
Origin, Evolution and Diversity of φ29-like Phages—Review and Bioinformatic Analysis

 ,
,  ,
,
Abstract
:1. Introduction
2. Results
2.1. Brief Review of Published Studies on Phage φ29 Genomics and Structural Features
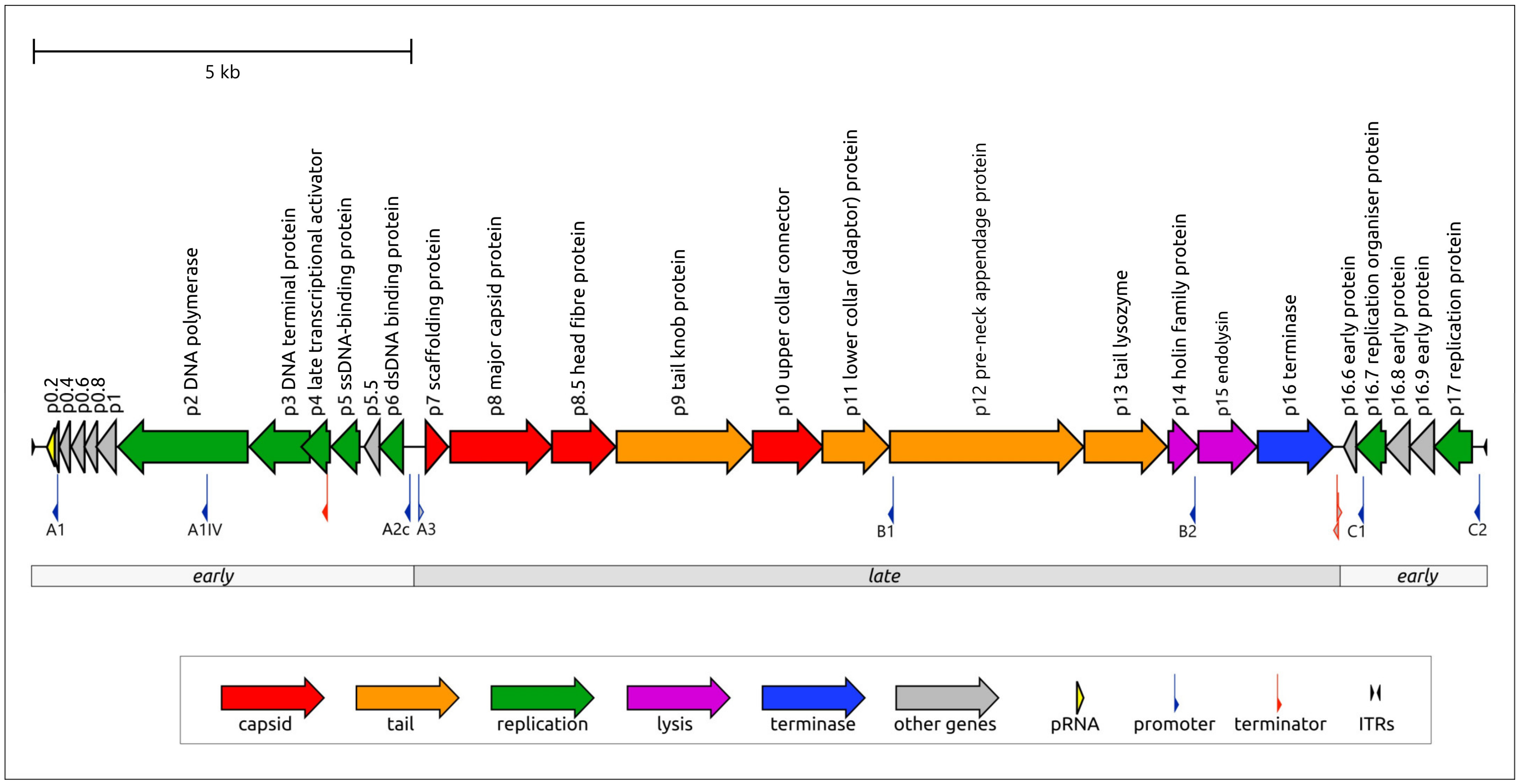
2.1.1. General Genomic Features of Phage φ29
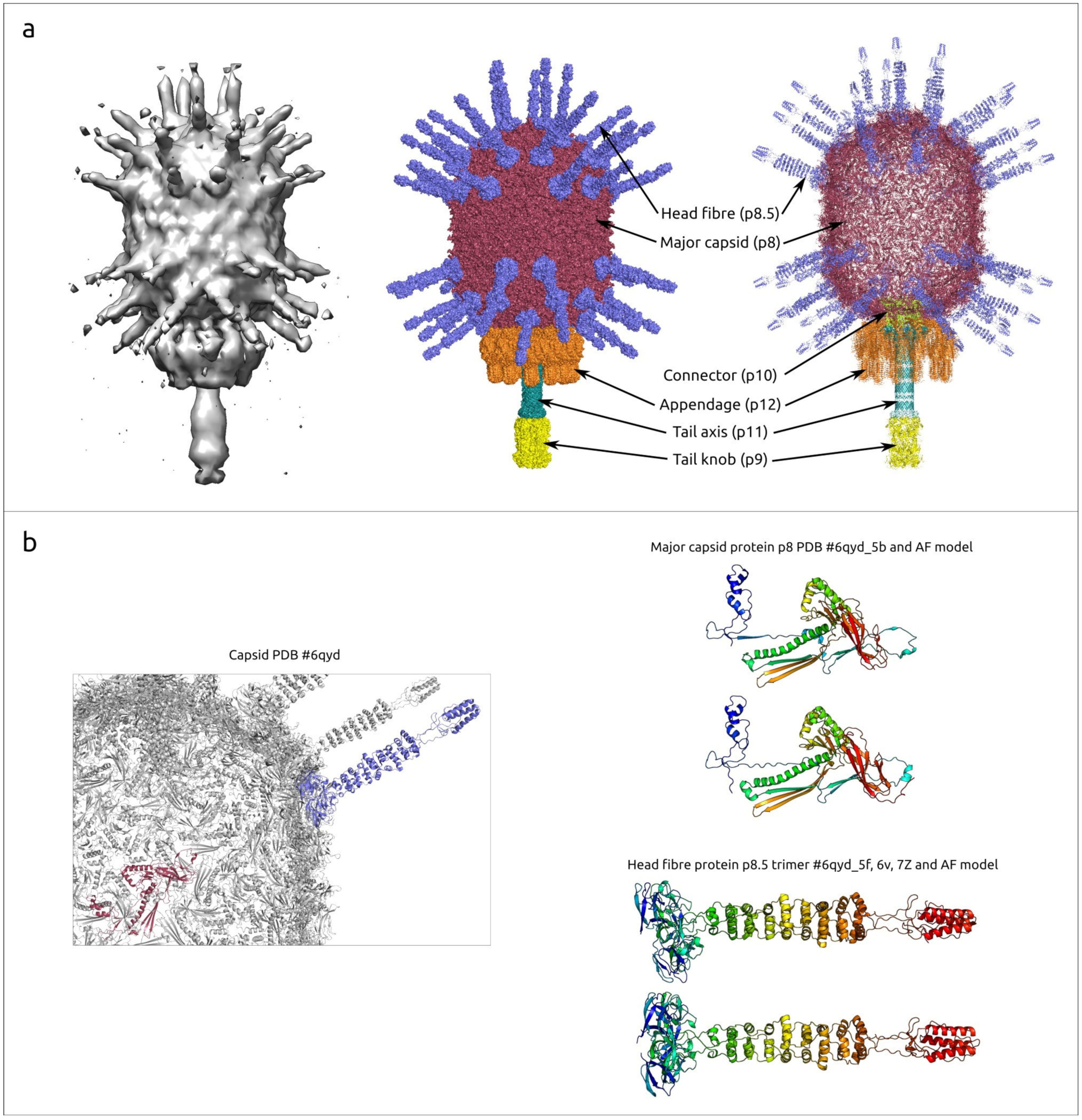
2.1.2. General Characterisation of the φ29 Virion and Proteins
2.2. Searches for Similar Proteins Using Sequences and Structures of φ29 Proteins
2.2.1. Major Capsid Protein (p8)
2.2.2. Head Fibre Protein (p8.5)
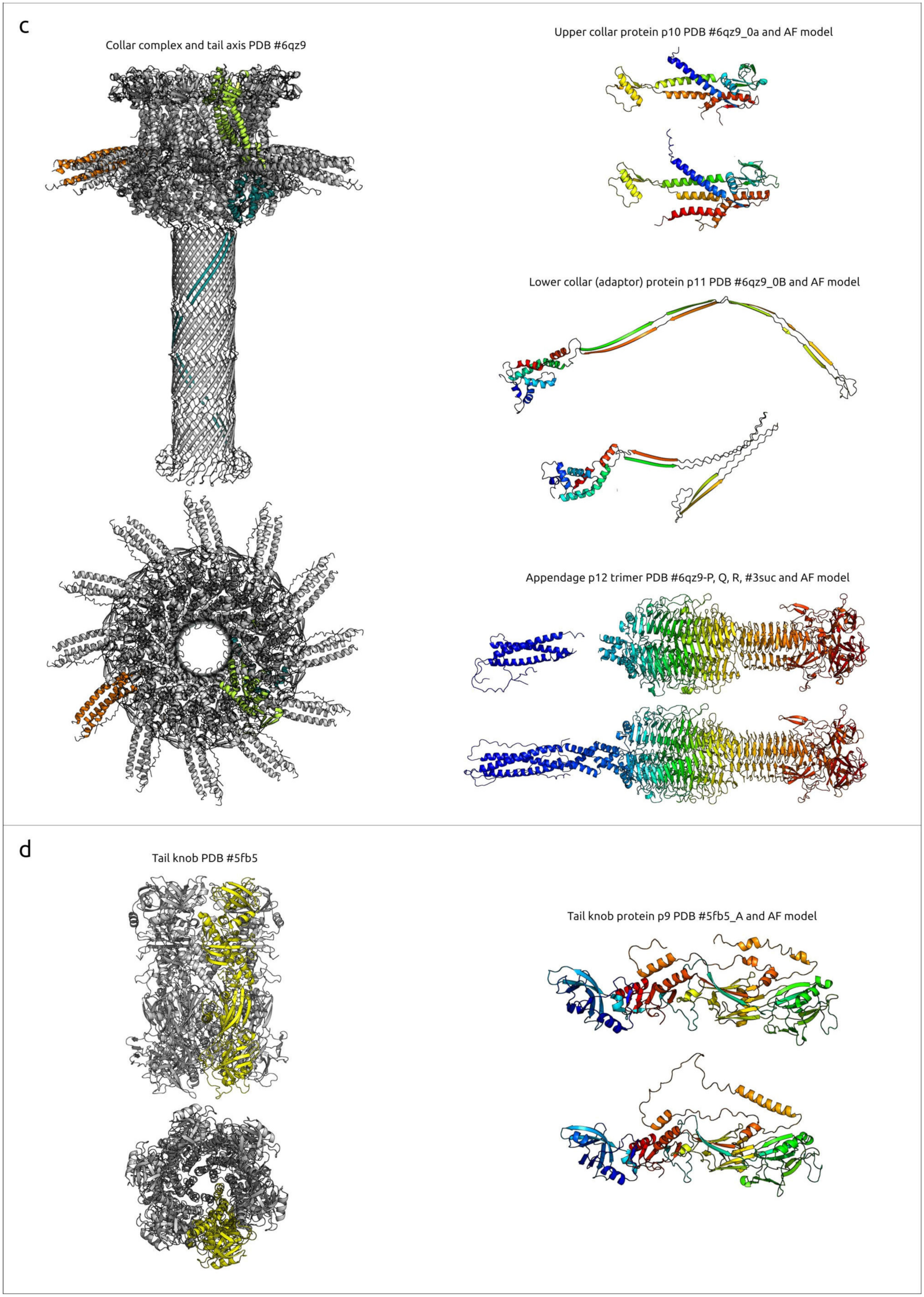
2.2.3. Tail Knob Protein (p9), Upper Collar Protein (p10) and Lower Collar Protein (p11)
2.2.4. Appendage Protein (p12)
2.2.5. DNA Polymerase (p2), Terminal Protein (p3) and “Histone-like Protein” (p6)
2.2.6. Terminase (p16)
2.2.7. Lysozyme (Endolysin) (p15)
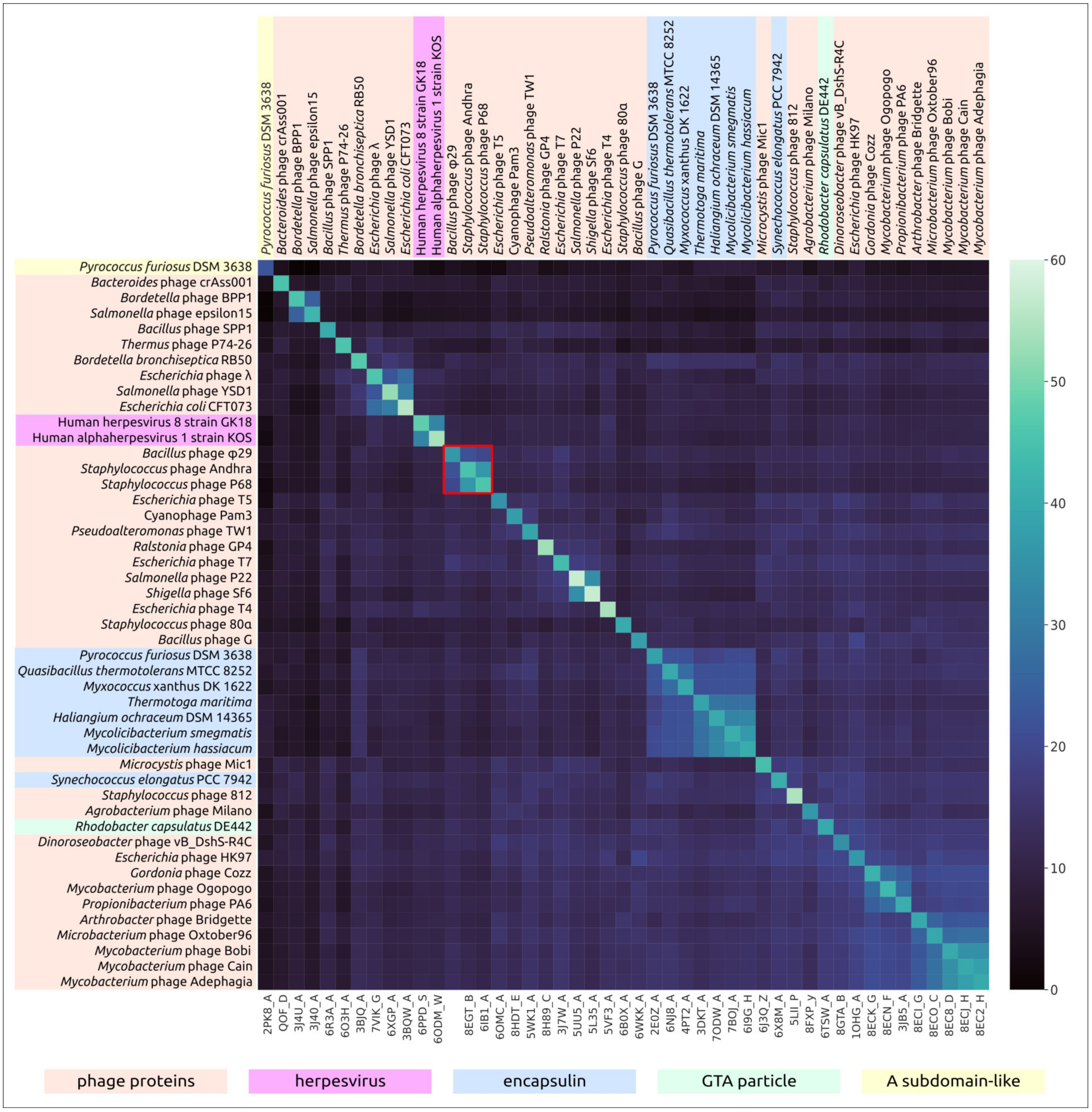
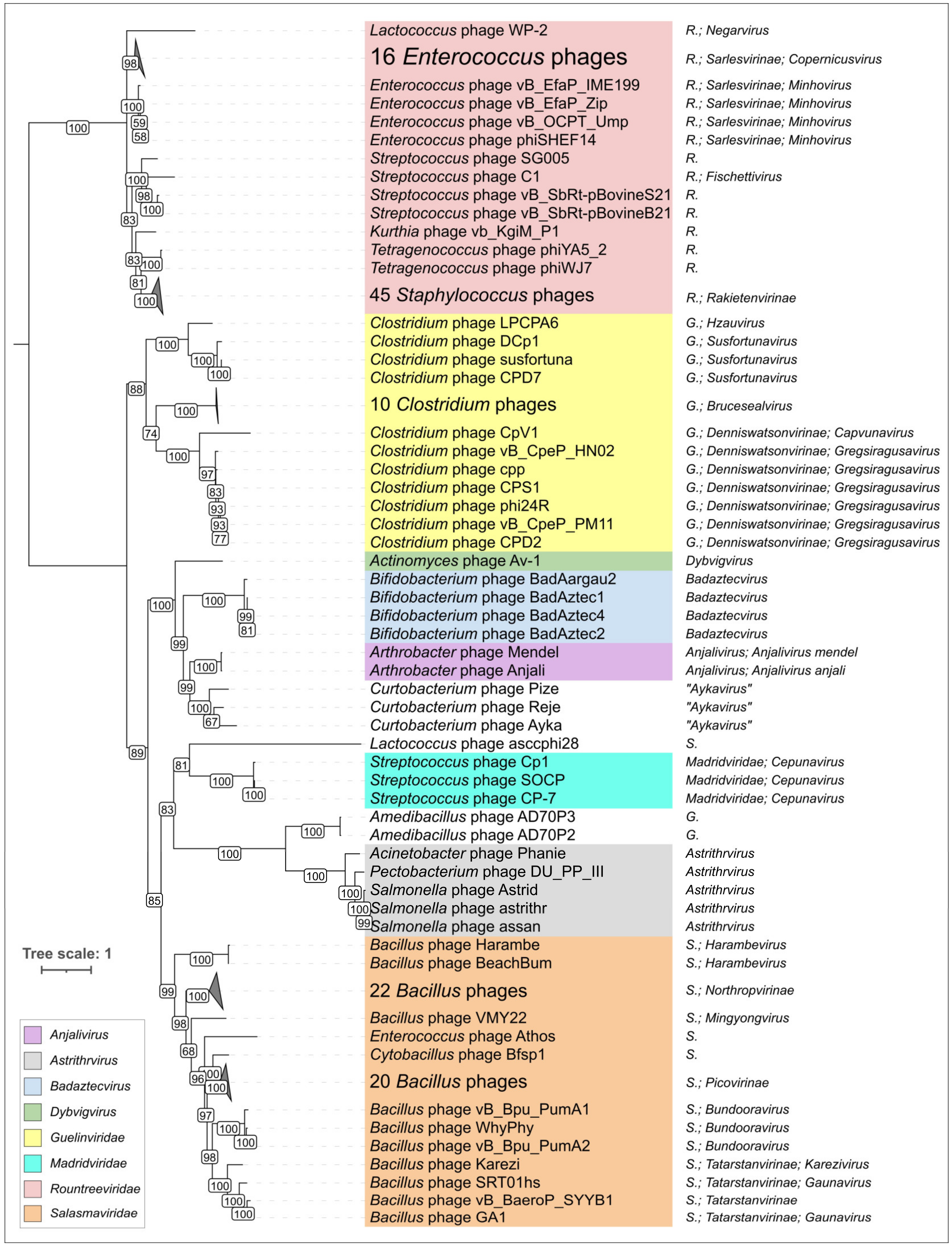
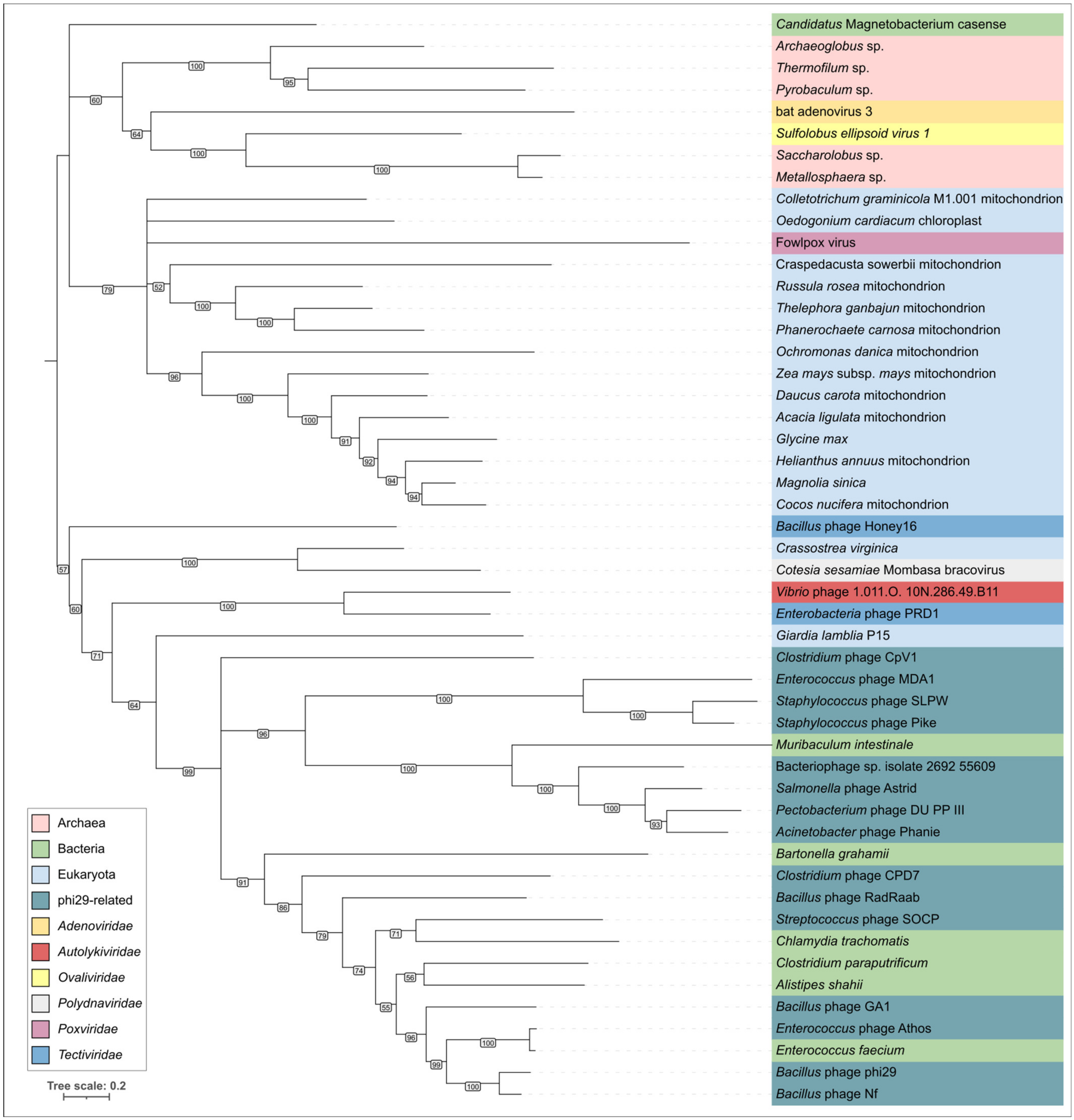
2.3. Search for φ29-Related Viruses
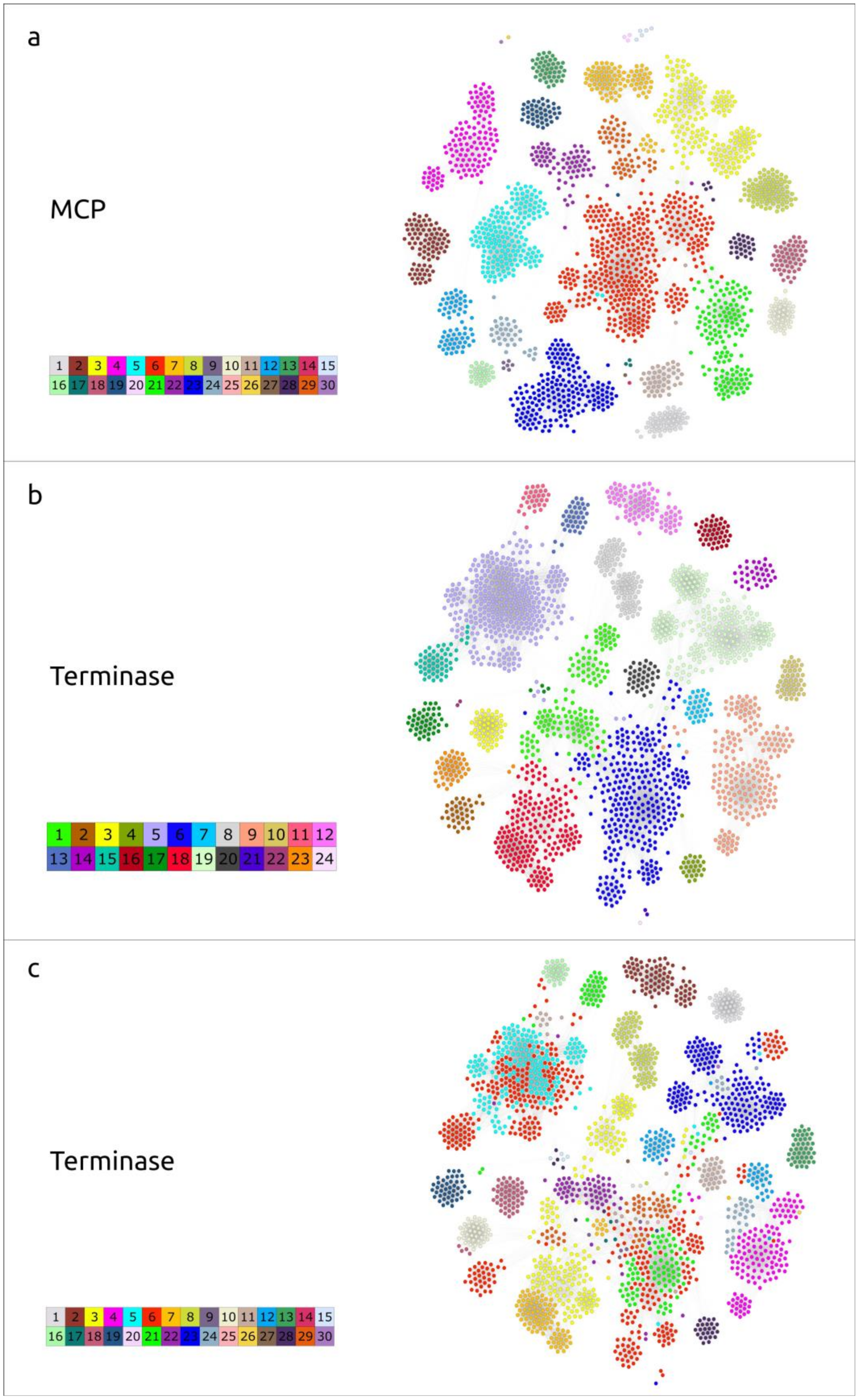
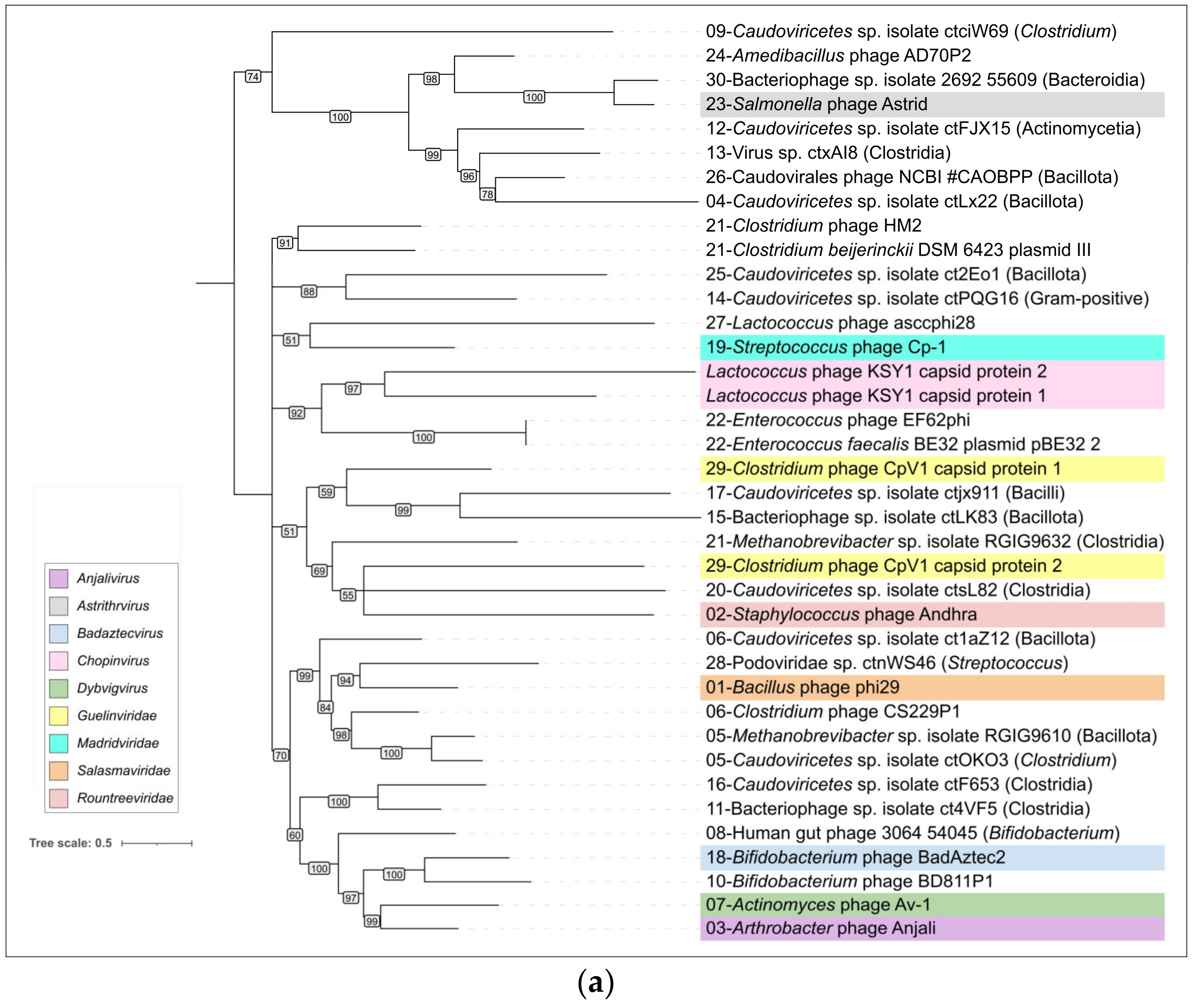
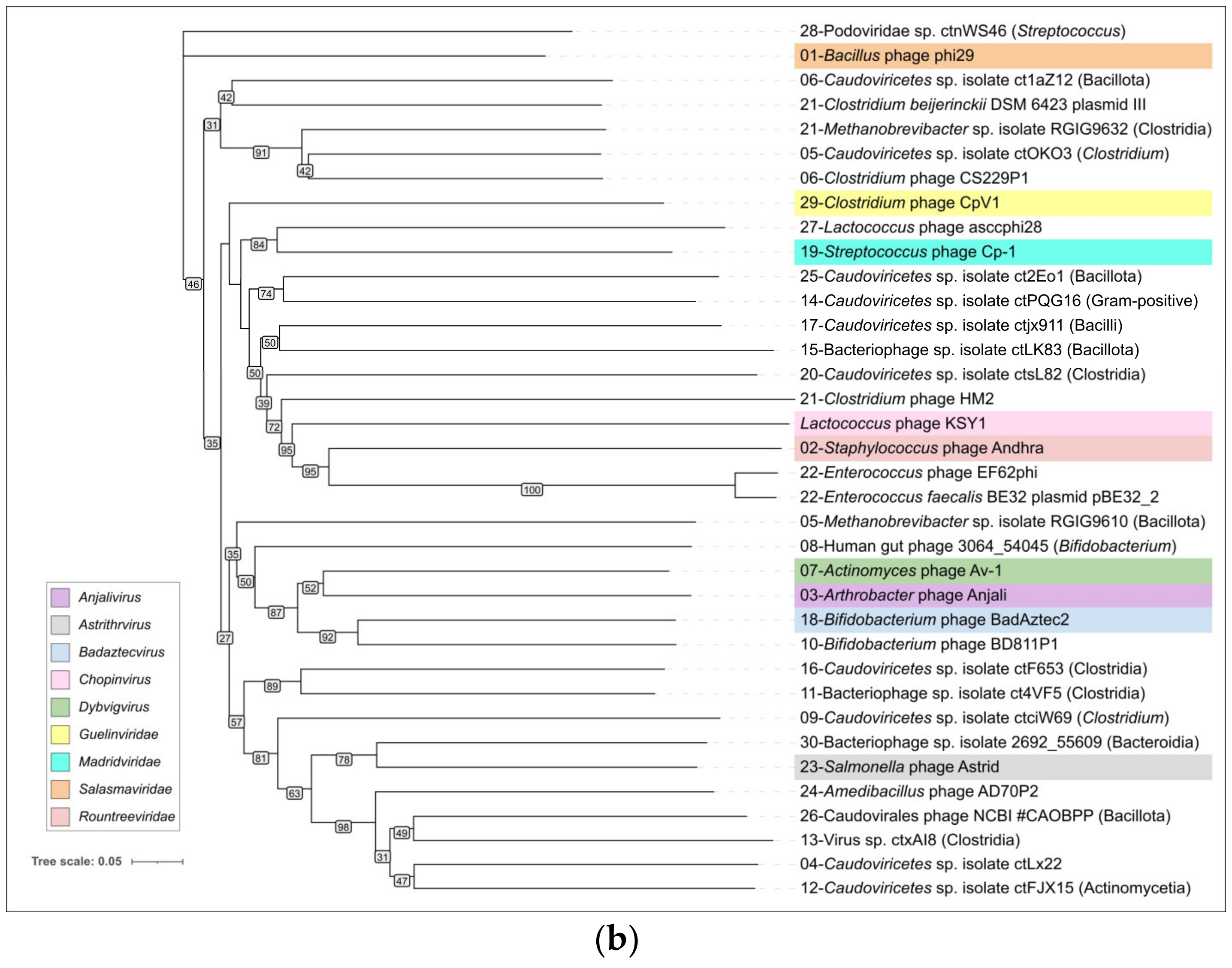
2.4. Clustering of φ29-Related Viruses
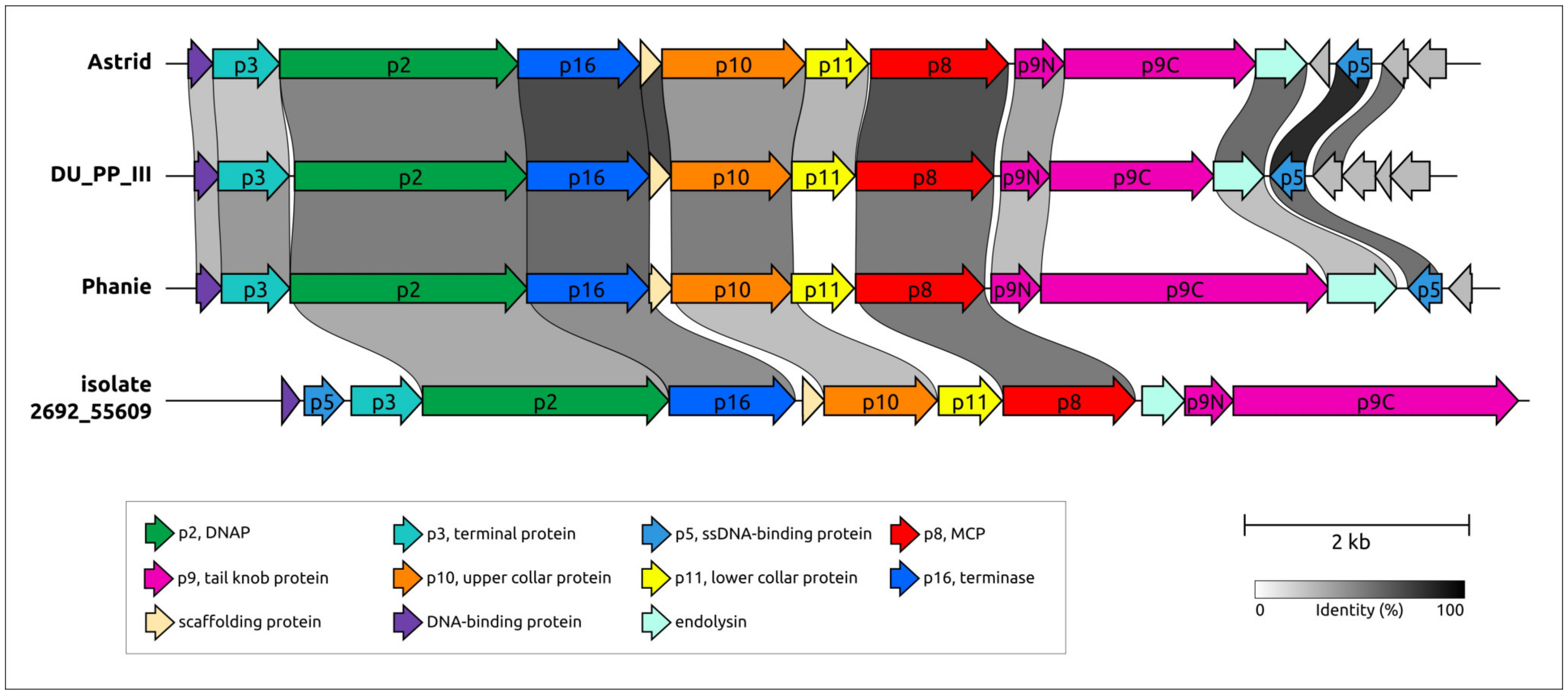
2.5. Genome Organisation of φ29-Related Viruses: Main Structural Proteins
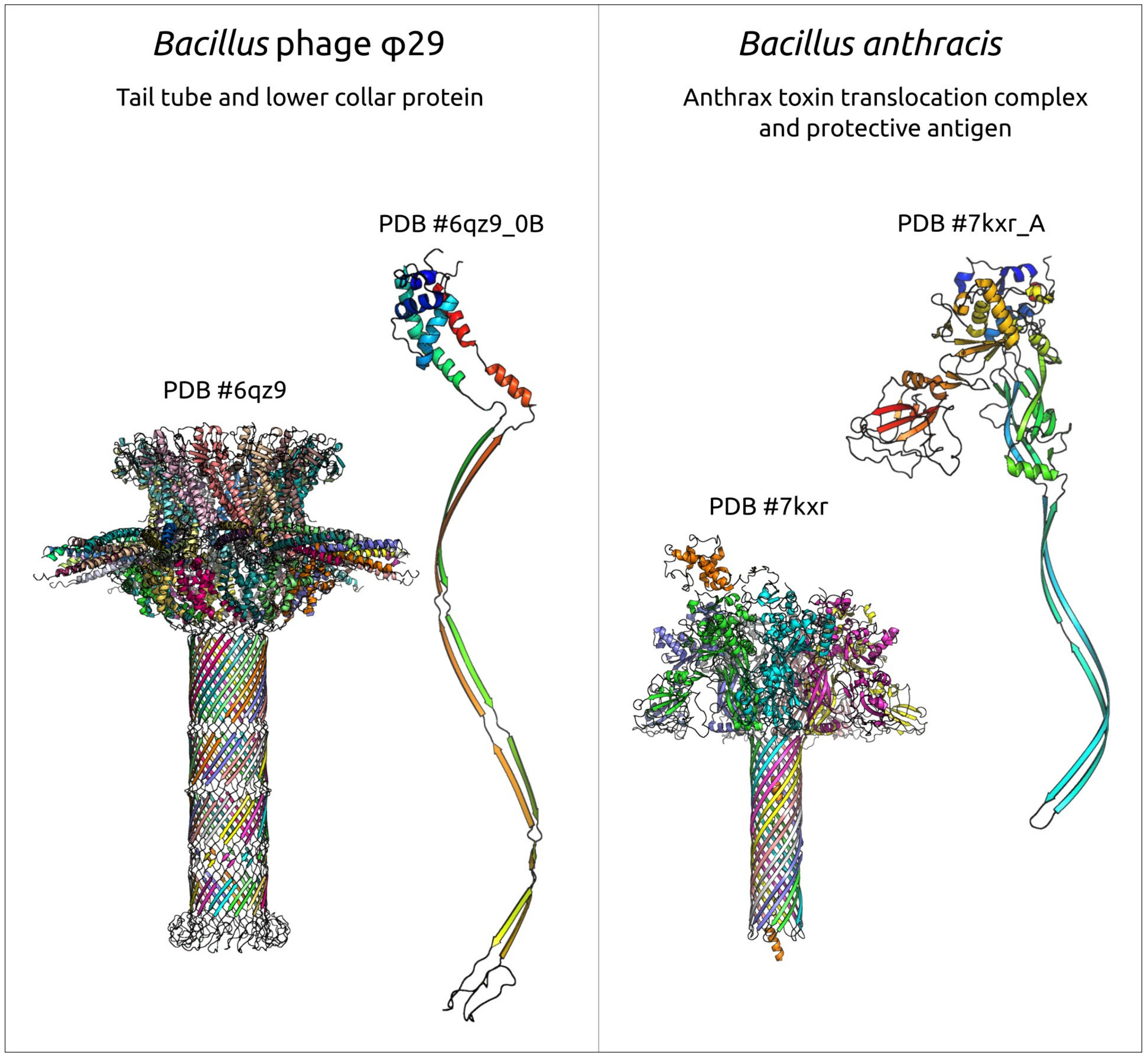
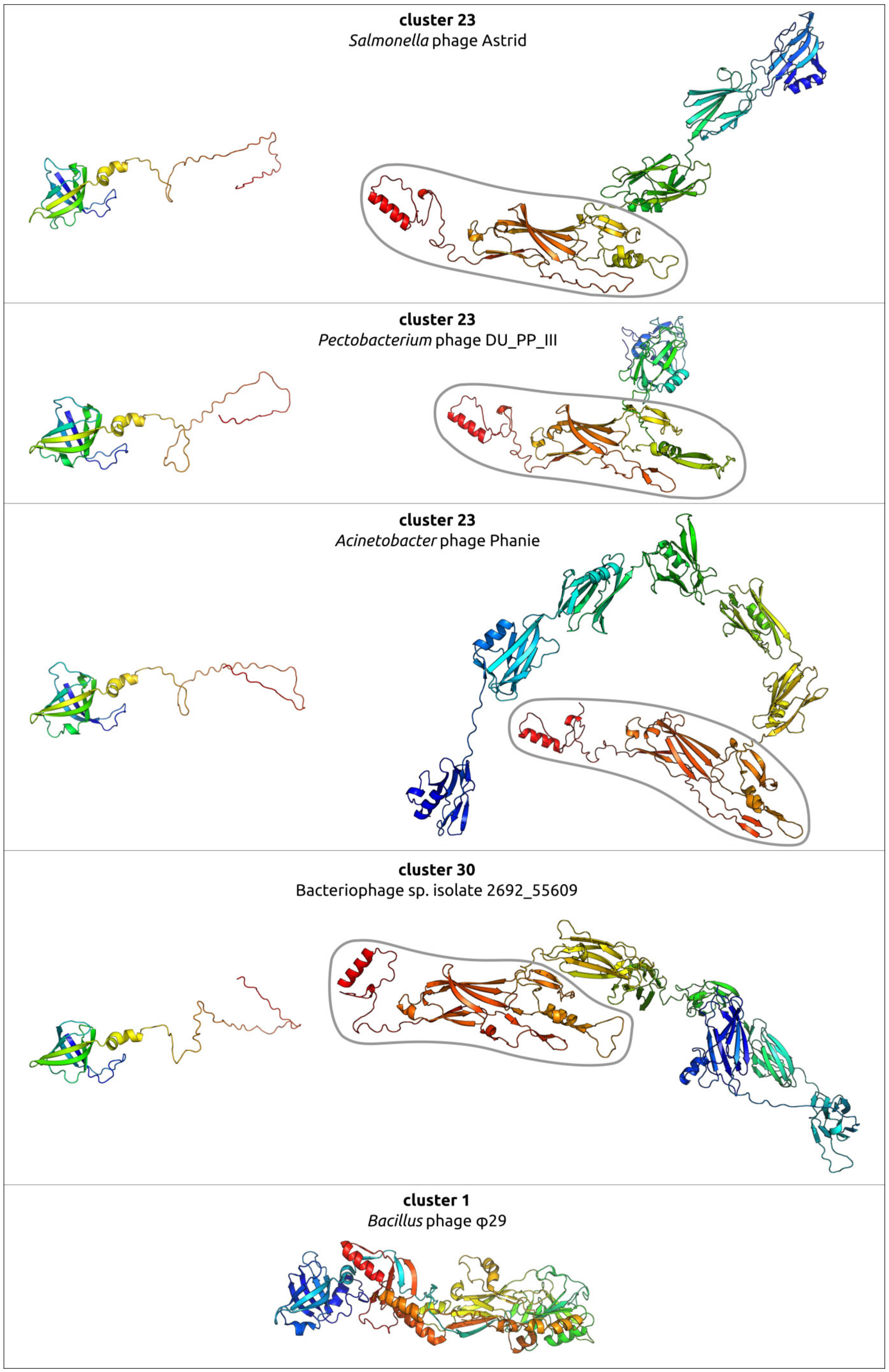
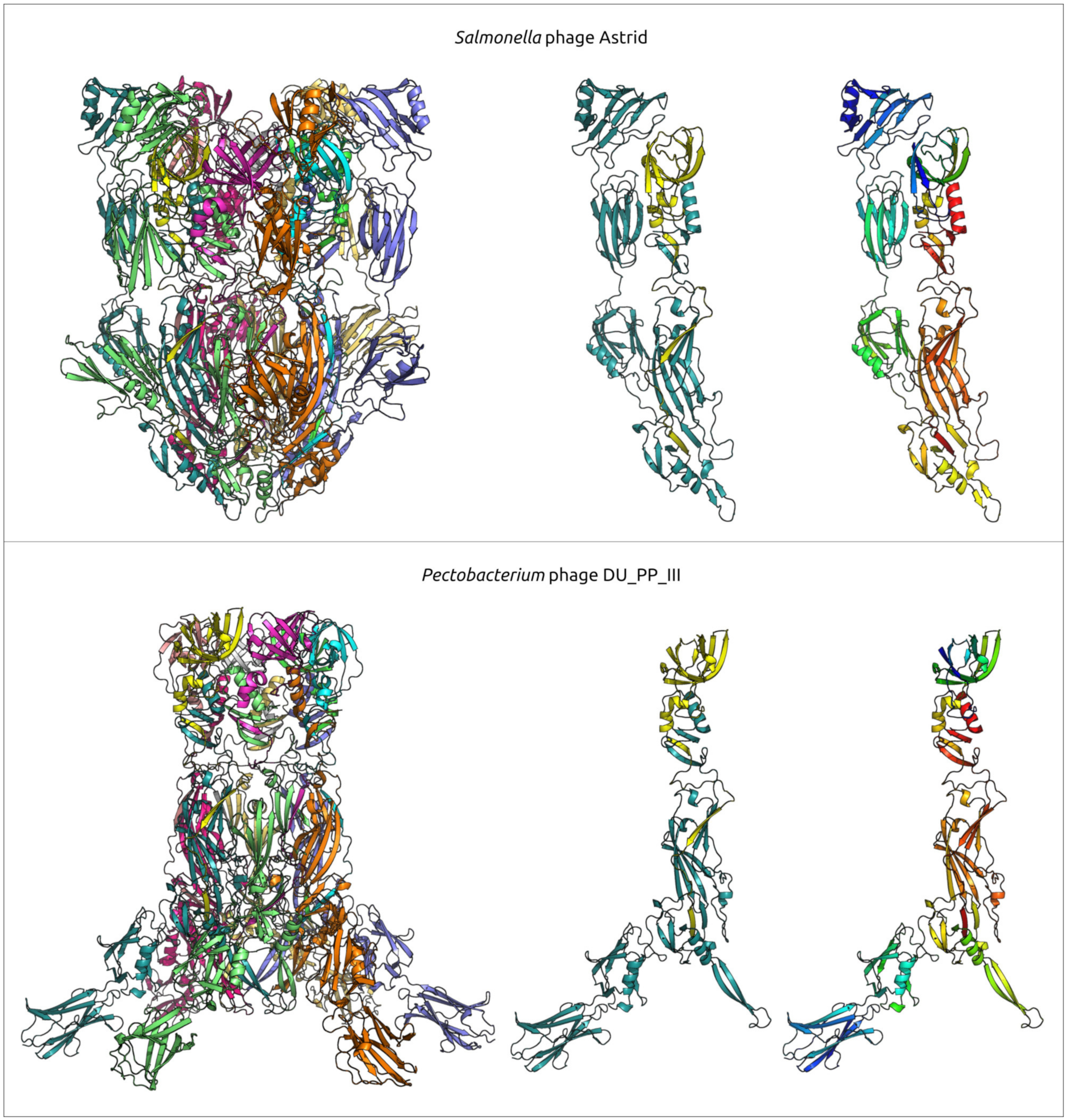
2.6. Variations in the Structural Architecture of Morphogenetic Proteins of φ29-Related Phages Infecting Gram-Negative Bacteria
2.7. Lactococcus Phage KSY1: General Genomic Features and Relatedness to φ29-like Phages
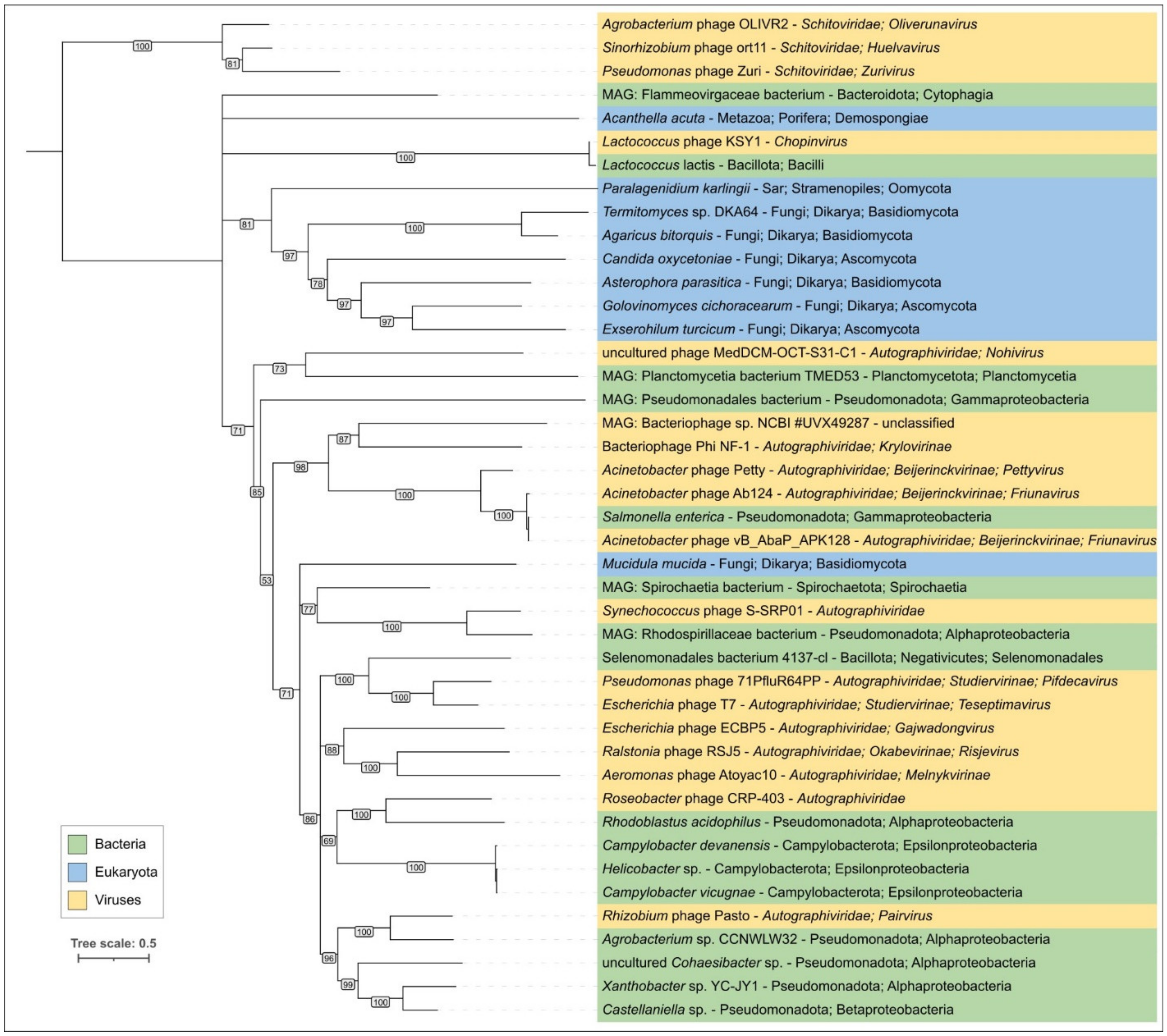
2.8. ViPtree Proteome Phylogeny and VIRIDIC Clustering
3. Discussion
4. Materials and Methods
4.1. Data Collection and Database Construction
4.2. Protein Structure Modelling
4.3. Functional Annotation
4.4. Phylogenetic Analysis, Structural Comparison and VIRIDIC Analysis
4.5. Similarity Clustering Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reilly, B.E. A Study of the Bacteriophages of Bacillus subtilis and Their Infectious Nucleic Acids; Case Western Reserve University: Cleveland, OH, USA, 1965. [Google Scholar]
- Anderson, D.L.; Hickman, D.D.; Reilly, B.E. Structure of Bacillus subtilis Bacteriophage Φ29 and the Length of Φ29 Deoxyribonucleic Acid. J. Bacteriol. 1966, 91, 2081–2089. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.L.; Reilly, B.E. Analysis of Bacteriophage Φ29 Gene Function: Protein Synthesis in Suppressor-Sensitive Mutant Infection of Bacillus subtilis. J. Virol. 1974, 13, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Reilly, B.E.; Nelson, R.A.; Anderson, D.L. Morphogenesis of Bacteriophage Φ29 of Bacillus subtilis: Mapping and Functional Analysis of the Head Fiber Gene. J. Virol. 1977, 24, 363–377. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Dolja, V.V.; Krupovic, M.; Varsani, A.; Wolf, Y.I.; Yutin, N.; Zerbini, F.M.; Kuhn, J.H. Global Organization and Proposed Megataxonomy of the Virus World. Microbiol. Mol. Biol. Rev. MMBR 2020, 84, e00061-19. [Google Scholar] [CrossRef]
- Turner, D.; Kropinski, A.M.; Adriaenssens, E.M. A Roadmap for Genome-Based Phage Taxonomy. Viruses 2021, 13, 506. [Google Scholar] [CrossRef]
- Volozhantsev, N.V.; Oakley, B.B.; Morales, C.A.; Verevkin, V.V.; Bannov, V.A.; Krasilnikova, V.M.; Popova, A.V.; Zhilenkov, E.L.; Garrish, J.K.; Schegg, K.M.; et al. Molecular Characterization of Podoviral Bacteriophages Virulent for Clostridium Perfringens and Their Comparison with Members of the Picovirinae. PLoS ONE 2012, 7, e38283. [Google Scholar] [CrossRef]
- Martín, A.C.; López, R.; García, P. Pneumococcal Bacteriophage Cp-1 Encodes Its Own Protease Essential for Phage Maturation. J. Virol. 1998, 72, 3491–3494. [Google Scholar] [CrossRef]
- Hawkins, N.C.; Kizziah, J.L.; Hatoum-Aslan, A.; Dokland, T. Structure and Host Specificity of Staphylococcus Epidermidis Bacteriophage Andhra. Sci. Adv. 2022, 8, eade0459. [Google Scholar] [CrossRef] [PubMed]
