
Chemokine Receptor N-Terminus Charge Dictates Reliance on Post-Translational Modifications for Effective Ligand Capture and Following Boosting by Defense Peptides

 , , , ,
, , , ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
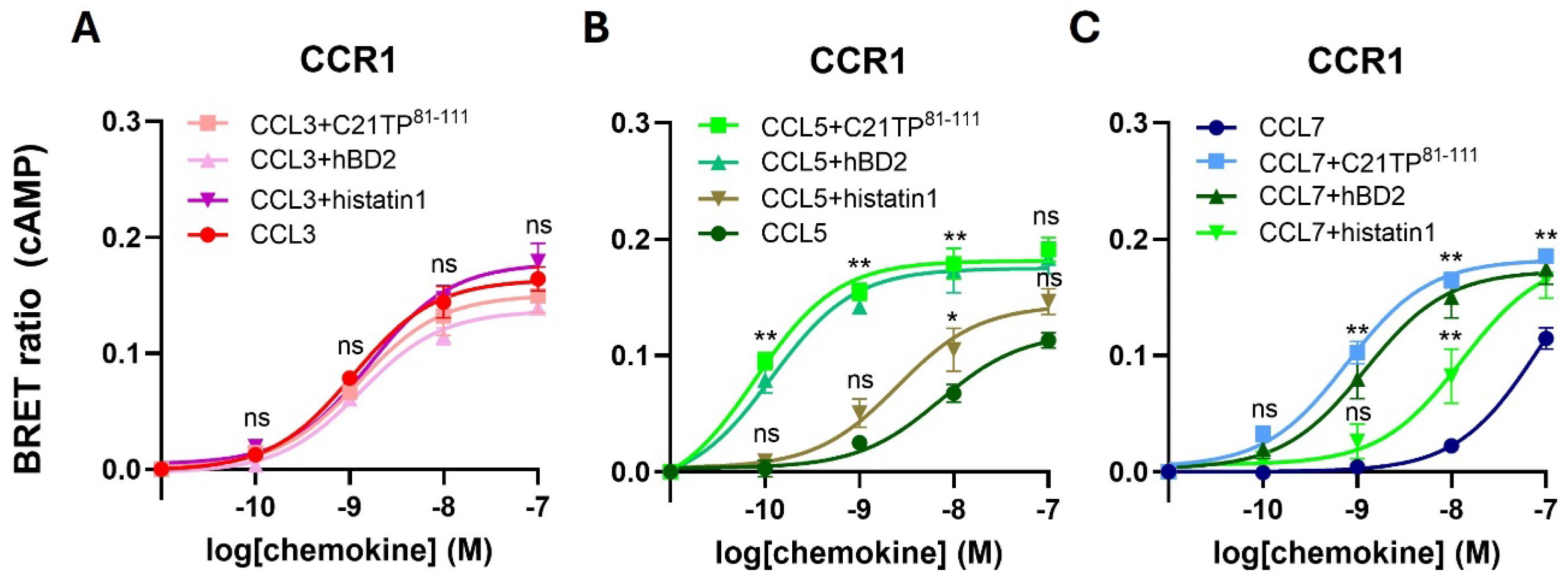
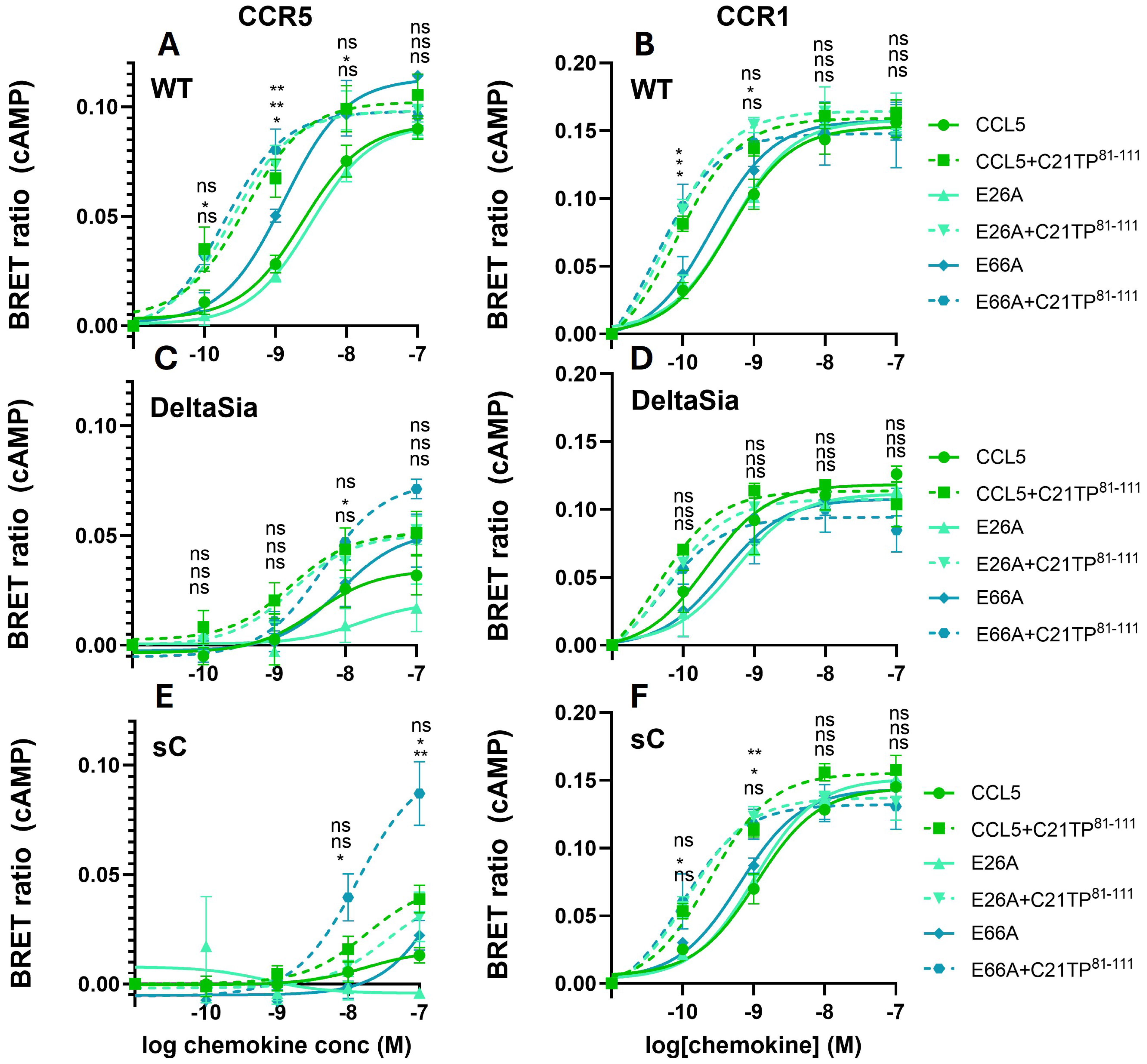
2.1. Basic Peptide Boosting of Ligand-Induced Signaling at CCR1 and CCR5
