
Pleiotrophin Activates cMet- and mTORC1-Dependent Protein Synthesis through PTPRZ1—The Role of ανβ3 Integrin

 ,
,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
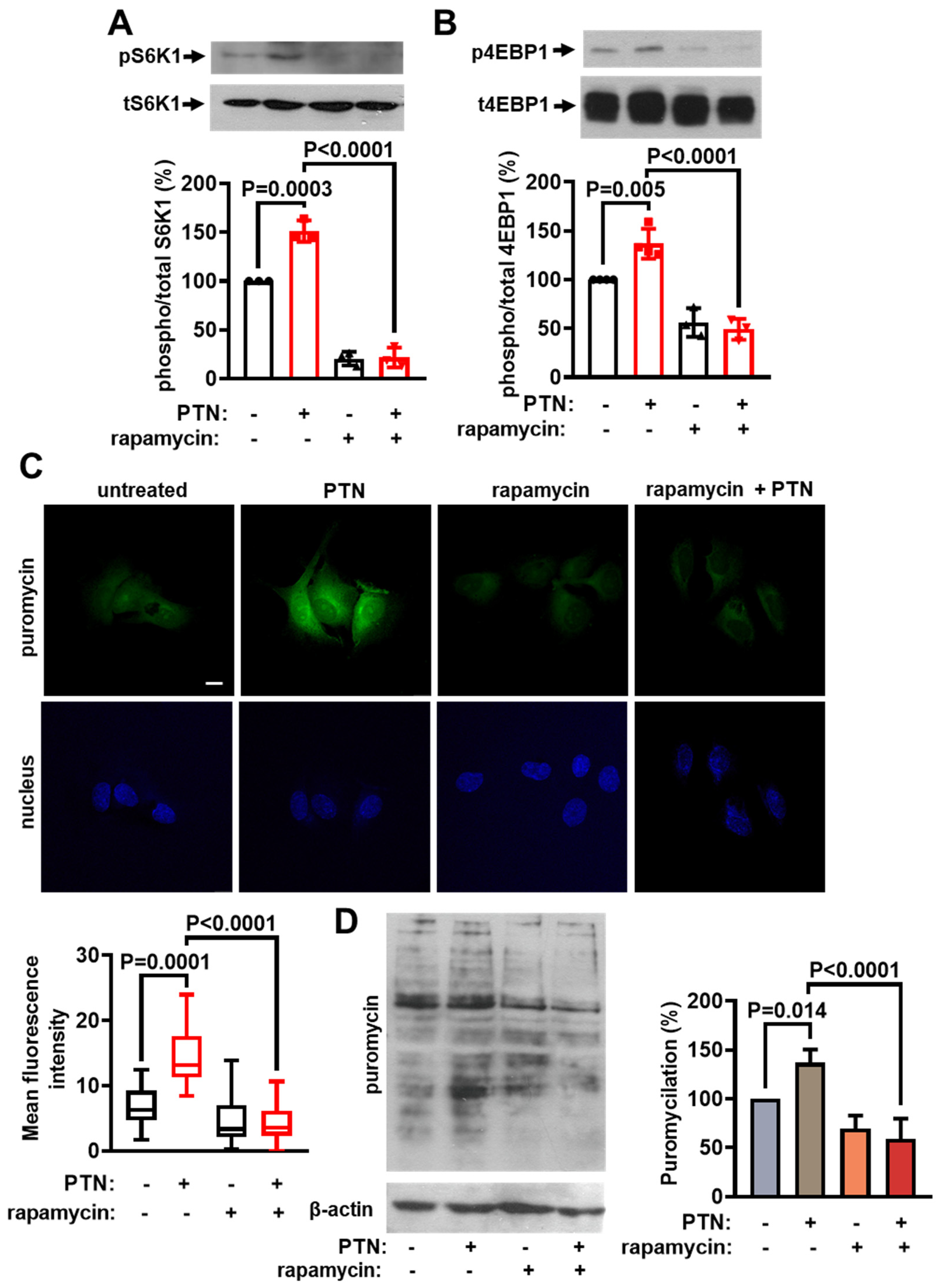
2.1. PTN Activates mTORC1 and Protein Synthesis in Endothelial Cells
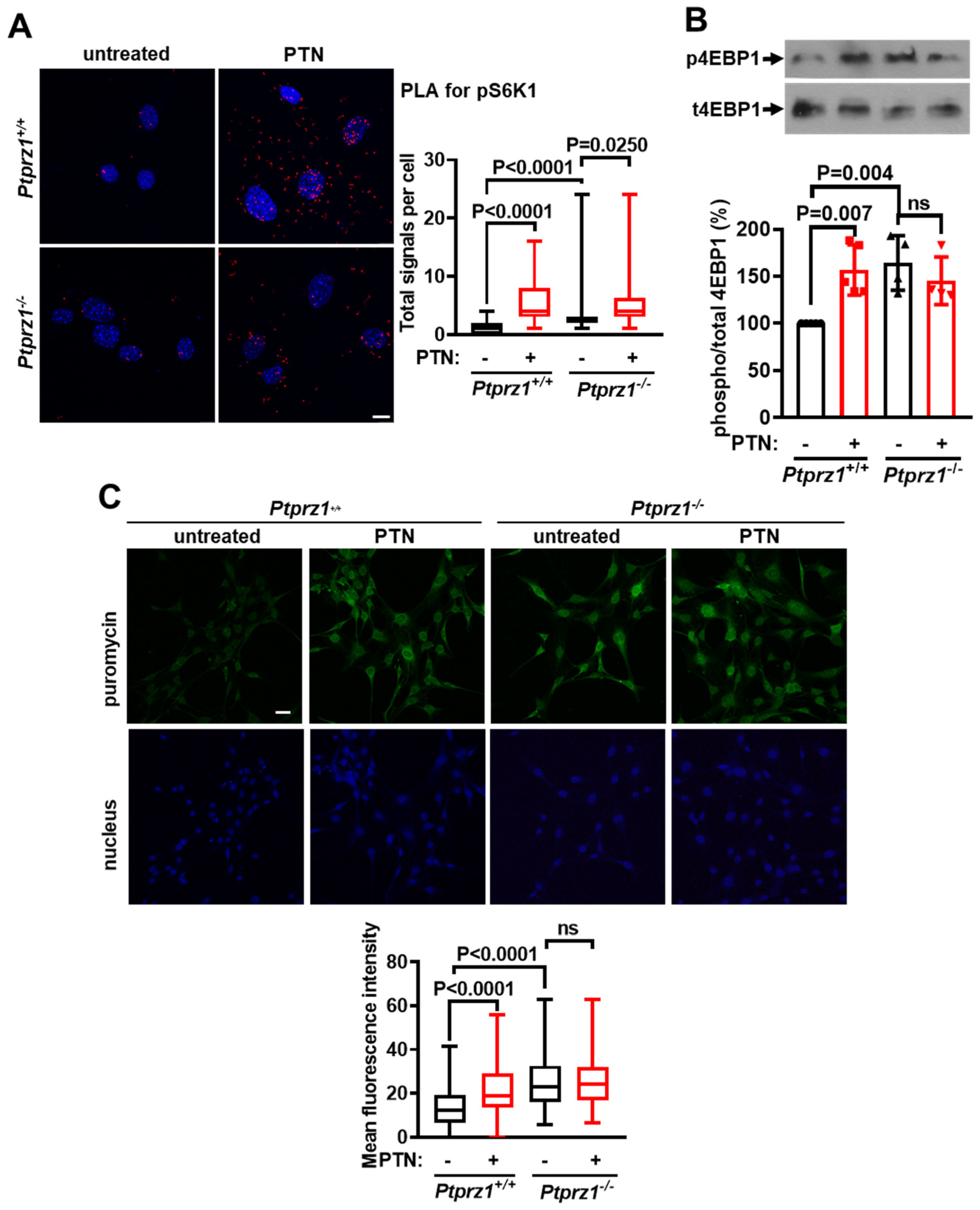
2.2. Ptprz1 Genetic Deletion Activates mTORC1 and Protein Synthesis in Endothelial Cells
2.3. PTN Activates mTORC1 and Protein Synthesis in Endothelial Cells through PTPRZ1
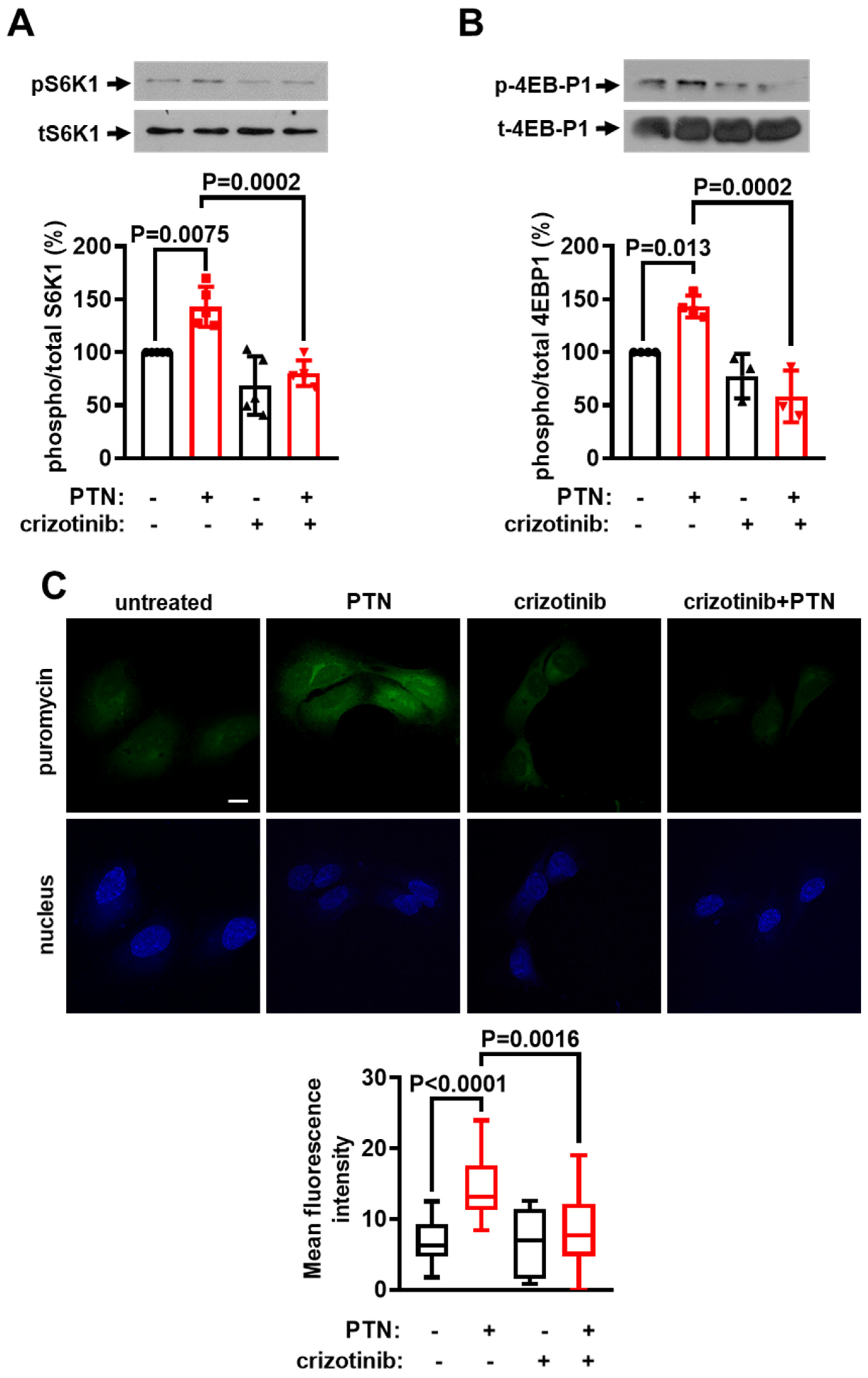
2.4. cMet Activation Mediates the PTN-Induced mTORC1 Activation Downstream of PTPRZ1
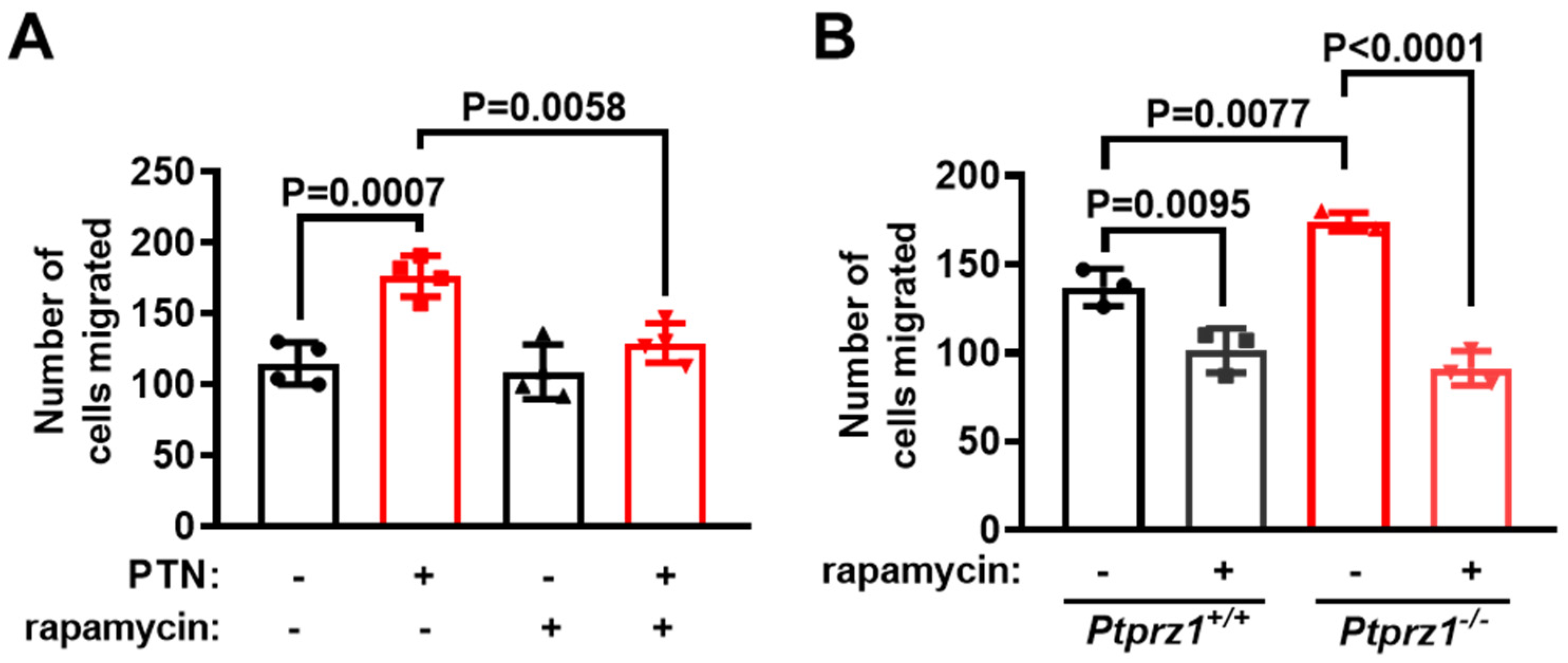
2.5. mTORC1 Is Required for Endothelial Cell Migration Downstream of the PTN/PTPRZ1 Axis
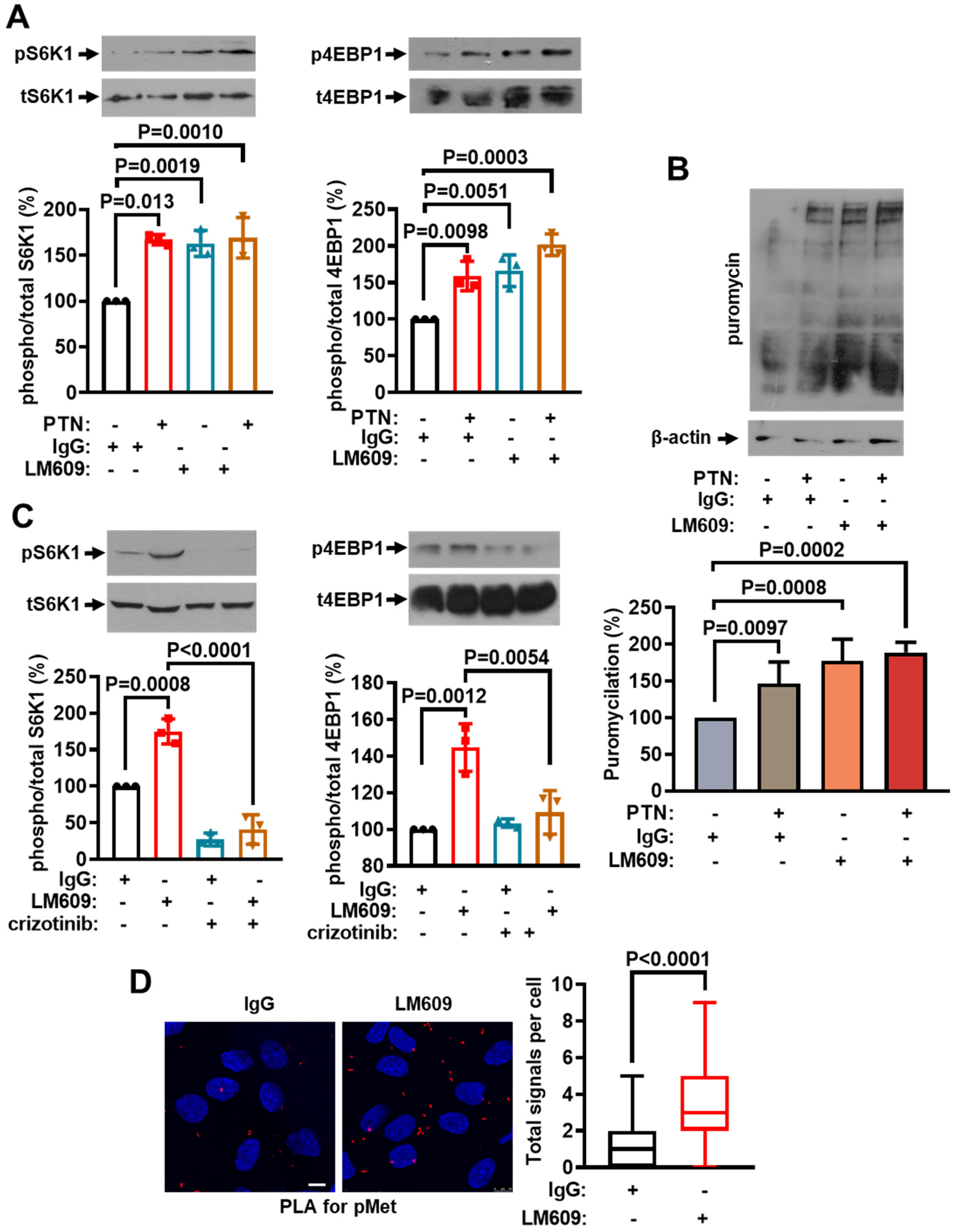
2.6. The Role of ανβ3 Integrin on mTORC1 Activation
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Migration Assay
4.4. Protein Synthesis Assessment
4.5. Proximity Ligation Assay (PLA)
4.6. Western Blot Analysis
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Papadimitriou, E.; Mourkogianni, E.; Ntenekou, D.; Christopoulou, M.; Koutsioumpa, M.; Lamprou, M. On the Role of Pleiotrophin and Its Receptors in Development and Angiogenesis. Int. J. Dev. Biol. 2021, 66, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Papadimitriou, E.; Pantazaka, E.; Castana, P.; Tsalios, T.; Polyzos, A.; Beis, D. Pleiotrophin and Its Receptor Protein Tyrosine Phosphatase Beta/Zeta as Regulators of Angiogenesis and Cancer. Biochim. Biophys. Acta-Rev. Cancer 2016, 9, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Kastana, P.; Ntenekou, D.; Mourkogianni, E.; Enake, M.K.; Xanthopoulos, A.; Choleva, E.; Marazioti, A.; Nikou, S.; Akwii, R.G.; Papadaki, E.; et al. Genetic Deletion or Tyrosine Phosphatase Inhibition of PTPRZ1 Activates C-Met to up-Regulate Angiogenesis and Lung Adenocarcinoma Growth. Int. J. Cancer 2023, 153, 1051–1066. [Google Scholar] [CrossRef] [PubMed]