- Tarakanov, R.I.; Lukianova, A.A.; Evseev, P.V.; Pilik, R.I.; Tokmakova, A.D.; Kulikov, E.E.; Toshchakov, S.V.; Ignatov, A.N.; Dzhalilov, F.S.-U.; Miroshnikov, K.A. Ayka, a Novel Curtobacterium Bacteriophage, Provides Protection against Soybean Bacterial Wilt and Tan Spot. Int. J. Mol. Sci. 2022, 23, 10913. [Google Scholar] [CrossRef] [PubMed]
- Alanin, K.W.S.; Olsen, N.S.; Djurhuus, A.M.; Carstens, A.B.; Nielsen, T.K.; Rothgardt, M.M.; Russel, A.M.; Wagner, N.; Lametsch, R.; Bak, F.; et al. Four Novel Curtobacterium Phages Isolated from Environmental Samples. Arch. Virol. 2023, 168, 89. [Google Scholar] [CrossRef]
- Current ICTV Taxonomy Release|ICTV. Available online: https://ictv.global/taxonomy (accessed on 9 November 2022).
- Pourcel, C.; Essoh, C.; Ouldali, M.; Tavares, P. Acinetobacter Baumannii Satellite Phage Aci01-2-Phanie Depends on a Helper Myophage for Its Multiplication. J. Virol. 2024, 98, e00667-24. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Dolja, V.V. Virus World as an Evolutionary Network of Viruses and Capsidless Selfish Elements. Microbiol. Mol. Biol. Rev. MMBR 2014, 78, 278–303. [Google Scholar] [CrossRef] [PubMed]
- Botstein, D. A Theory of Modular Evolution for Bacteriophages. Ann. N. Y. Acad. Sci. 1980, 354, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, R.W.; Smith, M.C.M.; Burns, R.N.; Ford, M.E.; Hatfull, G.F. Evolutionary Relationships among Diverse Bacteriophages and Prophages: All the World’s a Phage. Proc. Natl. Acad. Sci. USA 1999, 96, 2192–2197. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Koonin, E.V. Multiple Origins of Viral Capsid Proteins from Cellular Ancestors. Proc. Natl. Acad. Sci. USA 2017, 114, E2401–E2410. [Google Scholar] [CrossRef]
- Evseev, P.; Gutnik, D.; Shneider, M.; Miroshnikov, K. Use of an Integrated Approach Involving AlphaFold Predictions for the Evolutionary Taxonomy of Duplodnaviria Viruses. Biomolecules 2023, 13, 110. [Google Scholar] [CrossRef]
- Aksyuk, A.A.; Bowman, V.D.; Kaufmann, B.; Fields, C.; Klose, T.; Holdaway, H.A.; Fischetti, V.A.; Rossmann, M.G. Structural Investigations of a Podoviridae Streptococcus Phage C1, Implications for the Mechanism of Viral Entry. Proc. Natl. Acad. Sci. USA 2012, 109, 14001–14006. [Google Scholar] [CrossRef]
- Yoshikawa, H.; Ito, J. Terminal Proteins and Short Inverted Terminal Repeats of the Small Bacillus Bacteriophage Genomes. Proc. Natl. Acad. Sci. USA 1981, 78, 2596–2600. [Google Scholar] [CrossRef]
- Ito, J. Bacteriophage Phi29 Terminal Protein: Its Association with the 5’ Termini of the Phi29 Genome. J. Virol. 1978, 28, 895–904. [Google Scholar] [CrossRef]
- Guo, P.X.; Erickson, S.; Anderson, D. A Small Viral RNA Is Required for in Vitro Packaging of Bacteriophage Phi 29 DNA. Science 1987, 236, 690–694. [Google Scholar] [CrossRef]
- Hill, A.C.; Bartley, L.E.; Schroeder, S.J. Prohead RNA: A Noncoding Viral RNA of Novel Structure and Function. Wiley Interdiscip. Rev. RNA 2016, 7, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Escarmís, C.; Salas, M. Nucleotide Sequence of the Early Genes 3 and 4 of Bacteriophage Phi 29. Nucleic Acids Res. 1982, 10, 5785–5798. [Google Scholar] [CrossRef] [PubMed]
- Kohm, K.; Basu, S.; Nawaz, M.M.; Hertel, R. Chances and Limitations When Uncovering Essential and Non-Essential Genes of Bacillus subtilis Phages with CRISPR-Cas9. Environ. Microbiol. Rep. 2021, 13, 934–944. [Google Scholar] [CrossRef] [PubMed]
- Meijer, W.J.J.; Horcajadas, J.A.; Salas, M. Φ29 Family of Phages. Microbiol. Mol. Biol. Rev. 2001, 65, 261–287. [Google Scholar] [CrossRef]
- Monsalve, M.; Mencía, M.; Rojo, F.; Salas, M. Transcription Regulation in Bacillus subtilis Phage Phi 29: Expression of the Viral Promoters throughout the Infection Cycle. Virology 1995, 207, 23–31. [Google Scholar] [CrossRef]
- Barthelemy, I.; Salas, M.; Mellado, R.P. In Vivo Transcription of Bacteriophage Phi 29 DNA: Transcription Termination. J. Virol. 1987, 61, 1751–1755. [Google Scholar] [CrossRef]