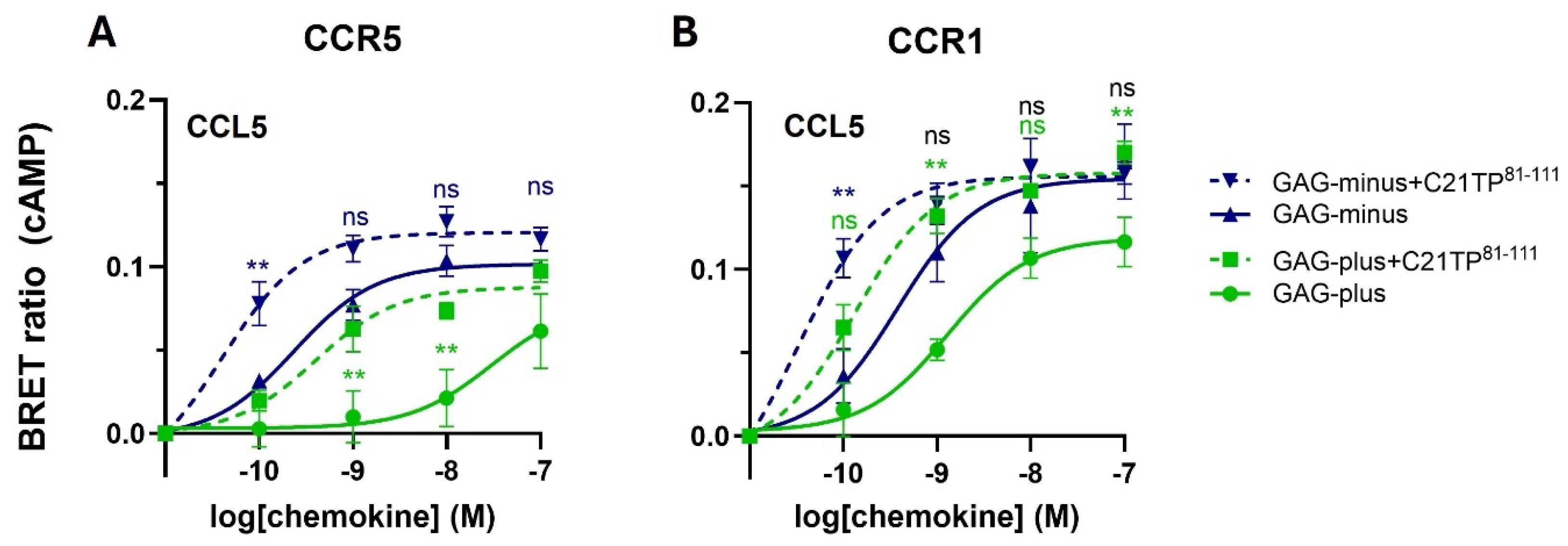
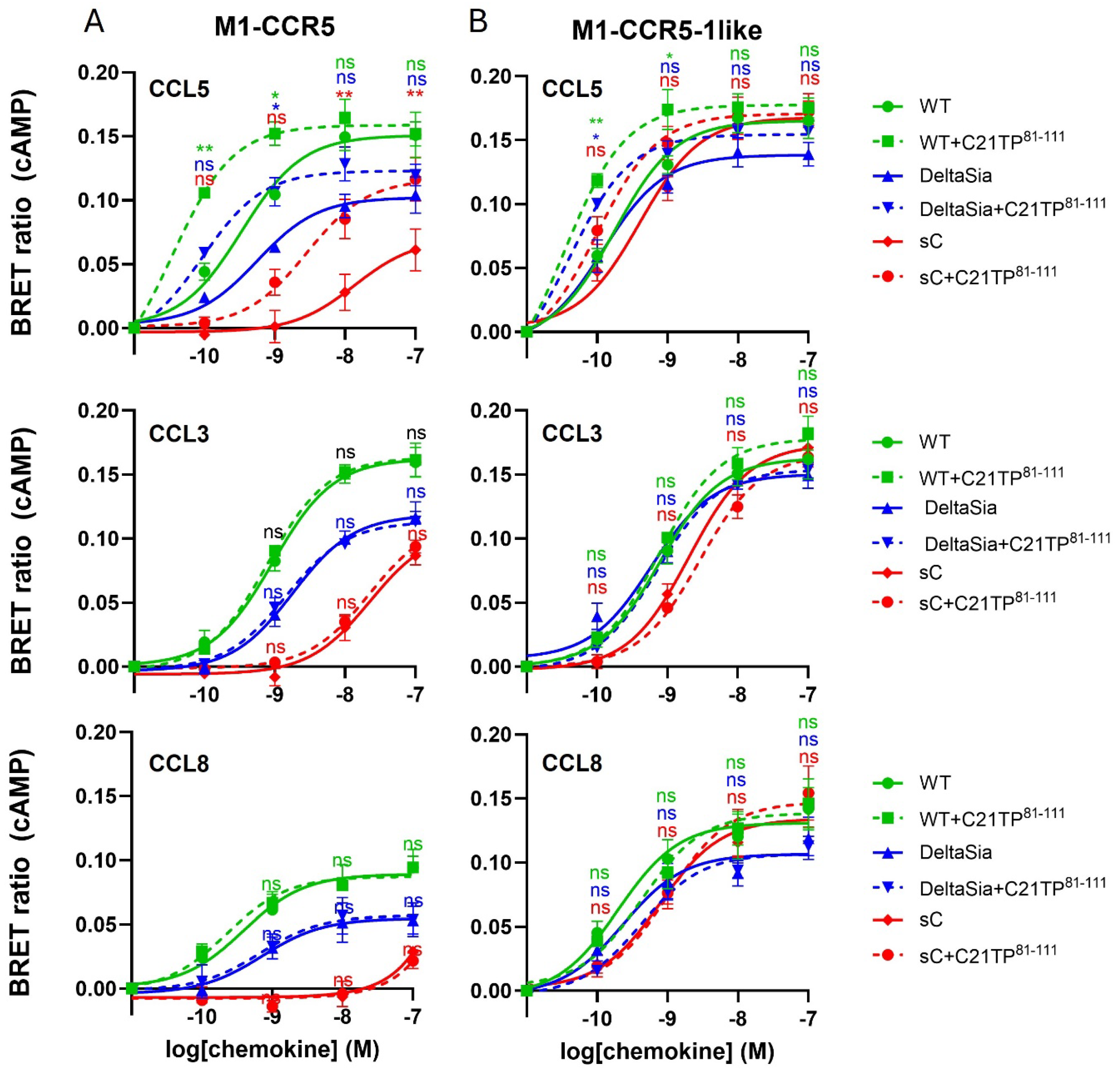
2.2. GAG Interference with Ligand-Induced Signaling at CCR5, Not at CCR1
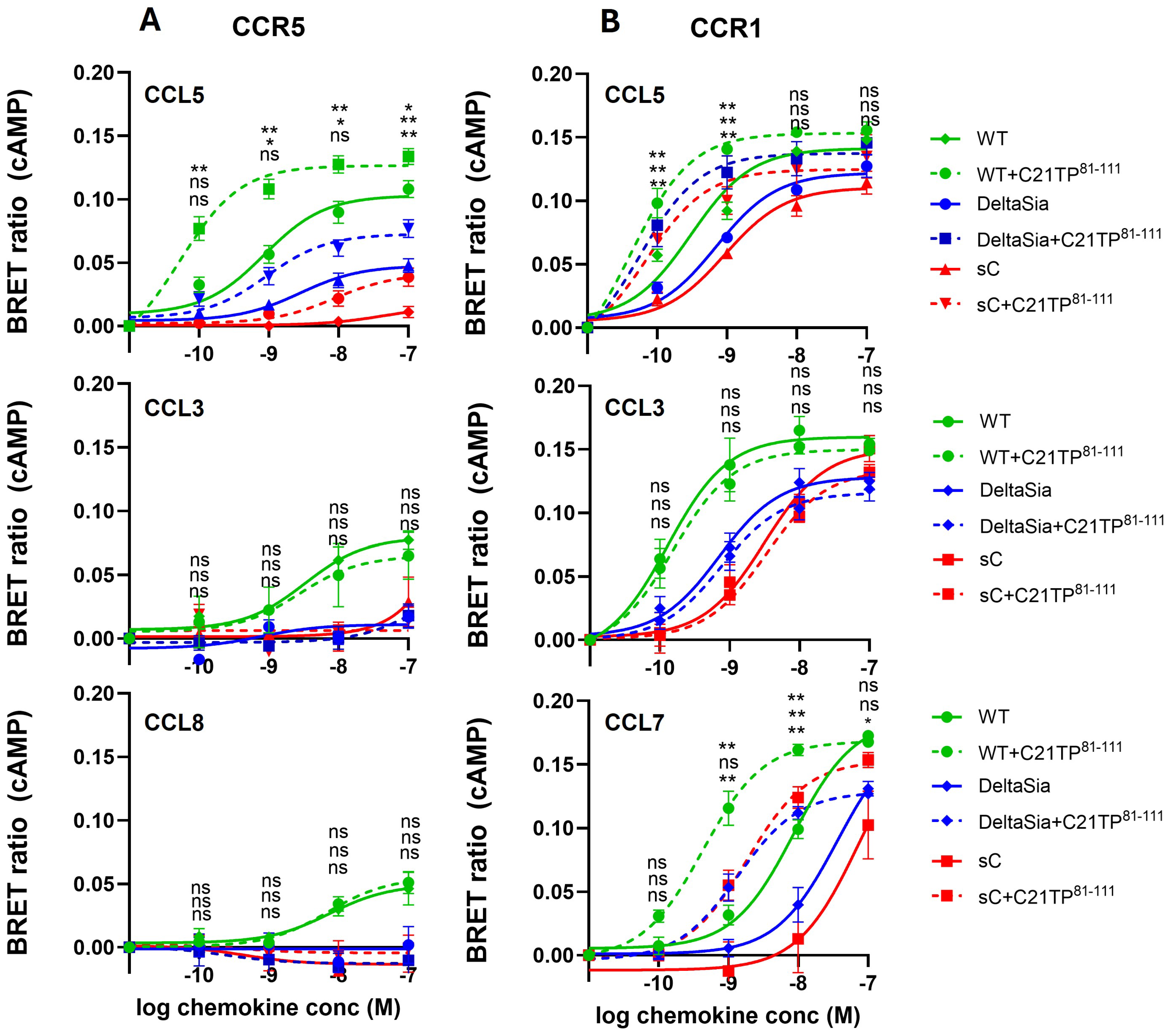
2.3. PTMS Are Needed for Ligand-Induced Signaling at CCR5, Not at CCR1
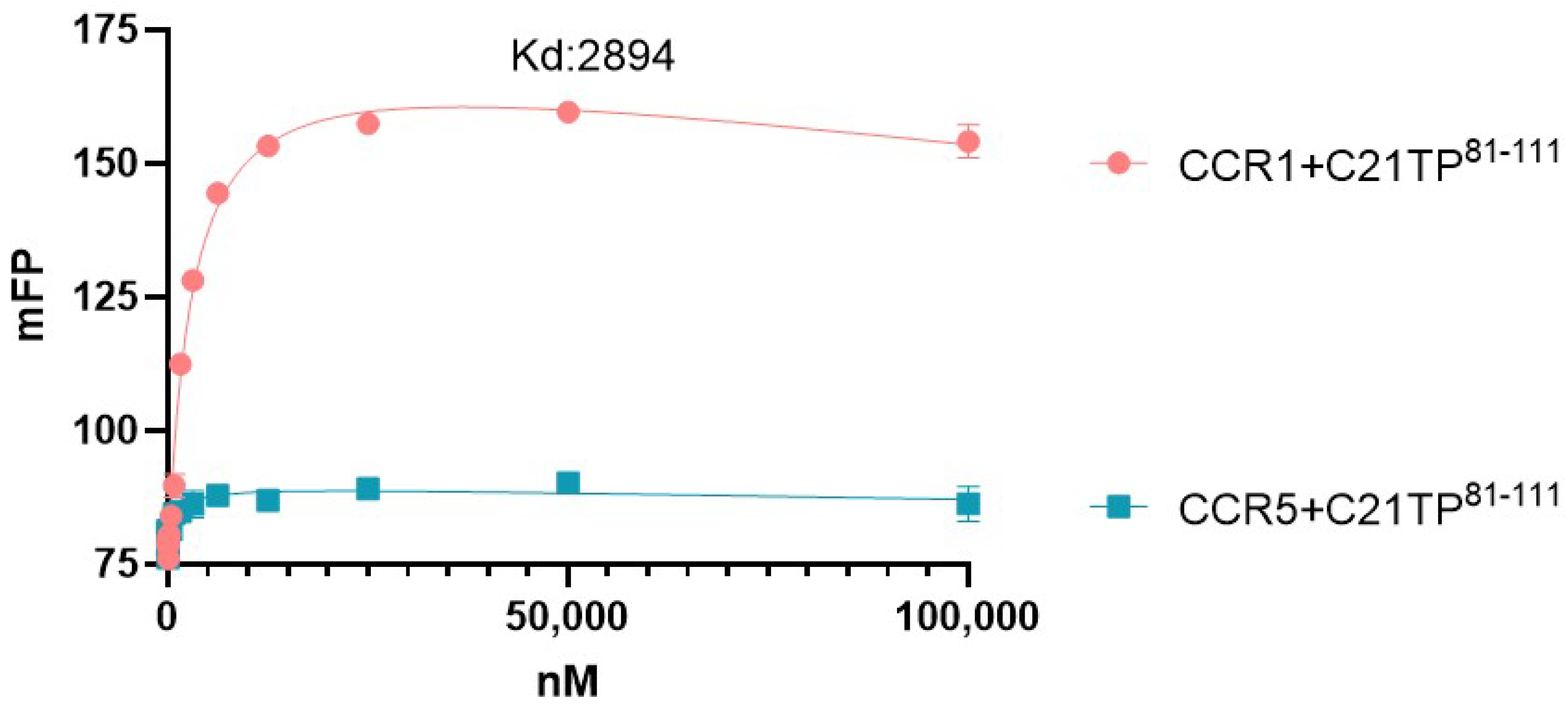
2.4. PTMs Are Needed for Peptide Interaction with CCR5, Not at CCR1 N-Terminus