- Lamprou, M.; Kastana, P.; Kofina, F.; Tzoupis, H.; Barmpoutsi, S.; Sajib, M.S.; Koutsioumpa, M.; Poimenidi, E.; Zompra, A.A.; Tassopoulos, D.; et al. Pleiotrophin Selectively Binds to Vascular Endothelial Growth Factor Receptor 2 and Inhibits or Stimulates Cell Migration Depending on ανβ3 Integrin Expression. Angiogenesis 2020, 23, 621–636. [Google Scholar] [CrossRef]
- Katraki-Pavlou, S.; Kastana, P.; Bousis, D.; Ntenekou, D.; Varela, A.; Davos, C.H.; Nikou, S.; Papadaki, E.; Tsigkas, G.; Athanasiadis, E.; et al. Protein Tyrosine Phosphatase Receptor-F1 Deletion Triggers Defective Heart Morphogenesis in Mice and Zebrafish. Am. J. Physiol.-Heart Circ. Physiol. 2022, 322, H8–H24. [Google Scholar] [CrossRef]
- Jhanwar-Uniyal, M.; Wainwright, J.V.; Mohan, A.L.; Tobias, M.E.; Murali, R.; Gandhi, C.D.; Schmidt, M.H. Diverse Signaling Mechanisms of MTOR Complexes: MTORC1 and MTORC2 in Forming a Formidable Relationship. Adv. Biol. Regul. 2019, 72, 51–62. [Google Scholar] [CrossRef]
- Szwed, A.; Kim, E.; Jacinto, E. Regulation and metabolic functions of mTORC1 and mTORC2. Physiol. Rev. 2021, 101, 1371–1426. [Google Scholar] [CrossRef]
- Liu, X.; Guo, B.; Li, Q.; Nie, J. mTOR in Metabolic Homeostasis and Disease. Exp. Cell Res. 2024, 441, 114173. [Google Scholar] [CrossRef]
- Melick, C.H.; Jewell, J.L. Regulation of Mtorc1 by Upstream Stimuli. Genes 2020, 11, 989. [Google Scholar] [CrossRef]
- Panwar, V.; Singh, A.; Bhatt, M.; Tonk, R.K.; Azizov, S.; Raza, A.S.; Sengupta, S.; Kumar, D.; Garg, M. Multifaceted Role of mTOR (Mammalian Target of Rapamycin) Signaling Pathway in Human Health and Disease. Signal Transduct. Target. Ther. 2023, 8, 375. [Google Scholar] [CrossRef]