- Badia, D.; Camacho, A.; Pérez-Lago, L.; Escandón, C.; Salas, M.; Coll, M. The Structure of Phage Φ29 Transcription Regulator P4-DNA Complex Reveals an N-Hook Motif for DNA Binding. Mol. Cell 2006, 22, 73–81. [Google Scholar] [CrossRef]
- Monsalve, M.; Mencia, M.; Rojo, F.; Salas, M. Activation and Repression of Transcription at Two Different Phage Phi29 Promoters Are Mediated by Interaction of the Same Residues of Regulatory Protein P4 with RNA Polymerase. EMBO J. 1996, 15, 383–391. [Google Scholar] [CrossRef]
- Camacho, A.; Salas, M. Molecular Interplay between RNA Polymerase and Two Transcriptional Regulators in Promoter Switch. J. Mol. Biol. 2004, 336, 357–368. [Google Scholar] [CrossRef]
- Bravo, A.; Illana, B.; Salas, M. Compartmentalization of Phage Φ29 DNA Replication: Interaction between the Primer Terminal Protein and the Membrane-Associated Protein P1. EMBO J. 2000, 19, 5575–5584. [Google Scholar] [CrossRef]
- de Jong, R.N.; van der Vliet, P.C.; Brenkman, A.B. Adenovirus DNA Replication: Protein Priming, Jumping Back and the Role of the DNA Binding Protein DBP. Curr. Top. Microbiol. Immunol. 2003, 272, 187–211. [Google Scholar] [CrossRef] [PubMed]
- Ravantti, J.J.; Gaidelyte, A.; Bamford, D.H.; Bamford, J.K.H. Comparative Analysis of Bacterial Viruses Bam35, Infecting a Gram-Positive Host, and PRD1, Infecting Gram-Negative Hosts, Demonstrates a Viral Lineage. Virology 2003, 313, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Guo, Z.; Feng, H.; Chen, Y.; Chen, X.; Li, Z.; Hernández-Ascencio, W.; Dai, X.; Zhang, Z.; Zheng, X.; et al. Novel Sulfolobus Virus with an Exceptional Capsid Architecture. J. Virol. 2018, 92, e01727-17. [Google Scholar] [CrossRef] [PubMed]
- Bao, K.; Cohen, S.N. Terminal Proteins Essential for the Replication of Linear Plasmids and Chromosomes in Streptomyces. Genes Dev. 2001, 15, 1518–1527. [Google Scholar] [CrossRef] [PubMed]
- Klassen, R.; Meinhardt, F. Linear Protein-Primed Replicating Plasmids in Eukaryotic Microbes. In Microbial Linear Plasmids; Meinhardt, F., Klassen, R., Eds.; Microbiology Monographs; Springer: Berlin/Heidelberg, Germany, 2007; pp. 187–226. ISBN 978-3-540-72025-6. [Google Scholar]
- Mendez, J.; Blanco, L.; Salas, M. Protein-Primed DNA Replication: A Transition between Two Modes of Priming by a Unique DNA Polymerase. EMBO J. 1997, 16, 2519–2527. [Google Scholar] [CrossRef]
- Calles, B.; Salas, M.; Rojo, F. The Φ29 Transcriptional Regulator Contacts the Nucleoid Protein P6 to Organize a Repression Complex. EMBO J. 2002, 21, 6185–6194. [Google Scholar] [CrossRef] [PubMed]
- Camacho, A.; Salas, M. Mechanism for the Switch of Φ29 DNA Early to Late Transcription by Regulatory Protein P4 and Histone-like Protein P6. EMBO J. 2001, 20, 6060–6070. [Google Scholar] [CrossRef]
- González-Huici, V.; Alcorlo, M.; Salas, M.; Hermoso, J.M. Binding of Phage Phi29 Architectural Protein P6 to the Viral Genome: Evidence for Topological Restriction of the Phage Linear DNA. Nucleic Acids Res. 2004, 32, 3493–3502. [Google Scholar] [CrossRef]
- Crucitti, P.; Lázaro, J.M.; Benes, V.; Salas, M. Bacteriophage Phi29 Early Protein P17 Is Conditionally Required for the First Rounds of Viral DNA Replication. Gene 1998, 223, 135–142. [Google Scholar] [CrossRef]
- Xu, J.; Wang, D.; Gui, M.; Xiang, Y. Structural Assembly of the Tailed Bacteriophage Φ29. Nat. Commun. 2019, 10, 2366. [Google Scholar] [CrossRef]
- Cohen, D.N.; Erickson, S.E.; Xiang, Y.; Rossmann, M.G.; Anderson, D.L. Multifunctional Roles of a Bacteriophage Φ29 Morphogenetic Factor in Assembly and Infection. J. Mol. Biol. 2008, 378, 802–815. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.H.; Morais, M.C.; Anderson, D.L.; Rossmann, M.G. Determinants of Bacteriophage Phi29 Head Morphology. Structure 2006, 14, 1723–1727. [Google Scholar] [CrossRef]