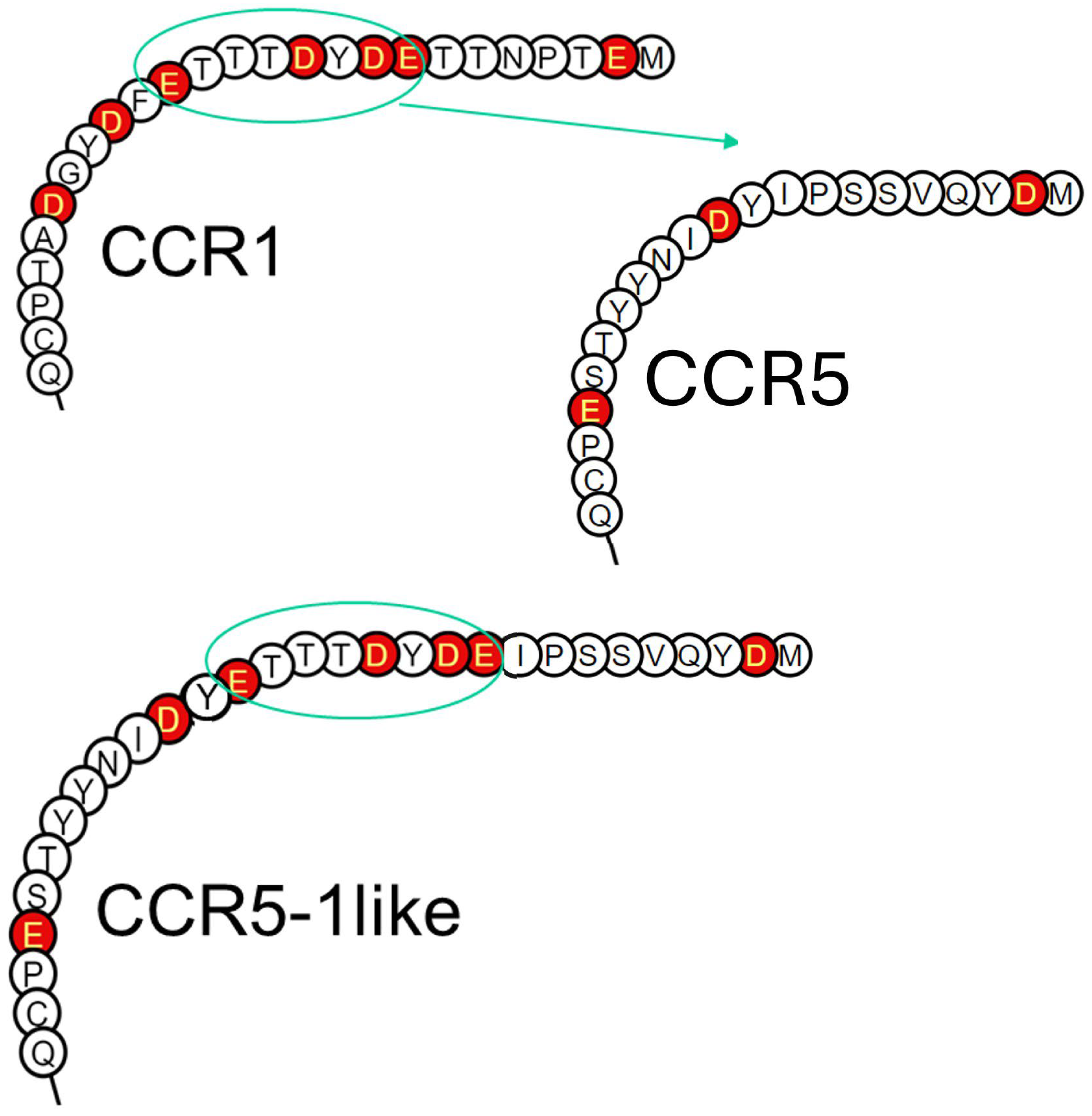
2.5. Inherent Charge of Receptor N-Terminus Dictates Dependence on PTMs for Ligand-Induced Signaling
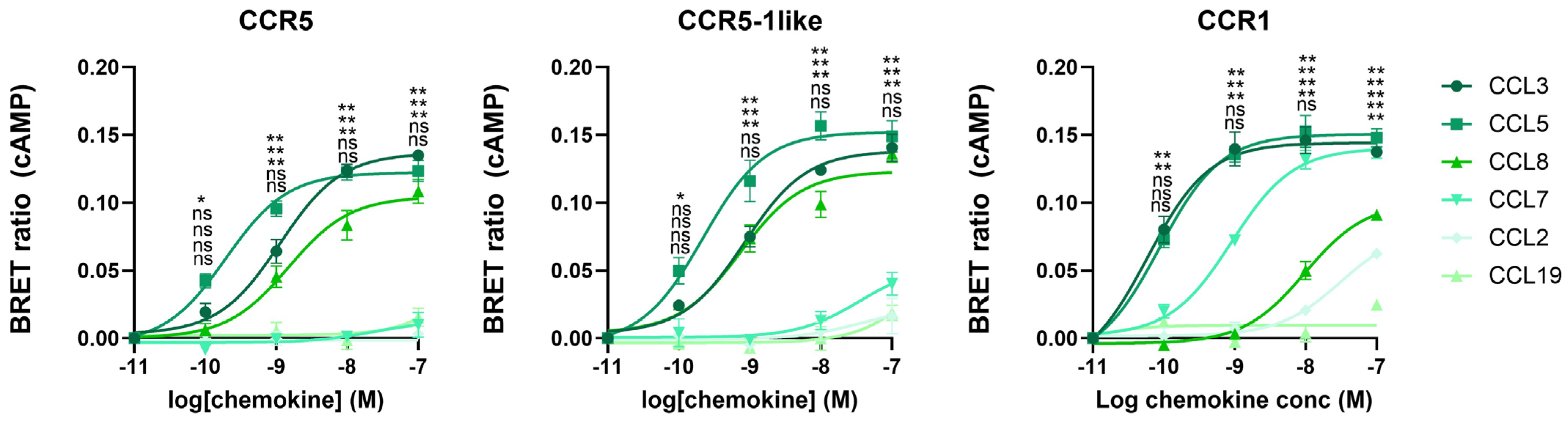
2.6. Receptor N-Terminus Serves to Capture Ligands, Not to Dictate Fruitful Ligand Docking and Receptor Activation