- Gentilella, A.; Kozma, S.C.; Thomas, G. A Liaison between MTOR Signaling, Ribosome Biogenesis and Cancer. Biochim. Biophys. Acta-Gene Regul. Mech. 2015, 7, 812–820. [Google Scholar] [CrossRef] [PubMed]
- Kim, L.C.; Cook, R.S.; Chen, J. MTORC1 and MTORC2 in Cancer and the Tumor Microenvironment. Oncogene 2017, 36, 2191–2201. [Google Scholar] [CrossRef] [PubMed]
- Mannick, J.B.; Lamming, D.W. Targeting the Biology of Aging with mTOR Inhibitors. Nat. Aging 2023, 3, 642–666. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Barilli, A.; Visigalli, R.; Sala, R.; Gazzola, G.C.; Parolari, A.; Tremoli, E.; Bonomini, S.; Simon, A.; Closs, E.I.; Dall’Asta, V.; et al. In human endothelial cells rapamycin causes mTORC2 inhibition and impairs cell viability and function. Cardiovasc. Res. 2008, 78, 563–571. [Google Scholar] [CrossRef]
- Schreiber, K.H.; Ortiz, D.; Academia, E.C.; Anies, A.C.; Liao, C.Y.; Kennedy, B.K. Rapamycin-mediated mTORC2 inhibition is determined by the relative expression of FK506-binding proteins. Aging Cell 2015, 14, 265–273. [Google Scholar] [CrossRef]
- Lamming, D.W.; Ye, L.; Katajisto, P.; Goncalves, M.D.; Saitoh, M.; Stevens, D.M.; Davis, J.G.; Salmon, A.B.; Richardson, A.; Ahima, R.S.; et al. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science 2012, 335, 1638–1643. [Google Scholar] [CrossRef]
- Tanga, N.; Kuboyama, K.; Kishimoto, A.; Kiyonari, H.; Shiraishi, A.; Suzuki, R.; Watanabe, T.; Fujikawa, A.; Noda, M. The PTN-PTPRZ Signal Activates the AFAP1L2-Dependent PI3K-AKT Pathway for Oligodendrocyte Differentiation: Targeted Inactivation of PTPRZ Activity in Mice. Glia 2019, 67, 967–984. [Google Scholar] [CrossRef]