- Xiang, Y.; Rossmann, M.G. Structure of Bacteriophage Φ29 Head Fibers Has a Supercoiled Triple Repeating Helix-Turn-Helix Motif. Proc. Natl. Acad. Sci. USA 2011, 108, 4806–4810. [Google Scholar] [CrossRef] [PubMed]
- Jing, P.; Burris, B.; Cortes, M. The PLB Measurement for the Connector in Phi29 Bacteriophage Reveals the Function of Its Channel Loop. Biophys. J. 2021, 120, 1650–1664. [Google Scholar] [CrossRef]
- Xiang, Y.; Morais, M.C.; Cohen, D.N.; Bowman, V.D.; Anderson, D.L.; Rossmann, M.G. Crystal and cryoEM Structural Studies of a Cell Wall Degrading Enzyme in the Bacteriophage Phi29 Tail. Proc. Natl. Acad. Sci. USA 2008, 105, 9552–9557. [Google Scholar] [CrossRef]
- Xu, J.; Gui, M.; Wang, D.; Xiang, Y. The Bacteriophage Φ29 Tail Possesses a Pore-Forming Loop for Cell Membrane Penetration. Nature 2016, 534, 544–547. [Google Scholar] [CrossRef]
- Xiang, Y.; Leiman, P.G.; Li, L.; Grimes, S.; Anderson, D.L.; Rossmann, M.G. Crystallographic Insights into the Autocatalytic Assembly Mechanism of a Bacteriophage Tail Spike. Mol. Cell 2009, 34, 375–386. [Google Scholar] [CrossRef]
- Bourassa, N.; Major, F. Implication of the Prohead RNA in Phage Phi29 DNA Packaging. Biochimie 2002, 84, 945–951. [Google Scholar] [CrossRef]
- Woodson, M.; Pajak, J.; Mahler, B.P.; Zhao, W.; Zhang, W.; Arya, G.; White, M.A.; Jardine, P.J.; Morais, M.C. A Viral Genome Packaging Motor Transitions between Cyclic and Helical Symmetry to Translocate dsDNA. Sci. Adv. 2021, 7, eabc1955. [Google Scholar] [CrossRef]
- Grimes, S.; Anderson, D. Cleaving the Prohead RNA of Bacteriophage Φ29 Alters the in Vitro Packaging of Restriction Fragments of DNA-Gp3. J. Mol. Biol. 1989, 209, 101–108. [Google Scholar] [CrossRef]
- Wichitwechkarn, J.; Bailey, S.; Bodley, J.W.; Anderson, D. Prohead RNA of Bacteriophage Phi 29: Size, Stoichiometry and Biological Activity. Nucleic Acids Res. 1989, 17, 3459–3468. [Google Scholar] [CrossRef] [PubMed]
- Ismail, A.; Saini, T.; Al Qaffas, A.; Erill, I.; Caruso, S.M.; Temple, L.; Johnson, A.A. Evidence of a Set of Core-Function Genes in 16 Bacillus Podoviral Genomes with Considerable Genomic Diversity. Viruses 2023, 15, 276. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.; Wichitwechkarn, J.; Johnson, D.; Reilly, B.E.; Anderson, D.L.; Bodley, J.W. Phylogenetic Analysis and Secondary Structure of the Bacillus subtilis Bacteriophage RNA Required for DNA Packaging. J. Biol. Chem. 1990, 265, 22365–22370. [Google Scholar] [CrossRef] [PubMed]
- Hilbert, B.J.; Hayes, J.A.; Stone, N.P.; Xu, R.-G.; Kelch, B.A. The Large Terminase DNA Packaging Motor Grips DNA with Its ATPase Domain for Cleavage by the Flexible Nuclease Domain. Nucleic Acids Res. 2017, 45, 3591–3605. [Google Scholar] [CrossRef]
- Zhao, W.; Jardine, P.J.; Grimes, S. An RNA Domain Imparts Specificity and Selectivity to a Viral DNA Packaging Motor. J. Virol. 2015, 89, 12457–12466. [Google Scholar] [CrossRef]
- Mahler, B.P.; Bujalowski, P.J.; Mao, H.; Dill, E.A.; Jardine, P.J.; Choi, K.H.; Morais, M.C. NMR Structure of a Vestigial Nuclease Provides Insight into the Evolution of Functional Transitions in Viral dsDNA Packaging Motors. Nucleic Acids Res. 2020, 48, 11737. [Google Scholar] [CrossRef]
- Helgstrand, C.; Wikoff, W.R.; Duda, R.L.; Hendrix, R.W.; Johnson, J.E.; Liljas, L. The Refined Structure of a Protein Catenane: The HK97 Bacteriophage Capsid at 3.44 A Resolution. J. Mol. Biol. 2003, 334, 885–899. [Google Scholar] [CrossRef]
- Wiryaman, T.; Toor, N. Cryo-EM Structure of a Thermostable Bacterial Nanocompartment. IUCrJ 2021, 8, 342–350. [Google Scholar] [CrossRef]
- Morais, M.C.; Kanamaru, S.; Badasso, M.O.; Koti, J.S.; Owen, B.A.L.; McMurray, C.T.; Anderson, D.L.; Rossmann, M.G. Bacteriophage Phi29 Scaffolding Protein Gp7 before and after Prohead Assembly. Nat. Struct. Biol. 2003, 10, 572–576. [Google Scholar] [CrossRef]
- del Prado, A.; Rodríguez, I.; Lázaro, J.M.; Moreno-Morcillo, M.; de Vega, M.; Salas, M. New Insights into the Coordination between the Polymerization and 3′-5′ Exonuclease Activities in Φ29 DNA Polymerase. Sci. Rep. 2019, 9, 923. [Google Scholar] [CrossRef]
