadav, A. Efficacy and safety of hydroxychloroquine/chloroquine against SARS-CoV-2 infection: A systematic review and meta-analysis. J. Infect. Chemother. 2021, 27, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Llanos-Cuentas, A.; Schwalb, A.; Quintana, J.L.; Delfin, B.; Alvarez, F.; Ugarte-Gil, C.; Guerra Gronerth, R.I.; Lucchetti, A.; Grogl, M.; Gotuzzo, E. Hydroxychloroquine to prevent SARS-CoV-2 infection among healthcare workers: Early termination of a phase 3, randomised, open-label, controlled clinical trial. BMC Res. Notes 2023, 16, 22. [Google Scholar] [CrossRef]
- Brevini, T.; Maes, M.; Webb, G.J.; John, B.V.; Fuchs, C.D.; Buescher, G.; Wang, L.; Griffiths, C.; Brown, M.L.; Scott, W.E.; et al. FXR inhibition may protect from SARS-CoV-2 infection by reducing ACE2. Nature 2023, 615, 134–142. [Google Scholar] [CrossRef]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkrüys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Hurtado Del Pozo, C.; Prosper, F.; et al. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell 2020, 181, 905–913.e907. [Google Scholar] [CrossRef]
- Karoyan, P.; Vieillard, V.; Gómez-Morales, L.; Odile, E.; Guihot, A.; Luyt, C.E.; Denis, A.; Grondin, P.; Lequin, O. Human ACE2 peptide-mimics block SARS-CoV-2 pulmonary cells infection. Commun. Biol. 2021, 4, 197. [Google Scholar] [CrossRef]
- Shin, Y.-H.; Jeong, K.; Lee, J.; Lee, H.J.; Yim, J.; Kim, J.; Kim, S.; Park, S.B. Inhibition of ACE2-Spike Interaction by an ACE2 Binder Suppresses SARS-CoV-2 Entry. Angew. Chem. Int. Ed. 2022, 61, e202115695. [Google Scholar] [CrossRef] [PubMed]
- Essalmani, R.; Jain, J.; Susan-Resiga, D.; Andréo, U.; Evagelidis, A.; Derbali, R.M.; Huynh, D.N.; Dallaire, F.; Laporte, M.; Delpal, A.; et al. Distinctive Roles of Furin and TMPRSS2 in SARS-CoV-2 Infectivity. J. Virol. 2022, 96, e0012822. [Google Scholar] [CrossRef]
- Bestle, D.; Heindl, M.R.; Limburg, H.; Van Lam van, T.; Pilgram, O.; Moulton, H.; Stein, D.A.; Hardes, K.; Eickmann, M.; Dolnik, O.; et al. TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life Sci. Alliance 2020, 3, e202000786. [Google Scholar] [CrossRef]
- Pászti-Gere, E.; Szentkirályi-Tóth, A.; Szabó, P.; Steinmetzer, T.; Fliszár-Nyúl, E.; Poór, M. In vitro characterization of the furin inhibitor MI-1851: Albumin binding, interaction with cytochrome P450 enzymes and cytotoxicity. Biomed. Pharmacother. 2022, 151, 113124. [Google Scholar] [CrossRef]
- Li, K.; Meyerholz, D.K.; Bartlett, J.A.; McCray, P.B., Jr. The TMPRSS2 Inhibitor Nafamostat Reduces SARS-CoV-2 Pulmonary Infection in Mouse Models of COVID-19. mBio 2021, 12, e0097021. [Google Scholar] [CrossRef]
- Kinoshita, T.; Shinoda, M.; Nishizaki, Y.; Shiraki, K.; Hirai, Y.; Kichikawa, Y.; Tsushima, K.; Shinkai, M.; Komura, N.; Yoshida, K.; et al. A multicenter, double-blind, randomized, parallel-group, placebo-controlled study to evaluate the efficacy and safety of camostat mesilate in patients with COVID-19 (CANDLE study). BMC Med. 2022, 20, 342. [Google Scholar] [CrossRef]