2.7. Boosting by Basic Peptides Is Independent of Ligand Oligomerization State
3. Discussion
3.1. Basic Peptides Boost the Action of Basic but Not Acidic Chemokines
3.2. Signaling via CCR5 Not CCR1 Is Inhibited by GAGs
3.3. CCR5 Is Highly Dependent on N-Terminal O-Glycosylation for Proper Signaling
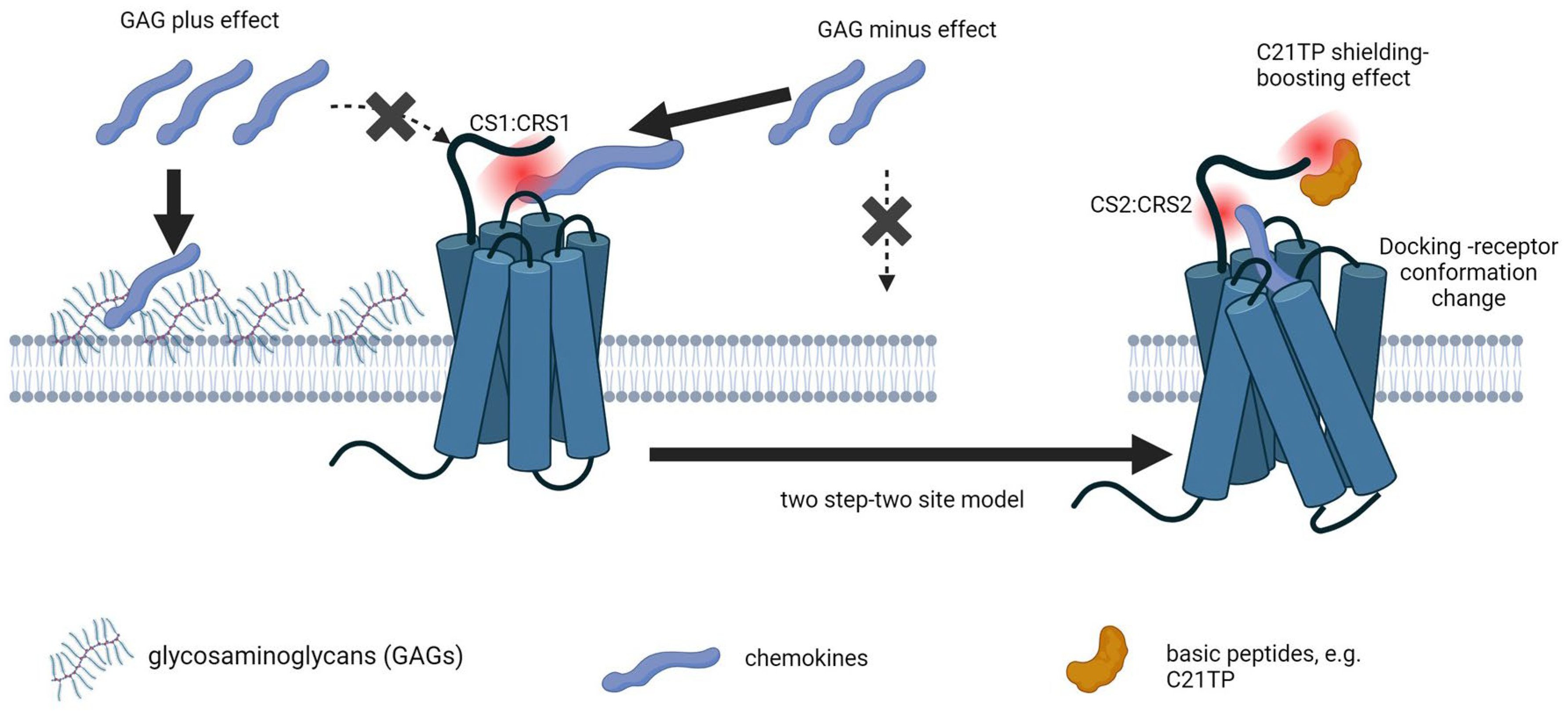
3.4. The Initial Contact between a Chemokine and Its Receptor Depends on Non-Selective Electrostatic Interactions Not Definable for Ligand Specificity
3.5. PTMs Could Add Another Layer in the Control of Chemokine Receptor Activity
3.6. Boosting Effect of Basic Peptides Is Not through a Disaggregating Effect on Chemokine Oligomers
4. Materials and Methods
- CCR1: (5,6-FAM)-QEDYDTTTEFDYGDATPCQKVNER,
- CCR5: (5,6-FAM)-ADYQVSSPIYDINYYTSEP.