- de Vega, M.; Lázaro, J.M.; Mencía, M.; Blanco, L.; Salas, M. Improvement of Φ29 DNA Polymerase Amplification Performance by Fusion of DNA Binding Motifs. Proc. Natl. Acad. Sci. USA 2010, 107, 16506–16511. [Google Scholar] [CrossRef] [PubMed]
- Hermoso, J.M.; Salas, M. Protein P3 Is Linked to the DNA of Phage Phi 29 through a Phosphoester Bond between Serine and 5’-dAMP. Proc. Natl. Acad. Sci. USA 1980, 77, 6425–6428. [Google Scholar] [CrossRef] [PubMed]
- Ballesteros-Plaza, D.; Holguera, I.; Scheffers, D.-J.; Salas, M.; Muñoz-Espín, D. Phage Φ29 Protein P1 Promotes Replication by Associating with the FtsZ Ring of the Divisome in Bacillus subtilis. Proc. Natl. Acad. Sci. USA 2013, 110, 12313–12318. [Google Scholar] [CrossRef] [PubMed]
- Martín, G.; Lázaro, J.M.; Méndez, E.; Salas, M. Characterization of the Phage Phi 29 Protein P5 as a Single-Stranded DNA Binding Protein. Function in Phi 29 DNA-Protein P3 Replication. Nucleic Acids Res. 1989, 17, 3663–3672. [Google Scholar] [CrossRef] [PubMed]
- Meijer, W.J.J.; Serna-Rico, A.; Salas, M. Characterization of the Bacteriophage Φ29-Encoded Protein P16.7: A Membrane Protein Involved in Phage DNA Replication. Mol. Microbiol. 2001, 39, 731–746. [Google Scholar] [CrossRef]
- Asensio, J.L.; Albert, A.; Muñoz-Espín, D.; Gonzalez, C.; Hermoso, J.; Villar, L.; Jiménez-Barbero, J.; Salas, M.; Meijer, W.J.J. Structure of the Functional Domain of Phi29 Replication Organizer: Insights into Oligomerization and Dna Binding. J. Biol. Chem. 2005, 280, 20730–20739. [Google Scholar] [CrossRef] [PubMed]
- Albert, A.; Muñoz-Espín, D.; Jiménez, M.; Asensio, J.L.; Hermoso, J.A.; Salas, M.; Meijer, W.J.J. Structural Basis for Membrane Anchorage of Viral Phi29 DNA during Replication. J. Biol. Chem. 2005, 280, 42486–42488. [Google Scholar] [CrossRef] [PubMed]
- Young, R. Phage Lysis: Three Steps, Three Choices, One Outcome. J. Microbiol. Seoul Korea 2014, 52, 243–258. [Google Scholar] [CrossRef] [PubMed]
- Steiner, M.; Lubitz, W.; Bläsi, U. The Missing Link in Phage Lysis of Gram-Positive Bacteria: Gene 14 of Bacillus subtilis Phage Phi 29 Encodes the Functional Homolog of Lambda S Protein. J. Bacteriol. 1993, 175, 1038–1042. [Google Scholar] [CrossRef]
- Saedi, M.S.; Garvey, K.J.; Ito, J. Cloning and Purification of a Unique Lysozyme Produced by Bacillus Phage Phi 29. Proc. Natl. Acad. Sci. USA 1987, 84, 955–958. [Google Scholar] [CrossRef]
- Hrebík, D.; Štveráková, D.; Škubník, K.; Füzik, T.; Pantůček, R.; Plevka, P. Structure and Genome Ejection Mechanism of Staphylococcus Aureus Phage P68. Sci. Adv. 2019, 5, eaaw7414. [Google Scholar] [CrossRef] [PubMed]
- RCSC Protein Data Bank. RCSB PDB-7R3T: Crystal Structure of the Dimeric C-Terminal Big_2-CBM56 Domains from Paenibacillus Illinoisensis (Bacillus circulans IAM1165) Beta-1,3-Glucanase H. Available online: https://www.rcsb.org/structure/7R3T (accessed on 25 August 2024).
- Kumar, P.; Vyas, P.; Faisal, S.M.; Chang, Y.-F.; Akif, M. Crystal Structure of a Variable Region Segment of Leptospira Host-Interacting Outer Surface Protein, LigA, Reveals the Orientation of Ig-like Domains. Int. J. Biol. Macromol. 2023, 244, 125445. [Google Scholar] [CrossRef] [PubMed]
- Fioravanti, A.; Van Hauwermeiren, F.; Van der Verren, S.E.; Jonckheere, W.; Goncalves, A.; Pardon, E.; Steyaert, J.; De Greve, H.; Lamkanfi, M.; Remaut, H. Structure of S-Layer Protein Sap Reveals a Mechanism for Therapeutic Intervention in Anthrax. Nat. Microbiol. 2019, 4, 1805–1814. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, C.-A.; Effantin, G.; Vivès, C.; Engilberge, S.; Bacia, M.; Boulanger, P.; Girard, E.; Schoehn, G.; Breyton, C. Bacteriophage T5 Tail Tube Structure Suggests a Trigger Mechanism for Siphoviridae DNA Ejection. Nat. Commun. 2017, 8, 1953. [Google Scholar] [CrossRef]