- Qiu, X.; Zhou, R.; Su, X.; Ying, J.; Qu, Y.; Mu, D. Pleiotrophin Ameliorates White Matter Injury of Neonatal Rats by Activating the mTOR/YY1/Id4 Signaling Pathway. FASEB J. 2023, 37, e23082. [Google Scholar] [CrossRef]
- Tian, Z.; Du, Z.; Bai, G.; Gong, Q.; You, Y.; Xu, G.; Liu, J.; Xiao, M.; Wang, Y.; He, Y. Schwann Cell Derived Pleiotrophin Stimulates Fibroblast for Proliferation and Excessive Collagen Deposition in Plexiform Neurofibroma. Cancer Gene Ther. 2024, 31, 627–640. [Google Scholar] [CrossRef]
- Mikelis, C.; Sfaelou, E.; Koutsioumpa, M.; Kieffer, N.; Papadimitriou, E. Integrin α v β 3 Is a Pleiotrophin Receptor Required for Pleiotrophin-induced Endothelial Cell Migration through Receptor Protein Tyrosine Phosphatase β/ζ. FASEB J. 2009, 23, 1459–1469. [Google Scholar] [CrossRef] [PubMed]
- Koutsioumpa, M.; Polytarchou, C.; Courty, J.; Zhang, Y.; Kieffer, N.; Mikelis, C.; Skandalis, S.S.; Hellman, U.; Iliopoulos, D.; Papadimitriou, E. Interplay between Avβ3 Integrin and Nucleolin Regulates Human Endothelial and Glioma Cell Migration. J. Biol. Chem. 2013, 288, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Mikelis, C.; Lamprou, M.; Koutsioumpa, M.; Koutsioubas, A.G.; Spyranti, Z.; Zompra, A.A.; Spiliopoulos, N.; Vradis, A.A.; Katsoris, P.; Spyroulias, G.A.; et al. A Peptide Corresponding to the C-Terminal Region of Pleiotrophin Inhibits Angiogenesis in Vivo and in Vitro. J. Cell. Biochem. 2011, 112, 1532–1543. [Google Scholar] [CrossRef] [PubMed]
- Borst, A.J.; James, Z.M.; Zagotta, W.N.; Ginsberg, M.; Rey, F.A.; DiMaio, F.; Backovic, M.; Veesler, D. The Therapeutic Antibody LM609 Selectively Inhibits Ligand Binding to Human αVβ3 Integrin via Steric Hindrance. Structure 2017, 25, 1732–1739.e5. [Google Scholar] [CrossRef]