- Gunst, J.D.; Staerke, N.B.; Pahus, M.H.; Kristensen, L.H.; Bodilsen, J.; Lohse, N.; Dalgaard, L.S.; Brønnum, D.; Fröbert, O.; Hønge, B.; et al. Efficacy of the TMPRSS2 inhibitor camostat mesilate in patients hospitalized with Covid-19-a double-blind randomized controlled trial. EClinicalMedicine 2021, 35, 100849. [Google Scholar] [CrossRef] [PubMed]
- Zhuravel, S.V.; Khmelnitskiy, O.K.; Burlaka, O.O.; Gritsan, A.I.; Goloshchekin, B.M.; Kim, S.; Hong, K.Y. Nafamostat in hospitalized patients with moderate to severe COVID-19 pneumonia: A randomised Phase II clinical trial. EClinicalMedicine 2021, 41, 101169. [Google Scholar] [CrossRef] [PubMed]
- Shapira, T.; Monreal, I.A.; Dion, S.P.; Buchholz, D.W.; Imbiakha, B.; Olmstead, A.D.; Jager, M.; Désilets, A.; Gao, G.; Martins, M.; et al. A TMPRSS2 inhibitor acts as a pan-SARS-CoV-2 prophylactic and therapeutic. Nature 2022, 605, 340–348. [Google Scholar] [CrossRef]
- Moumbock, A.F.A.; Tran, H.T.T.; Lamy, E.; Günther, S. BC-11 is a covalent TMPRSS2 fragment inhibitor that impedes SARS-CoV-2 host cell entry. Arch. Der Pharm. 2023, 356, 2200371. [Google Scholar] [CrossRef]
- Padmanabhan, P.; Dixit, N.M. Modelling how increased Cathepsin B/L and decreased TMPRSS2 usage for cell entry by the SARS-CoV-2 Omicron variant may affect the efficacy and synergy of TMPRSS2 and Cathepsin B/L inhibitors. J. Theor. Biol. 2023, 572, 111568. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.; Pan, T.; Zhang, J.; Li, Q.; Zhang, X.; Bai, C.; Huang, F.; Peng, T.; Zhang, J.; Liu, C.; et al. Glycopeptide Antibiotics Potently Inhibit Cathepsin L in the Late Endosome/Lysosome and Block the Entry of Ebola Virus, Middle East Respiratory Syndrome Coronavirus (MERS-CoV), and Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV). J. Biol. Chem. 2016, 291, 9218–9232. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Pan, T.; Huang, F.; Ying, R.; Liu, J.; Fan, H.; Zhang, J.; Liu, W.; Lin, Y.; Yuan, Y.; et al. Glycopeptide Antibiotic Teicoplanin Inhibits Cell Entry of SARS-CoV-2 by Suppressing the Proteolytic Activity of Cathepsin L. Front. Microbiol. 2022, 13, 884034. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liu, L.; Wang, C.; Wang, Z.; Duan, X.; Chen, G.; Zhou, H.; Shao, H. Establishment of a pseudovirus neutralization assay based on SARS-CoV-2 S protein incorporated into lentiviral particles. Biosaf. Health 2022, 4, 38–44. [Google Scholar] [CrossRef]
- Ma, X.R.; Alugubelli, Y.R.; Ma, Y.; Vatansever, E.C.; Scott, D.A.; Qiao, Y.; Yu, G.; Xu, S.; Liu, W.R. MPI8 is Potent against SARS-CoV-2 by Inhibiting Dually and Selectively the SARS-CoV-2 Main Protease and the Host Cathepsin L. ChemMedChem 2022, 17, e202100456. [Google Scholar] [CrossRef]
- Rodon, J.; Muñoz-Basagoiti, J.; Perez-Zsolt, D.; Noguera-Julian, M.; Paredes, R.; Mateu, L.; Quiñones, C.; Perez, C.; Erkizia, I.; Blanco, I.; et al. Identification of Plitidepsin as Potent Inhibitor of SARS-CoV-2-Induced Cytopathic Effect After a Drug Repurposing Screen. Front. Pharmacol. 2021, 12, 646676. [Google Scholar] [CrossRef]
- Costanzi, E.; Kuzikov, M.; Esposito, F.; Albani, S.; Demitri, N.; Giabbai, B.; Camasta, M.; Tramontano, E.; Rossetti, G.; Zaliani, A.; et al. Structural and Biochemical Analysis of the Dual Inhibition of MG-132 against SARS-CoV-2 Main Protease (Mpro/3CLpro) and Human Cathepsin-L. Int. J. Mol. Sci. 2021, 22, 11779. [Google Scholar] [CrossRef]