- Nichols, R.J.; LaFrance, B.; Phillips, N.R.; Radford, D.R.; Oltrogge, L.M.; Valentin-Alvarado, L.E.; Bischoff, A.J.; Nogales, E.; Savage, D.F. Discovery and Characterization of a Novel Family of Prokaryotic Nanocompartments Involved in Sulfur Metabolism. eLife 2021, 10, e59288. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Dolja, V.V.; Krupovic, M. The Logic of Virus Evolution. Cell Host Microbe 2022, 30, 917–929. [Google Scholar] [CrossRef]
- Clancy Kelley, L.-L.; Dillard, B.D.; Tempel, W.; Chen, L.; Shaw, N.; Lee, D.; Newton, M.G.; Sugar, F.J.; Jenney, F.E.; Lee, H.S.; et al. Structure of the Hypothetical Protein PF0899 from Pyrococcus Furiosus at 1.85 Å Resolution. Acta Crystallogr. Sect. F 2007, 63, 549–552. [Google Scholar] [CrossRef]
- Hardy, J.M.; Dunstan, R.A.; Grinter, R.; Belousoff, M.J.; Wang, J.; Pickard, D.; Venugopal, H.; Dougan, G.; Lithgow, T.; Coulibaly, F. The Architecture and Stabilisation of Flagellotropic Tailed Bacteriophages. Nat. Commun. 2020, 11, 3748. [Google Scholar] [CrossRef]
- Wang, Z.; Hardies, S.C.; Fokine, A.; Klose, T.; Jiang, W.; Cho, B.C.; Rossmann, M.G. Structure of the Marine Siphovirus TW1: Evolution of Capsid-Stabilizing Proteins and Tail Spikes. Structure 2018, 26, 238–248.e3. [Google Scholar] [CrossRef]
- Bayfield, O.W.; Shkoporov, A.N.; Yutin, N.; Khokhlova, E.V.; Smith, J.L.R.; Hawkins, D.E.D.P.; Koonin, E.V.; Hill, C.; Antson, A.A. Structural Atlas of a Human Gut Crassvirus. Nature 2023, 617, 409–416. [Google Scholar] [CrossRef]
- Javed, A.; Villanueva, H.; Shataer, S.; Vasciaveo, S.; Savva, R.; Orlova, E.V. Cryo-EM Structures of Two Bacteriophage Portal Proteins Provide Insights for Antimicrobial Phage Engineering. Viruses 2021, 13, 2532. [Google Scholar] [CrossRef] [PubMed]
- Fàbrega-Ferrer, M.; Cuervo, A.; Fernández, F.J.; Machón, C.; Pérez-Luque, R.; Pous, J.; Vega, M.C.; Carrascosa, J.L.; Coll, M. Using a Partial Atomic Model from Medium-Resolution Cryo-EM to Solve a Large Crystal Structure. Acta Crystallogr. Sect. Struct. Biol. 2021, 77, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Tan, L.; Tan, Z.; Zhang, Y.; Chen, W.; Li, X.; Song, J.; Cheng, L.; Liu, H. Structure of the Siphophage Neck–Tail Complex Suggests That Conserved Tail Tip Proteins Facilitate Receptor Binding and Tail Assembly. PLOS Biol. 2023, 21, e3002441. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhang, X.; Gao, S.; Rao, P.A.; Padilla-Sanchez, V.; Chen, Z.; Sun, S.; Xiang, Y.; Subramaniam, S.; Rao, V.B.; et al. Cryo-EM Structure of the Bacteriophage T4 Portal Protein Assembly at near-Atomic Resolution. Nat. Commun. 2015, 6, 7548. [Google Scholar] [CrossRef]
- Bárdy, P.; Füzik, T.; Hrebík, D.; Pantůček, R.; Thomas Beatty, J.; Plevka, P. Structure and Mechanism of DNA Delivery of a Gene Transfer Agent. Nat. Commun. 2020, 11, 3034. [Google Scholar] [CrossRef]
- Hussain, T.; Llácer, J.L.; Wimberly, B.T.; Kieft, J.S.; Ramakrishnan, V. Large-Scale Movements of IF3 and tRNA during Bacterial Translation Initiation. Cell 2016, 167, 133–144.e13. [Google Scholar] [CrossRef]
- Bubunenko, M.; Korepanov, A.; Court, D.L.; Jagannathan, I.; Dickinson, D.; Chaudhuri, B.R.; Garber, M.B.; Culver, G.M. 30S Ribosomal Subunits Can Be Assembled in Vivo without Primary Binding Ribosomal Protein S15. RNA 2006, 12, 1229–1239. [Google Scholar] [CrossRef]
- Machen, A.J.; Fisher, M.T.; Freudenthal, B.D. Anthrax Toxin Translocation Complex Reveals Insight into the Lethal Factor Unfolding and Refolding Mechanism. Sci. Rep. 2021, 11, 13038. [Google Scholar] [CrossRef]
- Xu, X.; Godoy-Ruiz, R.; Adipietro, K.A.; Peralta, C.; Ben-Hail, D.; Varney, K.M.; Cook, M.E.; Roth, B.M.; Wilder, P.T.; Cleveland, T.; et al. Structure of the Cell-Binding Component of the Clostridium Difficile Binary Toxin Reveals a Di-Heptamer Macromolecular Assembly. Proc. Natl. Acad. Sci. USA 2020, 117, 1049–1058. [Google Scholar] [CrossRef]
- RCSC Protein Data Bank. RCSB PDB-7AL8: Structure of ATSI with Bovine Trypsin. Available online: https://www.rcsb.org/structure/7AL8 (accessed on 25 August 2024).