- McClain, C.R.; Sim, F.J.; Goldman, S.A. Pleiotrophin Suppression of Receptor Protein Tyrosine Phosphatase-/β/ζ Maintains the Self-Renewal Competence of Fetal Human Oligodendrocyte Progenitor Cells. J. Neurosci. 2012, 32, 15066–15075. [Google Scholar] [CrossRef]
- Liu, L.; Inoki, A.; Fan, K.; Mao, F.; Shi, G.; Jin, X.; Zhao, M.; Ney, G.; Jones, M.; Sun, S.; et al. ER-Associated Degradation Preserves Hematopoietic Stem Cell Quiescence and Self-Renewal by Restricting MTOR Activity. Blood 2020, 136, 2975–2986. [Google Scholar] [CrossRef]
- Himburg, H.A.; Harris, J.R.; Ito, T.; Daher, P.; Russell, J.L.; Quarmyne, M.; Doan, P.L.; Helms, K.; Nakamura, M.; Fixsen, E.; et al. Pleiotrophin Regulates the Retention and Self-Renewal of Hematopoietic Stem Cells in the Bone Marrow Vascular Niche. Cell Rep. 2012, 2, 964–975. [Google Scholar] [CrossRef]
- Soh, B.S.; Song, C.M.; Vallier, L.; Li, P.; Choong, C.; Yeo, B.H.; Lim, E.H.; Pedersen, R.A.; Yang, H.H.; Rao, M.; et al. Pleiotrophin Enhances Clonal Growth and Long-Term Expansion of Human Embryonic Stem Cells. Stem Cells 2007, 25, 3029–3037. [Google Scholar] [CrossRef]
- Easley, C.A.; Ben-Yehudah, A.; Redinger, C.J.; Oliver, S.L.; Varum, S.T.; Eisinger, V.M.; Carlisle, D.L.; Donovan, P.J.; Schatten, G.P. MTOR-Mediated Activation of P70 S6K Induces Differentiation of Pluripotent Human Embryonic Stem Cells. Cell. Reprogram. 2010, 12, 263–273. [Google Scholar] [CrossRef]
- Shi, Y.; Ping, Y.F.; Zhou, W.; He, Z.C.; Chen, C.; Bian, B.S.J.; Zhang, L.; Chen, L.; Lan, X.; Zhang, X.C.; et al. Tumour-Associated Macrophages Secrete Pleiotrophin to Promote PTPRZ1 Signalling in Glioblastoma Stem Cells for Tumour Growth. Nat. Commun. 2017, 8, 15080. [Google Scholar] [CrossRef]