- Kumar, P.; Mathayan, M.; Smieszek, S.P.; Przychodzen, B.P.; Koprivica, V.; Birznieks, G.; Polymeropoulos, M.H.; Prabhakar, B.S. Identification of potential COVID-19 treatment compounds which inhibit SARS Cov2 prototypic, Delta and Omicron variant infection. Virology 2022, 572, 64–71. [Google Scholar] [CrossRef]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a008656. [Google Scholar] [CrossRef]
- Chu, H.; Hou, Y.; Yang, D.; Wen, L.; Shuai, H.; Yoon, C.; Shi, J.; Chai, Y.; Yuen, T.T.-T.; Hu, B.; et al. Coronaviruses exploit a host cysteine-aspartic protease for replication. Nature 2022, 609, 785–792. [Google Scholar] [CrossRef]
- Samelson, A.J.; Tran, Q.D.; Robinot, R.; Carrau, L.; Rezelj, V.V.; Kain, A.M.; Chen, M.; Ramadoss, G.N.; Guo, X.; Lim, S.A.; et al. BRD2 inhibition blocks SARS-CoV-2 infection by reducing transcription of the host cell receptor ACE2. Nat. Cell Biol. 2022, 24, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Braga, L.; Ali, H.; Secco, I.; Chiavacci, E.; Neves, G.; Goldhill, D.; Penn, R.; Jimenez-Guardeño, J.M.; Ortega-Prieto, A.M.; Bussani, R.; et al. Drugs that inhibit TMEM16 proteins block SARS-CoV-2 spike-induced syncytia. Nature 2021, 594, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Verma, A.; Garcia, G., Jr.; Ramage, H.; Lucas, A.; Myers, R.L.; Michaelson, J.J.; Coryell, W.; Kumar, A.; Charney, A.W.; et al. Targeting the coronavirus nucleocapsid protein through GSK-3 inhibition. Proc. Natl. Acad. Sci. USA 2021, 118, e2113401118. [Google Scholar] [CrossRef] [PubMed]
- Levin, M.J.; Ustianowski, A.; De Wit, S.; Launay, O.; Avila, M.; Templeton, A.; Yuan, Y.; Seegobin, S.; Ellery, A.; Levinson, D.J.; et al. Intramuscular AZD7442 (Tixagevimab-Cilgavimab) for Prevention of Covid-19. N. Engl. J. Med. 2022, 386, 2188–2200. [Google Scholar] [CrossRef]
- Loo, Y.M.; McTamney, P.M.; Arends, R.H.; Abram, M.E.; Aksyuk, A.A.; Diallo, S.; Flores, D.J.; Kelly, E.J.; Ren, K.; Roque, R.; et al. The SARS-CoV-2 monoclonal antibody combination, AZD7442, is protective in nonhuman primates and has an extended half-life in humans. Sci. Transl. Med. 2022, 14, eabl8124. [Google Scholar] [CrossRef]
- Bruel, T.; Hadjadj, J.; Maes, P.; Planas, D.; Seve, A.; Staropoli, I.; Guivel-Benhassine, F.; Porrot, F.; Bolland, W.H.; Nguyen, Y.; et al. Serum neutralization of SARS-CoV-2 Omicron sublineages BA.1 and BA.2 in patients receiving monoclonal antibodies. Nat. Med. 2022, 28, 1297–1302. [Google Scholar] [CrossRef]
- Case, J.B.; Mackin, S.; Errico, J.M.; Chong, Z.; Madden, E.A.; Whitener, B.; Guarino, B.; Schmid, M.A.; Rosenthal, K.; Ren, K.; et al. Resilience of S309 and AZD7442 monoclonal antibody treatments against infection by SARS-CoV-2 Omicron lineage strains. Nat. Commun. 2022, 13, 3824. [Google Scholar] [CrossRef]
- Biosciences, B. Brii Bio Announces Amubarvimab/Romlusevimab Combination Retains Neutralizing Activity against Omicron SARS-CoV-2 Variant. Available online: https://www.briibio.com/en/media/press-release/ (accessed on 12 December 2021).