4.1. Primers
- CCR5-S6A-FP: ATG GAT TAT CAA GTG GCA AGT CCA ATC TAT GAC;
- CCR5-S6A-RP: GTC ATA GAT TGG ACT TGC CAC TTG ATA ATC CAT;
- CCR5-S7A-FP: ATG GAT TAT CAA GTG TCA GCT CCA ATC TAT GAC;
- CCR5-S7A-RP: GTC ATA GAT TGG AGC TGA CAC TTG ATA ATC CAT;
- CCR5-S6S7AA-FP: ATG GAT TAT CAA GTG GCA GCT CCA ATC TAT GAC;
- CCR5-S6S7AA-RP: GTC ATA GAT TGG AGC TGC CAC TTG ATA ATC CAT;
- CCR5-T16S17AA-FP: GAC ATC AAT TAT TAT GCA GCG GAG CCC TGC CAA;
- CCR5-T16S17AA-RP: TTG GCA GGG CTC CGC TGC ATA ATA ATT GAT GTC.
- CCR5-Y10A-FP: GTG TCA AGT CCA ATC GCT GAC ATC AAT TAT TAT ACA TCG;
- CCR5-Y10A-RP: CGA TGT ATA ATA ATT GAT GTC AGC GAT TGG ACT TGA CAC;
- CCR5-Y14Y15AA-FP: GCA GGG CTC CGA TGT AGC AGC ATT GAT GTC ATA GAT;
- CCR5-Y14Y15AA-RP: ATC TAT GAC ATC AAT GCT GCT ACA TCG GAG CCC TGC.
4.2. N-Terminus Swap-In
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Bachelerie, F.; Ben-Baruch, A.; Burkhardt, A.M.; Combadiere, C.; Farber, J.M.; Graham, G.J.; Horuk, R.; Sparre-Ulrich, A.H.; Locati, M.; Luster, A.D.; et al. International Union of Basic and Clinical Pharmacology. [corrected]. LXXXIX. Update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharmacol. Rev. 2013, 66, 1–79. [Google Scholar] [CrossRef] [PubMed]
- Steen, A.; Larsen, O.; Thiele, S.; Rosenkilde, M.M. Biased and g protein-independent signaling of chemokine receptors. Front. Immunol. 2014, 5, 277. [Google Scholar] [CrossRef] [PubMed]
- Corbisier, J.; Gales, C.; Huszagh, A.; Parmentier, M.; Springael, J.Y. Biased signaling at chemokine receptors. J. Biol. Chem. 2015, 290, 9542–9554. [Google Scholar] [CrossRef] [PubMed]
- Metzemaekers, M.; Van, D.J.; Mortier, A.; Proost, P. Regulation of Chemokine Activity—A Focus on the Role of Dipeptidyl Peptidase IV/CD26. Front. Immunol. 2016, 7, 483. [Google Scholar] [CrossRef]
- Leach, K.; Charlton, S.J.; Strange, P.G. Analysis of second messenger pathways stimulated by different chemokines acting at the chemokine receptor CCR5. Biochem. Pharmacol. 2007, 74, 881–890. [Google Scholar] [CrossRef]
- Verhallen, L.; Lackman, J.J.; Wendt, R.; Gustavsson, M.; Yang, Z.; Narimatsu, Y.; Sørensen, D.M.; Lafferty, K.M.; Gouwy, M.; Marques, P.E.; et al. “Glyco-sulfo barcodes” regulate chemokine receptor function. Cell Mol. Life 2023, 80, 55. [Google Scholar] [CrossRef]
- Sanchez, J.; Lane, J.R.; Canals, M.; Stone, M.J. Influence of Chemokine N-Terminal Modification on Biased Agonism at the Chemokine Receptor CCR1. Int. J. Mol. Sci. 2019, 20, 2417. [Google Scholar] [CrossRef]
- Sanchez, J.; Huma, Z.E.; Lane, J.R.; Liu, X.; Bridgford, J.L.; Payne, R.J.; Canals, M.; Stone, M.J. Evaluation and extension of the two-site, two-step model for binding and activation of the chemokine receptor CCR1. J. Biol. Chem. 2019, 294, 3464–3475. [Google Scholar] [CrossRef]
- Verkaar, F.; Van Offenbeek, J.; Van Der Lee, M.M.C.; Van Lith, L.H.C.J.; Watts, A.O.; Rops, A.L.W.M.M.; Aguilar, D.C.; Ziarek, J.J.; Van Der Vlag, J.; Handel, T.M.; et al. Chemokine cooperativity is caused by competitive glycosaminoglycan binding. J. Immunol. 2014, 192, 3908–3914. [Google Scholar] [CrossRef]
- de Paz, J.L.; Moseman, E.A.; Noti, C.; Polito, L.; von Andrian, U.H.; Seeberger, P.H. Profiling heparin-chemokine interactions using synthetic tools. ACS Chem. Biol. 2007, 2, 735–744. [Google Scholar] [CrossRef]
- Patel, D.D.; Koopmann, W.; Imai, T.; Whichard, L.P.; Yoshie, O.; Krangel, M.S. Chemokines have diverse abilities to form solid phase gradients. Clin. Immunol. 2001, 99, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Crump, M.P.; Gong, J.H.; Loetscher, P.; Rajarathnam, K.; Amara, A.; Arenzana-Seisdedos, F.; Virelizier, J.L.; Baggiolini, M.; Sykes, B.D.; Clark-Lewis, I. Solution structure and basis for functional activity of stromal cell-derived factor-1; dissociation of CXCR4 activation from binding and inhibition of HIV-1. EMBO 1997, 16, 6996–7007. [Google Scholar] [CrossRef]
- Hancock, R.E.; Haney, E.F.; Gill, E.E. The immunology of host defence peptides: Beyond antimicrobial activity. Nat. Rev. Immunol. 2016, 16, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.Y.; Destoumieux, D.; Wong, A.V.; Park, C.H.; Valore, E.V.; Liu, L.; Ganz, T. Human beta-defensin-2 production in keratinocytes is regulated by interleukin-1, bacteria, and the state of differentiation. J. Investig. Dermatol. 2002, 118, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Schumann, K.; Lämmermann, T.; Bruckner, M.; Legler, D.F.; Polleux, J.; Spatz, J.P.; Schuler, G.; Förster, R.; Lutz, M.B.; Sorokin, L.; et al. Immobilized chemokine fields and soluble chemokine gradients cooperatively shape migration patterns of dendritic cells. Immunity 2010, 32, 703–713. [Google Scholar] [CrossRef]