- Zhou, W.; Liu, L.; Xue, Y.; Zheng, J.; Liu, X.; Ma, J.; Li, Z.; Liu, Y. Combination of Endothelial-Monocyte-Activating Polypeptide-Ii with Temozolomide Suppress Malignant Biological Behaviors of Human Glioblastoma Stem Cells via Mir-590-3p/Macc1 Inhibiting Pi3k/Akt/Mtor Signal Pathway. Front. Mol. Neurosci. 2017, 10, 68. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, Y.; Wang, L.; Li, M.; Yang, J.; Chen, P.; Zhu, J.; Li, X.; Zeng, Z.; Li, G.; et al. FOXA1 Prevents Nutrients Deprivation Induced Autophagic Cell Death through Inducing Loss of Imprinting of IGF2 in Lung Adenocarcinoma. Cell Death Dis. 2022, 13, 711. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, W.; Han, X.; Du, L.; Li, A.; Huang, G. Methyltransferase-like 1 Regulates Lung Adenocarcinoma A549 Cell Proliferation and Autophagy via the AKT/MTORC1 Signaling Pathway. Oncol. Lett. 2021, 21, 330. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Ren, W.; Zhang, Z.; Guan, J. DNA Repair/Recombination Protein 54L Promotes the Progression of Lung Adenocarcinoma by Activating MTORC1 Pathway. Hum. Cell 2023, 36, 421–433. [Google Scholar] [CrossRef]
- Yu, J.; Wu, C.; Wu, Q.; Huang, J.; Fu, W.; Xie, X.; Li, W.; Tang, W.; Xu, C.; Jin, G. PCYT1A Suppresses Proliferation and Migration via Inhibiting MTORC1 Pathway in Lung Adenocarcinoma. Biochem. Biophys. Res. Commun. 2020, 529, 353–361. [Google Scholar] [CrossRef]
- Liegl, R.; Koenig, S.; Siedlecki, J.; Haritoglou, C.; Kampik, A.; Kernt, M. Temsirolimus Inhibits Proliferation and Migration in Retinal Pigment Epithelial and Endothelial Cells via MTOR Inhibition and Decreases VEGF and PDGF Expression. PLoS ONE 2014, 9, e88203. [Google Scholar] [CrossRef]
- Nakao, T.; Shiota, M.; Tatemoto, Y.; Izumi, Y.; Iwao, H. Pravastatin Induces Rat Aortic Endothelial Cell Proliferation and Migration via Activation of PI3K/Akt/mTOR/P70 S6 Kinase Signaling. J. Pharmacol. Sci. 2007, 105, 334–341. [Google Scholar] [CrossRef]