- Wang, R.; Zhang, Q.; Zhang, R.; Aw, Z.Q.; Chen, P.; Wong, Y.H.; Hong, J.; Ju, B.; Shi, X.; Ding, Q.; et al. SARS-CoV-2 Omicron Variants Reduce Antibody Neutralization and Acquire Usage of Mouse ACE2. Front. Immunol. 2022, 13, 854952. [Google Scholar] [CrossRef]
- Efficacy and safety of two neutralising monoclonal antibody therapies, sotrovimab and BRII-196 plus BRII-198, for adults hospitalised with COVID-19 (TICO): A randomised controlled trial. Lancet Infect. Dis. 2022, 22, 622–635. [CrossRef]
- Westendorf, K.; Žentelis, S.; Wang, L.; Foster, D.; Vaillancourt, P.; Wiggin, M.; Lovett, E.; van der Lee, R.; Hendle, J.; Pustilnik, A.; et al. LY-CoV1404 (bebtelovimab) potently neutralizes SARS-CoV-2 variants. Cell Rep. 2022, 39, 110812. [Google Scholar] [CrossRef]
- Iketani, S.; Liu, L.; Guo, Y.; Liu, L.; Chan, J.F.W.; Huang, Y.; Wang, M.; Luo, Y.; Yu, J.; Chu, H.; et al. Antibody evasion properties of SARS-CoV-2 Omicron sublineages. Nature 2022, 604, 553–556. [Google Scholar] [CrossRef]
- Liu, L.; Wang, P.; Nair, M.S.; Yu, J.; Rapp, M.; Wang, Q.; Luo, Y.; Chan, J.F.; Sahi, V.; Figueroa, A.; et al. Potent neutralizing antibodies against multiple epitopes on SARS-CoV-2 spike. Nature 2020, 584, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Chi, X.; Yan, R.; Zhang, J.; Zhang, G.; Zhang, Y.; Hao, M.; Zhang, Z.; Fan, P.; Dong, Y.; Yang, Y.; et al. A neutralizing human antibody binds to the N-terminal domain of the Spike protein of SARS-CoV-2. Science 2020, 369, 650–655. [Google Scholar] [CrossRef]
- McCallum, M.; De Marco, A.; Lempp, F.A.; Tortorici, M.A.; Pinto, D.; Walls, A.C.; Beltramello, M.; Chen, A.; Liu, Z.; Zatta, F.; et al. N-terminal domain antigenic mapping reveals a site of vulnerability for SARS-CoV-2. Cell 2021, 184, 2332–2347.e2316. [Google Scholar] [CrossRef]
- Li, W.; Wang, F.; Li, Y.; Yan, L.; Liu, L.; Zhu, W.; Ma, P.; Shi, X.; Yang, G. Potent NTD-Targeting Neutralizing Antibodies against SARS-CoV-2 Selected from a Synthetic Immune System. Vaccines 2023, 11, 771. [Google Scholar] [CrossRef]
- Montgomerie, I.; Bird, T.W.; Palmer, O.R.; Mason, N.C.; Pankhurst, T.E.; Lawley, B.; Hernández, L.C.; Harfoot, R.; Authier-Hall, A.; Anderson, D.E.; et al. Incorporation of SARS-CoV-2 spike NTD to RBD protein vaccine improves immunity against viral variants. iScience 2023, 26, 106256. [Google Scholar] [CrossRef]
- Haslwanter, D.; Dieterle, M.E.; Wec, A.Z.; O’Brien, C.M.; Sakharkar, M.; Florez, C.; Tong, K.; Rappazzo, C.G.; Lasso, G.; Vergnolle, O.; et al. A Combination of Receptor-Binding Domain and N-Terminal Domain Neutralizing Antibodies Limits the Generation of SARS-CoV-2 Spike Neutralization-Escape Mutants. mBio 2021, 12, e0247321. [Google Scholar] [CrossRef]