- Lorenz, N.; Loef, E.J.; Kelch, I.D.; Verdon, D.J.; Black, M.M.; Middleditch, M.J.; Greenwood, D.R.; Graham, E.S.; Brooks, A.E.; Dunbar, P.R.; et al. Plasmin and regulators of plasmin activity control the migratory capacity and adhesion of human T cells and dendritic cells by regulating cleavage of the chemokine CCL21. Immunol. Cell Biol. 2016, 94, 955–963. [Google Scholar] [CrossRef]
- Loef, E.J.; Sheppard, H.M.; Birch, N.P.; Dunbar, P.R. Plasminogen and plasmin can bind to human T cells and generate truncated CCL21 that increases dendritic cell chemotactic responses. J. Biol. Chem. 2022, 298, 102112. [Google Scholar] [CrossRef]
- Hauser, M.A.; Kindinger, I.; Laufer, J.M.; Späte, A.-K.; Bucher, D.; Vanes, S.L.; Krueger, W.A.; Wittmann, V.; Legler, D.F. Distinct CCR7 glycosylation pattern shapes receptor signaling and endocytosis to modulate chemotactic responses. J. Leukoc. Biol. 2016, 99, 993–1007. [Google Scholar] [CrossRef]
- Jørgensen, A.S.; Brandum, E.P.; Mikkelsen, J.M.; Orfin, K.A.; Boilesen, D.R.; Egerod, K.L.; Moussouras, N.A.; Vilhardt, F.; Kalinski, P.; Basse, P.; et al. The C-terminal peptide of CCL21 drastically augments CCL21 activity through the dendritic cell lymph node homing receptor CCR7 by interaction with the receptor N-terminus. Cell Mol. Life Sci. 2021, 78, 6963–6978. [Google Scholar] [CrossRef]
- Brandum, E.P.; Jørgensen, A.S.; Calvo, M.B.; Spiess, K.; Peterson, F.C.; Yang, Z.; Volkman, B.F.; Veldkamp, C.T.; Rosenkilde, M.M.; Goth, C.K.; et al. Selective Boosting of CCR7-Acting Chemokines; Short Peptides Boost Chemokines with Short Basic Tails, Longer Peptides Boost Chemokines with Long Basic Tails. Int. J. Mol. Sci. 2022, 23, 1397. [Google Scholar] [CrossRef]
- Kiermaier, E.; Moussion, C.; Veldkamp, C.T.; Gerardy-Schahn, R.; De Vries, I.; Williams, L.G.; Chaffee, G.R.; Phillips, A.J.; Freiberger, F.; Imre, R.; et al. Polysialylation controls dendritic cell trafficking by regulating chemokine recognition. Science 2016, 351, 186–190. [Google Scholar] [CrossRef] [PubMed]
- Rey-Gallardo, A.; Delgado-Martin, C.; Gerardy-Schahn, R.; Rodriguez-Fernandez, J.L.; Vega, M.A. Polysialic acid is required for neuropilin-2a/b-mediated control of CCL21-driven chemotaxis of mature dendritic cells and for their migration in vivo. Glycobiology 2011, 21, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Rey-Gallardo, A.; Escribano, C.; Delgado-Martin, C.; Rodriguez-Fernandez, J.L.; Gerardy-Schahn, R.; Rutishauser, U.; Corbi, A.L.; Vega, M.A. Polysialylated neuropilin-2 enhances human dendritic cell migration through the basic C-terminal region of CCL21. Glycobiology 2010, 20, 1139–1146. [Google Scholar] [CrossRef] [PubMed]
- Goth, C.K.; Petaja-Repo, U.E.; Rosenkilde, M.M. G Protein-Coupled Receptors in the Sweet Spot: Glycosylation and other Post-translational Modifications. ACS Pharmacol. Transl. Sci. 2020, 3, 237–245. [Google Scholar] [CrossRef]
- Bannert, N.; Craig, S.; Farzan, M.; Sogah, D.; Santo, N.V.; Choe, H.; Sodroski, J. Sialylated O-glycans and sulfated tyrosines in the NH2-terminal domain of CC chemokine receptor 5 contribute to high affinity binding of chemokines. J. Exp. Med. 2001, 194, 1661–1674. [Google Scholar] [CrossRef]
- Kessler, N.; Akabayov, S.R.; Moseri, A.; Cohen, L.S.; Sakhapov, D.; Bolton, D.; Fridman, B.; Kay, L.E.; Naider, F.; Anglister, J. Allovalency observed by transferred NOE: Interactions of sulfated tyrosine residues in the N-terminal segment of CCR5 with the CCL5 chemokine. FEBS J. 2020, 288, 1648–1663. [Google Scholar] [CrossRef]
- Hemmerich, S.; Paavola, C.; Bloom, A.; Bhakta, S.; Freedman, R.; Grunberger, D.; Krstenansky, J.; Lee, S.; McCarley, D.; Mulkins, M.; et al. Identification of residues in the monocyte chemotactic protein-1 that contact the MCP-1 receptor, CCR2. Biochemistry 1999, 38, 13013–13025. [Google Scholar] [CrossRef]
- Jarnagin, K.; Grunberger, D.; Mulkins, M.; Wong, B.; Hemmerich, S.; Paavola, C.; Bloom, A.; Bhakta, S.; Diehl, F.; Freedman, R.; et al. Identification of surface residues of the monocyte chemotactic protein 1 that affect signaling through the receptor CCR2. Biochemistry 1999, 38, 16167–16177. [Google Scholar] [CrossRef]
- Tan, J.H.Y.; Ludeman, J.P.; Wedderburn, J.; Canals, M.; Hall, P.; Butler, S.J.; Taleski, D.; Christopoulos, A.; Hickey, M.J.; Payne, R.J.; et al. Tyrosine sulfation of chemokine receptor CCR2 enhances interactions with both monomeric and dimeric forms of the chemokine monocyte chemoattractant protein-1 (MCP-1). J. Biol. Chem. 2013, 288, 10024–10034. [Google Scholar] [CrossRef]
- Ludeman, J.P.; Stone, M.J. The structural role of receptor tyrosine sulfation in chemokine recognition. Brit. J. Pharmacol. 2014, 171, 1167–1179. [Google Scholar] [CrossRef]