- Ge, X.; Wang, J. Structural Mechanism of Bacteriophage Lambda Tail’s Interaction with the Bacterial Receptor. Nat. Commun. 2024, 15, 4185. [Google Scholar] [CrossRef]
- Müller, J.J.; Barbirz, S.; Heinle, K.; Freiberg, A.; Seckler, R.; Heinemann, U. An Intersubunit Active Site between Supercoiled Parallel Beta Helices in the Trimeric Tailspike Endorhamnosidase of Shigella Flexneri Phage Sf6. Structure 2008, 16, 766–775. [Google Scholar] [CrossRef]
- Greenfield, J.; Shang, X.; Luo, H.; Zhou, Y.; Linden, S.B.; Heselpoth, R.D.; Leiman, P.G.; Nelson, D.C.; Herzberg, O. Structure and Function of Bacteriophage CBA120 ORF211 (TSP2), the Determinant of Phage Specificity towards E. coli O157:H7. Sci. Rep. 2020, 10, 15402. [Google Scholar] [CrossRef] [PubMed]
- d’Acapito, A.; Roret, T.; Zarkadas, E.; Mocaër, P.-Y.; Lelchat, F.; Baudoux, A.-C.; Schoehn, G.; Neumann, E. Structural Study of the Cobetia Marina Bacteriophage 1 (Carin-1) by Cryo-EM. J. Virol. 2023, 97, e00248-23. [Google Scholar] [CrossRef] [PubMed]
- Pas, C.; Latka, A.; Fieseler, L.; Briers, Y. Phage Tailspike Modularity and Horizontal Gene Transfer Reveals Specificity towards E. Coli O-Antigen Serogroups. Virol. J. 2023, 20, 174. [Google Scholar] [CrossRef] [PubMed]
- Kamtekar, S.; Berman, A.J.; Wang, J.; Lázaro, J.M.; de Vega, M.; Blanco, L.; Salas, M.; Steitz, T.A. Insights into Strand Displacement and Processivity from the Crystal Structure of the Protein-Primed DNA Polymerase of Bacteriophage Phi29. Mol. Cell 2004, 16, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Berman, A.J.; Kamtekar, S.; Goodman, J.L.; Lázaro, J.M.; de Vega, M.; Blanco, L.; Salas, M.; Steitz, T.A. Structures of Phi29 DNA Polymerase Complexed with Substrate: The Mechanism of Translocation in B-Family Polymerases. EMBO J. 2007, 26, 3494–3505. [Google Scholar] [CrossRef]
- Coloma, J.; Johnson, R.E.; Prakash, L.; Prakash, S.; Aggarwal, A.K. Human DNA Polymerase α in Binary Complex with a DNA:DNA Template-Primer. Sci. Rep. 2016, 6, 23784. [Google Scholar] [CrossRef]
- Reid, S.P.; Cárdenas, W.B.; Basler, C.F. Homo-Oligomerization Facilitates the Interferon-Antagonist Activity of the Ebolavirus VP35 Protein. Virology 2005, 341, 179–189. [Google Scholar] [CrossRef]
- Bruhn, J.F.; Kirchdoerfer, R.N.; Urata, S.M.; Li, S.; Tickle, I.J.; Bricogne, G.; Saphire, E.O. Crystal Structure of the Marburg Virus VP35 Oligomerization Domain. J. Virol. 2017, 91, e01085-16. [Google Scholar] [CrossRef]
- Dilley, K.A.; Voorhies, A.A.; Luthra, P.; Puri, V.; Stockwell, T.B.; Lorenzi, H.; Basler, C.F.; Shabman, R.S. The Ebola Virus VP35 Protein Binds Viral Immunostimulatory and Host RNAs Identified through Deep Sequencing. PLoS ONE 2017, 12, e0178717. [Google Scholar] [CrossRef]
- Zhao, H.; Christensen, T.E.; Kamau, Y.N.; Tang, L. Structures of the Phage Sf6 Large Terminase Provide New Insights into DNA Translocation and Cleavage. Proc. Natl. Acad. Sci. USA 2013, 110, 8075–8080. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A.F.U.H.; Chan, C.; Guan, H.; Gong, B.; Guo, P.; Cheng, X.; Ouyang, S. Structural Insights into Gp16 ATPase in the Bacteriophage Φ29 DNA Packaging Motor. Biochemistry 2021, 60, 886–897. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Cingolani, G. Structure of P22 Headful Packaging Nuclease. J. Biol. Chem. 2012, 287, 28196–28205. [Google Scholar] [CrossRef] [PubMed]
- Cernooka, E.; Rumnieks, J.; Zrelovs, N.; Tars, K.; Kazaks, A. Diversity of the Lysozyme Fold: Structure of the Catalytic Domain from an Unusual Endolysin Encoded by Phage Enc34. Sci. Rep. 2022, 12, 5005. [Google Scholar] [CrossRef] [PubMed]
- Sykilinda, N.N.; Nikolaeva, A.Y.; Shneider, M.M.; Mishkin, D.V.; Patutin, A.A.; Popov, V.O.; Boyko, K.M.; Klyachko, N.L.; Miroshnikov, K.A. Structure of an Acinetobacter Broad-Range Prophage Endolysin Reveals a C-Terminal α-Helix with the Proposed Role in Activity against Live Bacterial Cells. Viruses 2018, 10, 309. [Google Scholar] [CrossRef]
- Randich, A.M.; Kysela, D.T.; Morlot, C.; Brun, Y.V. Origin of a Core Bacterial Gene via Co-Option and Detoxification of a Phage Lysin. Curr. Biol. CB 2019, 29, 1634–1646.e6. [Google Scholar] [CrossRef]
- Garvey, K.J.; Saedi, M.S.; Ito, J. Nucleotide Sequence of Bacillus Phage Phi 29 Genes 14 and 15: Homology of Gene 15 with Other Phage Lysozymes. Nucleic Acids Res. 1986, 14, 10001–10008. [Google Scholar] [CrossRef]
- Piligrimova, E.G.; Kazantseva, O.A.; Kazantsev, A.N.; Nikulin, N.A.; Skorynina, A.V.; Koposova, O.N.; Shadrin, A.M. Putative Plasmid Prophages of Bacillus Cereus Sensu Lato May Hold the Key to Undiscovered Phage Diversity. Sci. Rep. 2021, 11, 7611. [Google Scholar] [CrossRef]
- Olsen, N.S.; Hendriksen, N.B.; Hansen, L.H.; Kot, W. A New High-Throughput Screening Method for Phages: Enabling Crude Isolation and Fast Identification of Diverse Phages with Therapeutic Potential. Phage 2020, 1, 137–148. [Google Scholar] [CrossRef]
- Miroshnikov, K.A.; Evseev, P.V.; Lukianova, A.A.; Ignatov, A.N. Tailed Lytic Bacteriophages of Soft Rot Pectobacteriaceae. Microorganisms 2021, 9, 1819. [Google Scholar] [CrossRef]
- Brammer Basta, L.A.; Ghosh, A.; Pan, Y.; Jakoncic, J.; Lloyd, E.P.; Townsend, C.A.; Lamichhane, G.; Bianchet, M.A. Loss of a Functionally and Structurally Distinct Ld-Transpeptidase, LdtMt5, Compromises Cell Wall Integrity in Mycobacterium tuberculosis. J. Biol. Chem. 2015, 290, 25670–25685. [Google Scholar] [CrossRef] [PubMed]
- Chopin, A.; Deveau, H.; Ehrlich, S.D.; Moineau, S.; Chopin, M.-C. KSY1, a Lactococcal Phage with a T7-like Transcription. Virology 2007, 365, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Evseev, P.; Lukianova, A.; Sykilinda, N.; Gorshkova, A.; Bondar, A.; Shneider, M.; Kabilov, M.; Drucker, V.; Miroshnikov, K. Pseudomonas Phage MD8: Genetic Mosaicism and Challenges of Taxonomic Classification of Lambdoid Bacteriophages. Int. J. Mol. Sci. 2021, 22, 10350. [Google Scholar] [CrossRef] [PubMed]
- Fraser, J.S.; Yu, Z.; Maxwell, K.L.; Davidson, A.R. Ig-Like Domains on Bacteriophages: A Tale of Promiscuity and Deceit. J. Mol. Biol. 2006, 359, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Gao, M. Jumbo Bacteriophages: An Overview. Front. Microbiol. 2017, 8, 403. [Google Scholar] [CrossRef] [PubMed]
- Evseev, P.; Shneider, M.; Miroshnikov, K. Evolution of Phage Tail Sheath Protein. Viruses 2022, 14, 1148. [Google Scholar] [CrossRef]
- Sulakvelidze, A.; Alavidze, Z.; Morris, J.G. Bacteriophage Therapy. Antimicrob. Agents Chemother. 2001, 45, 649–659. [Google Scholar] [CrossRef]
- Peralta, B.; Gil-Carton, D.; Castaño-Díez, D.; Bertin, A.; Boulogne, C.; Oksanen, H.M.; Bamford, D.H.; Abrescia, N.G.A. Mechanism of Membranous Tunnelling Nanotube Formation in Viral Genome Delivery. PLoS Biol. 2013, 11, e1001667. [Google Scholar] [CrossRef]
- Leiman, P.G.; Basler, M.; Ramagopal, U.A.; Bonanno, J.B.; Sauder, J.M.; Pukatzki, S.; Burley, S.K.; Almo, S.C.; Mekalanos, J.J. Type VI Secretion Apparatus and Phage Tail-Associated Protein Complexes Share a Common Evolutionary Origin. Proc. Natl. Acad. Sci. USA 2009, 106, 4154–4159. [Google Scholar] [CrossRef]
- Veesler, D.; Cambillau, C. A Common Evolutionary Origin for Tailed-Bacteriophage Functional Modules and Bacterial Machineries. Microbiol. Mol. Biol. Rev. MMBR 2011, 75, 423–433. [Google Scholar] [CrossRef]
- Zoued, A.; Brunet, Y.R.; Durand, E.; Aschtgen, M.-S.; Logger, L.; Douzi, B.; Journet, L.; Cambillau, C.; Cascales, E. Architecture and Assembly of the Type VI Secretion System. Biochim. Biophys. Acta 2014, 1843, 1664–1673. [Google Scholar] [CrossRef] [PubMed]
- Fokine, A.; Rossmann, M.G. Common Evolutionary Origin of Procapsid Proteases, Phage Tail Tubes, and Tubes of Bacterial Type VI Secretion Systems. Structure 2016, 24, 1928–1935. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.; Fàbrega-Ferrer, M.; Machón, C.; Conesa, J.J.; Fernández, F.J.; Pérez-Luque, R.; Pérez-Ruiz, M.; Pous, J.; Vega, M.C.; Carrascosa, J.L.; et al. Structures of T7 Bacteriophage Portal and Tail Suggest a Viral DNA Retention and Ejection Mechanism. Nat. Commun. 2019, 10, 3746. [Google Scholar] [CrossRef] [PubMed]
- Pietrzyk-Brzezinska, A.J.; Absmeier, E.; Klauck, E.; Wen, Y.; Antelmann, H.; Wahl, M.C. Crystal Structure of the Escherichia Coli DExH-Box NTPase HrpB. Structure 2018, 26, 1462–1473.e4. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Meng, Y.; Campbell, J.L.; Shen, B. Multiple Roles of DNA2 Nuclease/Helicase in DNA Metabolism, Genome Stability and Human Diseases. Nucleic Acids Res. 2020, 48, 16–35. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate Structure Prediction of Biomolecular Interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef]
- AlQuraishi, M. AlphaFold at CASP13. Bioinforma. Oxf. Engl. 2019, 35, 4862–4865. [Google Scholar] [CrossRef] [PubMed]
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying Bacterial Genes and Endosymbiont DNA with Glimmer. Bioinforma. Oxf. Engl. 2007, 23, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, L.; Stephens, A.; Nam, S.-Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at Its Core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef]
- Holm, L. Dali Server: Structural Unification of Protein Families. Nucleic Acids Res. 2022, 50, W210–W215. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Holm, L.; Kääriäinen, S.; Rosenström, P.; Schenkel, A. Searching Protein Structure Databases with DaliLite v.3. Bioinformatics 2008, 24, 2780–2781. [Google Scholar] [CrossRef]