- Chen, J.R.; Zhao, J.T.; Xie, Z.Z. Integrin-Mediated Cancer Progression as a Specific Target in Clinical Therapy. Biomed. Pharmacother. 2022, 155, 113745. [Google Scholar] [CrossRef]
- Liu, F.; Wu, Q.; Dong, Z.; Liu, K. Integrins in Cancer: Emerging Mechanisms and Therapeutic Opportunities. Pharmacol. Ther. 2023, 247, 108458. [Google Scholar] [CrossRef]
- Bergonzini, C.; Kroese, K.; Zweemer, A.J.M.; Danen, E.H.J. Targeting Integrins for Cancer Therapy—Disappointments and Opportunities. Front. Cell Dev. Biol. 2022, 10, 863850. [Google Scholar] [CrossRef]
- Dodd, K.M.; Yang, J.; Shen, M.H.; Sampson, J.R.; Tee, A.R. MTORC1 Drives HIF-1α and VEGF-A Signalling via Multiple Mechanisms Involving 4E-BP1, S6K1 and STAT3. Oncogene 2015, 34, 2239–2250. [Google Scholar] [CrossRef] [PubMed]
- Oberkersch, R.E.; Pontarin, G.; Astone, M.; Spizzotin, M.; Arslanbaeva, L.; Tosi, G.; Panieri, E.; Ricciardi, S.; Allega, M.F.; Brossa, A.; et al. Aspartate Metabolism in Endothelial Cells Activates the MTORC1 Pathway to Initiate Translation during Angiogenesis. Dev. Cell 2022, 57, 1241–1256. [Google Scholar] [CrossRef] [PubMed]
- Kawasome, H.; Papst, P.; Webb, S.; Keller, G.M.; Johnson, G.L.; Gelfand, E.W.; Terada, N. Targeted Disruption of P70s6k Defines Its Role in Protein Synthesis and Rapamycin Sensitivity. Proc. Natl. Acad. Sci. USA 1998, 95, 5033–5038. [Google Scholar] [CrossRef] [PubMed]
- Koutsioumpa, M.; Papadimitriou, E. Cell Surface Nucleolin as a Target for Anti-Cancer Therapies. Recent Pat. Anti-Cancer Drug Discov. 2014, 9, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Chalabi-Dchar, M.; Cruz, E.; Mertani, H.C.; Diaz, J.J.; Courty, J.; Cascone, I.; Bouvet, P. Nucleolin Aptamer N6l Reprograms the Translational Machinery and Acts Synergistically with Mtori to Inhibit Pancreatic Cancer Proliferation. Cancers 2021, 13, 4957. [Google Scholar] [CrossRef]