- Blanpain, C.; Doranz, B.J.; Vakili, J.; Rucker, J.; Govaerts, C.; Baik, S.S.W.; Lorthioir, O.; Migeotte, I.; Libert, F.; Baleux, F.; et al. Multiple charged and aromatic residues in CCR5 amino-terminal domain are involved in high affinity binding of both chemokines and HIV-1 Env protein. J. Biol. Chem. 1999, 274, 34719–34727. [Google Scholar] [CrossRef] [PubMed]
- Farzan, M.; Chung, S.; Li, W.; Vasilieva, N.; Wright, P.L.; Schnitzler, C.E.; Marchione, R.J.; Gerard, C.; Gerard, N.P.; Sodroski, J.; et al. Tyrosine-sulfated peptides functionally reconstitute a CCR5 variant lacking a critical amino-terminal region. J. Biol. Chem. 2002, 277, 40397–40402. [Google Scholar] [CrossRef] [PubMed]
- Bax, M.; van Vliet, S.J.; Litjens, M.; Garcia-Vallejo, J.J.; van Kooyk, Y. Interaction of polysialic acid with CCL21 regulates the migratory capacity of human dendritic cells. PLoS ONE 2009, 4, e6987. [Google Scholar] [CrossRef] [PubMed]
- Sallusto, F.; Schaerli, P.; Loetscher, P.; Schaniel, C.; Lenig, D.; Mackay, C.R.; Qin, S.; Lanzavecchia, A. Rapid and coordinated switch in chemokine receptor expression during dendritic cell maturation. Eur. J. Immunol. 1998, 28, 2760–2769. [Google Scholar] [CrossRef]
- Sallusto, F.; Kremmer, E.; Palermo, B.; Hoy, A.; Ponath, P.; Qin, S.; Förster, R.; Lipp, M.; Lanzavecchia, A. Switch in chemokine receptor expression upon TCR stimulation reveals novel homing potential for recently activated T cells. Eur. J. Immunol. 1999, 29, 2037–2045. [Google Scholar] [CrossRef]
- Villanueva-Cabello, T.M.; Gutierrez-Valenzuela, L.D.; Lopez-Guerrero, D.V.; Cruz-Munoz, M.E.; Mora-Montes, H.M.; Martinez-Duncker, I. Polysialic acid is expressed in human naive CD4+ T cells and is involved in modulating activation. Glycobiology 2019, 29, 557–564. [Google Scholar] [CrossRef]
- Dyer, D.P.; Salanga, C.L.; Volkman, B.F.; Kawamura, T.; Handel, T.M. The dependence of chemokine-glycosaminoglycan interactions on chemokine oligomerization. Glycobiology 2015, 26, 312–326. [Google Scholar] [CrossRef]
- Wang, X.; Watson, C.; Sharp, J.S.; Handel, T.M.; Prestegard, J.H. Oligomeric structure of the chemokine CCL5/RANTES from NMR, MS, and SAXS data. Structure 2011, 19, 1138–1148. [Google Scholar] [CrossRef]
- Wang, X.; Sharp, J.S.; Handel, T.M.; Prestegard, J.H. Chemokine oligomerization in cell signaling and migration. Prog. Mol. Biol. Transl. Sci. 2013, 117, 531–578. [Google Scholar] [CrossRef]
- Esko, J.D.; Weinke, J.L.; Taylor, W.H.; Ekborg, G.; Roden, L.; Anantharamaiah, G.; Gawish, A. Inhibition of chondroitin and heparan sulfate biosynthesis in Chinese hamster ovary cell mutants defective in galactosyltransferase I. J. Biol. Chem. 1987, 262, 12189–12195. [Google Scholar] [CrossRef]
- Jiang, L.I.; Collins, J.; Davis, R.; Lin, K.M.; DeCamp, D.; Roach, T.; Hsueh, R.; Rebres, R.A.; Ross, E.M.; Taussig, R.; et al. Use of a cAMP BRET sensor to characterize a novel regulation of cAMP by the sphingosine 1-phosphate/G13 pathway. J. Biol. Chem. 2007, 282, 10576–10584. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, T.; Schou, A.S.; Lackman, J.J.; Barrio-Calvo, M.; Verhallen, L.; Goth, C.K.; Jensen, B.A.H.; Veldkamp, C.T.; Volkman, B.F.; Peterson, F.C.; et al. Chemokine Receptor N-Terminus Charge Dictates Reliance on Post-Translational Modifications for Effective Ligand Capture and Following Boosting by Defense Peptides. Int. J. Mol. Sci. 2024, 25, 10854. https://doi.org/10.3390/ijms251910854
Xu T, Schou AS, Lackman JJ, Barrio-Calvo M, Verhallen L, Goth CK, Jensen BAH, Veldkamp CT, Volkman BF, Peterson FC, et al. Chemokine Receptor N-Terminus Charge Dictates Reliance on Post-Translational Modifications for Effective Ligand Capture and Following Boosting by Defense Peptides. International Journal of Molecular Sciences. 2024; 25(19):10854. https://doi.org/10.3390/ijms251910854
Chicago/Turabian StyleXu, Ting, Anne Sophie Schou, Jarkko J. Lackman, Marina Barrio-Calvo, Lisa Verhallen, Christoffer Knak Goth, Benjamin Anderschou Holbech Jensen, Christopher T. Veldkamp, Brian F. Volkman, Francis C. Peterson, and et al. 2024. "Chemokine Receptor N-Terminus Charge Dictates Reliance on Post-Translational Modifications for Effective Ligand Capture and Following Boosting by Defense Peptides" International Journal of Molecular Sciences 25, no. 19: 10854. https://doi.org/10.3390/ijms251910854
APA StyleXu, T., Schou, A. S., Lackman, J. J., Barrio-Calvo, M., Verhallen, L., Goth, C. K., Jensen, B. A. H., Veldkamp, C. T., Volkman, B. F., Peterson, F. C., & Hjortø, G. M. (2024). Chemokine Receptor N-Terminus Charge Dictates Reliance on Post-Translational Modifications for Effective Ligand Capture and Following Boosting by Defense Peptides. International Journal of Molecular Sciences, 25(19), 10854. https://doi.org/10.3390/ijms251910854