- Moraru, C.; Varsani, A.; Kropinski, A.M. VIRIDIC—A Novel Tool to Calculate the Intergenomic Similarities of Prokaryote-Infecting Viruses. Viruses 2020, 12, 1268. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Göker, M. VICTOR: Genome-Based Phylogeny and Classification of Prokaryotic Viruses. Bioinformatics 2017, 33, 3396–3404. [Google Scholar] [CrossRef]
- Kim, J.; Na, S.-I.; Kim, D.; Chun, J. UBCG2: Up-to-Date Bacterial Core Genes and Pipeline for Phylogenomic Analysis. J. Microbiol. Seoul Korea 2021, 59, 609–615. [Google Scholar] [CrossRef]
- Gilchrist, C.L.M.; Chooi, Y.-H. Clinker & Clustermap.Js: Automatic Generation of Gene Cluster Comparison Figures. Bioinformatics 2021, 37, 2473–2475. [Google Scholar] [CrossRef]
- Buchfink, B.; Reuter, K.; Drost, H.-G. Sensitive Protein Alignments at Tree-of-Life Scale Using DIAMOND. Nat. Methods 2021, 18, 366–368. [Google Scholar] [CrossRef]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An Open Source Software for Exploring and Manipulating Networks. Proc. Int. AAAI Conf. Web Soc. Media 2009, 3, 361–362. [Google Scholar] [CrossRef]
- Jacomy, M.; Venturini, T.; Heymann, S.; Bastian, M. ForceAtlas2, a Continuous Graph Layout Algorithm for Handy Network Visualization Designed for the Gephi Software. PLoS ONE 2014, 9, e98679. [Google Scholar] [CrossRef] [PubMed]
- Blondel, V.D.; Guillaume, J.-L.; Lambiotte, R.; Lefebvre, E. Fast Unfolding of Communities in Large Networks. J. Stat. Mech. Theory Exp. 2008, 2008, P10008. [Google Scholar] [CrossRef]
- Lambiotte, R. Multi-Scale Modularity and Dynamics in Complex Networks. In Dynamics On and Of Complex Networks; Mukherjee, A., Choudhury, M., Peruani, F., Ganguly, N., Mitra, B., Eds.; Modeling and Simulation in Science, Engineering and Technology; Springer: New York, NY, USA, 2013; Volume 2, pp. 125–141. ISBN 978-1-4614-6728-1. [Google Scholar]
- Ding, F.; Lu, C.; Zhao, W.; Rajashankar, K.R.; Anderson, D.L.; Jardine, P.J.; Grimes, S.; Ke, A. Structure and Assembly of the Essential RNA Ring Component of a Viral DNA Packaging Motor. Proc. Natl. Acad. Sci. USA 2011, 108, 7357–7362. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cluster | Name | Sequence Length, bp | % GC | Host | NCBI Taxonomy | Isolation Source, NCBI |
|---|---|---|---|---|---|---|
| 01 | Bacillus phage phi29 | 19,282 | 40.0% | Bacillus subtilis | Viruses; Duplodnaviria; Heunggongvirae; Uroviricota; Caudoviricetes; Salasmaviridae; Picovirinae; Salasvirus; Salasvirus phi29 | |
| 02 | Staphylococcus phage Andhra | 18,546 | 29.8% | Staphylococcus epidermidis | Viruses; Duplodnaviria; Heunggongvirae; Uroviricota; Caudoviricetes; Rountreeviridae; Rakietenvirinae; Andhravirus; Andhravirus andhra | |
| 03 | Arthrobacter phage Anjali | 19,679 | 59.4% | Arthrobacter globiformis | Viruses; Duplodnaviria; Heunggongvirae; Uroviricota; Caudoviricetes; Anjalivirus; Anjalivirus anjali | Soil |
| 04 | Caudoviricetes sp. isolate ctLx22 | 14,372 | 35.7% | Bacillota (suggested) | Viruses; Duplodnaviria; Heunggongvirae; Uroviricota; Caudoviricetes | Human Metagenome |
| 05 | Caudoviricetes sp. isolate ctOKO3 | 17,065 | 35.7% | Clostridium (suggested) | Viruses; Duplodnaviria; Heunggongvirae; Uroviricota; Caudoviricetes | Human Metagenome |
| 05 | Methanobrevibacter sp. isolate RGIG9610 | 16,264 | 34.6% | Bacillota (suggested) | Archaea; Euryarchaeota; Methanomada group; Methanobacteria; Methanobacteriales; Methanobacteriaceae; Methanobrevibacter | Ruminant gastrointestinal tract |
| 06 | Caudoviricetes sp. isolate ct1aZ12 | 15,307 | 42.6% | Bacillota (suggested) | Viruses; Duplodnaviria; Heunggongvirae; Uroviricota; Caudoviricetes | Human Metagenome |
| 06 | Clostridium phage CS229P1 | 16,473 | 37.8% | Clostridium symbiosum | Viruses; Duplodnaviria; Heunggongvirae; Uroviricota; Caudoviricetes | |
| 07 | Actinomyces phage Av-1 | 17,171 | 49.5% | Actinomyces naeslundii | Viruses; Duplodnaviria; Heunggongvirae; Uroviricota; Caudoviricetes; Dybvigvirus; Dybvigvirus Av1 | |
| 08 | Human gut phage 3064_54045 | 15,891 | 51.2% | Bifidobacterium (suggested) | Viruses | Faeces |
| 09 | Caudoviricetes sp. isolate ctciW69 | 14,784 | 41.5% | Clostridium (suggested) | Viruses; Duplodnaviria; Heunggongvirae; Uroviricota; Caudoviricetes | Human Metagenome |
| 10 | Bifidobacterium phage BD811P1 | 17,657 | 54.8% | Bifidobacterium dentium | Viruses; Duplodnaviria; Heunggongvirae; Uroviricota; Caudoviricetes | Municipal sewage |
| 11 | Bacteriophage sp. isolate ct4VF5 | 16,226 | 38.7% | Clostridia (suggested) | Viruses | Human Metagenome |
| 12 | Caudoviricetes sp. isolate ctFJX15 | 12,830 | 38.7% | Actinomycetia (suggested) | Viruses; Duplodnaviria; Heunggongvirae; Uroviricota; Caudoviricetes | Human Metagenome |
| 13 | Virus sp. ctxAI8 | 13,382 | 49.8% | Clostridia (suggested) | Viruses | Human Metagenome |
| 14 | Caudoviricetes sp. isolate ctPQG16 | 12,323 | 53.5% | Gram-positive (suggested) | Viruses; Duplodnaviria; Heunggongvirae; Uroviricota; Caudoviricetes | Human Metagenome |
| 15 | Bacteriophage sp. isolate ctLK83 | 14,962 | 40.1% | Bacillota (suggested) | Viruses | Human Metagenome |
| 16 | Caudoviricetes sp. isolate ctF653 | 15,970 | 41.3% | Clostridia (suggested) | Viruses; Duplodnaviria; Heunggongvirae; Uroviricota; Caudoviricetes | Human Metagenome |
| 17 | Caudoviricetes sp. isolate ctjx911 | 13,168 | 37.8% | Bacilli (suggested) | Viruses; Duplodnaviria; Heunggongvirae; Uroviricota; Caudoviricetes | Human Metagenome |
| 18 | Bifidobacterium phage BadAztec2 | 18,689 | 48.7% | Bifidobacterium asteroides | Viruses; Duplodnaviria; Heunggongvirae; Uroviricota; Caudoviricetes; Badaztecvirus; Badaztecvirus badaztec1 | Honeybee gut |
| 19 | Streptococcus phage Cp-1 | 19,343 | 38.8% | Streptococcus pneumoniae | Viruses; Duplodnaviria; Heunggongvirae; Uroviricota; Caudoviricetes; Madridviridae; Cepunavirus; Cepunavirus Cp1 | |
| 20 | Caudoviricetes sp. isolate ctsL82 | 13,451 | 32.4% | Clostridia (suggested) | Viruses; Duplodnaviria; Heunggongvirae; Uroviricota; Caudoviricetes | Human Metagenome |
| 21 | Clostridium beijerinckii DSM 6423 plasmid III | 16,762 | 28.4% | Clostridium beijerinckii | Bacteria; Bacillota; Clostridia; Eubacteriales; Clostridiaceae; Clostridium | |
| 21 | Clostridium phage HM2 | 17,470 | 29.4% | Clostridium saccharoperbutylacetonicum | Viruses; Duplodnaviria; Heunggongvirae; Uroviricota; Caudoviricetes | |
| 21 | Methanobrevibacter sp. isolate RGIG9632 | 16,579 | 34.2% | Clostridia (suggested) | Archaea; Euryarchaeota; Methanomada group; Methanobacteria; Methanobacteriales; Methanobacteriaceae; Methanobrevibacter | Gut metagenome |
| 22 | Enterococcus faecalis BE32 plasmid pBE32_2 | 29,025 | 32.5% | Enterococcus faecalis | Bacteria; Bacillota; Bacilli; Lactobacillales; Enterococcaceae; Enterococcus | |
| 22 | Enterococcus phage EF62phi | 30,505 | 32.7% | Enterococcus faecalis | Viruses; Duplodnaviria; Heunggongvirae; Uroviricota; Caudoviricetes | |
| 23 | Salmonella phage Astrid | 11,713 | 39.8% | Salmonella enterica | Viruses; Duplodnaviria; Heunggongvirae; Uroviricota; Caudoviricetes; Astrithrvirus; Astrithrvirus astrithr | Wastewater |
| 24 | Amedibacillus phage AD70P2 | 12,320 | 26.7% | Amedibacillus sp. | Viruses; Duplodnaviria; Heunggongvirae; Uroviricota; Caudoviricetes; Guelinviridae | Municipal sewage |
| 25 | Caudoviricetes sp. isolate ct2Eo1 | 13,822 | 45.1% | Bacillota (suggested) | Viruses; Duplodnaviria; Heunggongvirae; Uroviricota; Caudoviricetes | Human Metagenome |
| 26 | Caudovirales phage NCBI #CAOBPP | 13,791 | 37.5% | Bacillota (suggested) | Viruses; Duplodnaviria; Heunggongvirae; Uroviricota; Caudoviricetes; environmental samples | Human faeces |
| 27 | Lactococcus phage asccphi28 | 18,762 | 33.7% | Lactococcus lactis | Viruses; Duplodnaviria; Heunggongvirae; Uroviricota; Caudoviricetes; Salasmaviridae | |
| 28 | Podoviridae sp. ctnWS46 | 19,654 | 32.3% | Streptococcus (suggested) | Viruses; Duplodnaviria; Heunggongvirae; Uroviricota; Caudoviricetes | Human Metagenome |
| 29 | Clostridium phage CpV1 | 16,748 | 30.5% | Clostridium perfringens | Viruses; Duplodnaviria; Heunggongvirae; Uroviricota; Caudoviricetes; Guelinviridae; Denniswatsonvirinae; Capvunavirus; Capvunavirus CpV1 | |
| 30 | Bacteriophage sp. isolate 2692_55609 | 12,148 | 42.5% | Bacteroidales (suggested) | Viruses | Faeces |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Evseev, P.; Gutnik, D.; Evpak, A.; Kasimova, A.; Miroshnikov, K. Origin, Evolution and Diversity of φ29-like Phages—Review and Bioinformatic Analysis. Int. J. Mol. Sci. 2024, 25, 10838. https://doi.org/10.3390/ijms251910838
Evseev P, Gutnik D, Evpak A, Kasimova A, Miroshnikov K. Origin, Evolution and Diversity of φ29-like Phages—Review and Bioinformatic Analysis. International Journal of Molecular Sciences. 2024; 25(19):10838. https://doi.org/10.3390/ijms251910838
Chicago/Turabian StyleEvseev, Peter, Daria Gutnik, Alena Evpak, Anastasia Kasimova, and Konstantin Miroshnikov. 2024. "Origin, Evolution and Diversity of φ29-like Phages—Review and Bioinformatic Analysis" International Journal of Molecular Sciences 25, no. 19: 10838. https://doi.org/10.3390/ijms251910838