- Deshmukh, L.; Meller, N.; Alder, N.; Byzova, T.; Vinogradova, O. Tyrosine Phosphorylation as a Conformational Switch: A Case Study of Integrin β 3 Cytoplasmic Tail. J. Biol. Chem. 2011, 286, 40943–40953. [Google Scholar] [CrossRef]
- Schmidt, E.K.; Clavarino, G.; Ceppi, M.; Pierre, P. SUnSET, a Nonradioactive Method to Monitor Protein Synthesis. Nat. Methods 2009, 6, 275–277. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mourkogianni, E.; Karavasili, K.; Xanthopoulos, A.; Enake, M.-K.; Menounou, L.; Papadimitriou, E. Pleiotrophin Activates cMet- and mTORC1-Dependent Protein Synthesis through PTPRZ1—The Role of ανβ3 Integrin. Int. J. Mol. Sci. 2024, 25, 10839. https://doi.org/10.3390/ijms251910839
Mourkogianni E, Karavasili K, Xanthopoulos A, Enake M-K, Menounou L, Papadimitriou E. Pleiotrophin Activates cMet- and mTORC1-Dependent Protein Synthesis through PTPRZ1—The Role of ανβ3 Integrin. International Journal of Molecular Sciences. 2024; 25(19):10839. https://doi.org/10.3390/ijms251910839
Chicago/Turabian StyleMourkogianni, Eleni, Katerina Karavasili, Athanasios Xanthopoulos, Michaela-Karina Enake, Lydia Menounou, and Evangelia Papadimitriou. 2024. "Pleiotrophin Activates cMet- and mTORC1-Dependent Protein Synthesis through PTPRZ1—The Role of ανβ3 Integrin" International Journal of Molecular Sciences 25, no. 19: 10839. https://doi.org/10.3390/ijms251910839
APA StyleMourkogianni, E., Karavasili, K., Xanthopoulos, A., Enake, M.-K., Menounou, L., & Papadimitriou, E. (2024). Pleiotrophin Activates cMet- and mTORC1-Dependent Protein Synthesis through PTPRZ1—The Role of ανβ3 Integrin. International Journal of Molecular Sciences, 25(19), 10839. https://doi.org/10.3390/ijms251910839