- Cox, M.; Peacock, T.P.; Harvey, W.T.; Hughes, J.; Wright, D.W.; Willett, B.J.; Thomson, E.; Gupta, R.K.; Peacock, S.J.; Robertson, D.L.; et al. SARS-CoV-2 variant evasion of monoclonal antibodies based on in vitro studies. Nat. Rev. Microbiol. 2023, 21, 112–124. [Google Scholar] [CrossRef]
- Piccoli, L.; Park, Y.J.; Tortorici, M.A.; Czudnochowski, N.; Walls, A.C.; Beltramello, M.; Silacci-Fregni, C.; Pinto, D.; Rosen, L.E.; Bowen, J.E.; et al. Mapping Neutralizing and Immunodominant Sites on the SARS-CoV-2 Spike Receptor-Binding Domain by Structure-Guided High-Resolution Serology. Cell 2020, 183, 1024–1042.e1021. [Google Scholar] [CrossRef] [PubMed]
- Dacon, C.; Tucker, C.; Peng, L.; Lee, C.D.; Lin, T.H.; Yuan, M.; Cong, Y.; Wang, L.; Purser, L.; Williams, J.K.; et al. Broadly neutralizing antibodies target the coronavirus fusion peptide. Science 2022, 377, 728–735. [Google Scholar] [CrossRef]
- Xia, S.; Zhu, Y.; Liu, M.; Lan, Q.; Xu, W.; Wu, Y.; Ying, T.; Liu, S.; Shi, Z.; Jiang, S.; et al. Fusion mechanism of 2019-nCoV and fusion inhibitors targeting HR1 domain in spike protein. Cell. Mol. Immunol. 2020, 17, 765–767. [Google Scholar] [CrossRef]
- Xia, S.; Chan, J.F.; Wang, L.; Jiao, F.; Chik, K.K.; Chu, H.; Lan, Q.; Xu, W.; Wang, Q.; Wang, C.; et al. Peptide-based pan-CoV fusion inhibitors maintain high potency against SARS-CoV-2 Omicron variant. Cell Res. 2022, 32, 404–406. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, K.S.; Geers, D.; Vries, R.D.d.; Bovier, T.F.; Mykytyn, A.Z.; Kessel, C.H.G.v.; Haagmans, B.L.; Porotto, M.; Swart, R.L.d.; Moscona, A. Potency of Fusion-Inhibitory Lipopeptides against SARS-CoV-2 Variants of Concern. mBio 2022, 13, e01249-01222. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, J.; Ma, Q.; Su, Z.; Cheng, X. Advancements in the Development of Anti-SARS-CoV-2 Therapeutics. Int. J. Mol. Sci. 2024, 25, 10820. https://doi.org/10.3390/ijms251910820
Huang J, Ma Q, Su Z, Cheng X. Advancements in the Development of Anti-SARS-CoV-2 Therapeutics. International Journal of Molecular Sciences. 2024; 25(19):10820. https://doi.org/10.3390/ijms251910820
Chicago/Turabian StyleHuang, Junjie, Qianqian Ma, Zhengding Su, and Xiyao Cheng. 2024. "Advancements in the Development of Anti-SARS-CoV-2 Therapeutics" International Journal of Molecular Sciences 25, no. 19: 10820. https://doi.org/10.3390/ijms251910820
APA StyleHuang, J., Ma, Q., Su, Z., & Cheng, X. (2024). Advancements in the Development of Anti-SARS-CoV-2 Therapeutics. International Journal of Molecular Sciences, 25(19), 10820. https://doi.org/10.3390/ijms251910820