
Dengue Envelope Protein as a Cytotoxic Factor Inducing Hemorrhage and Endothelial Cell Death in Mice


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
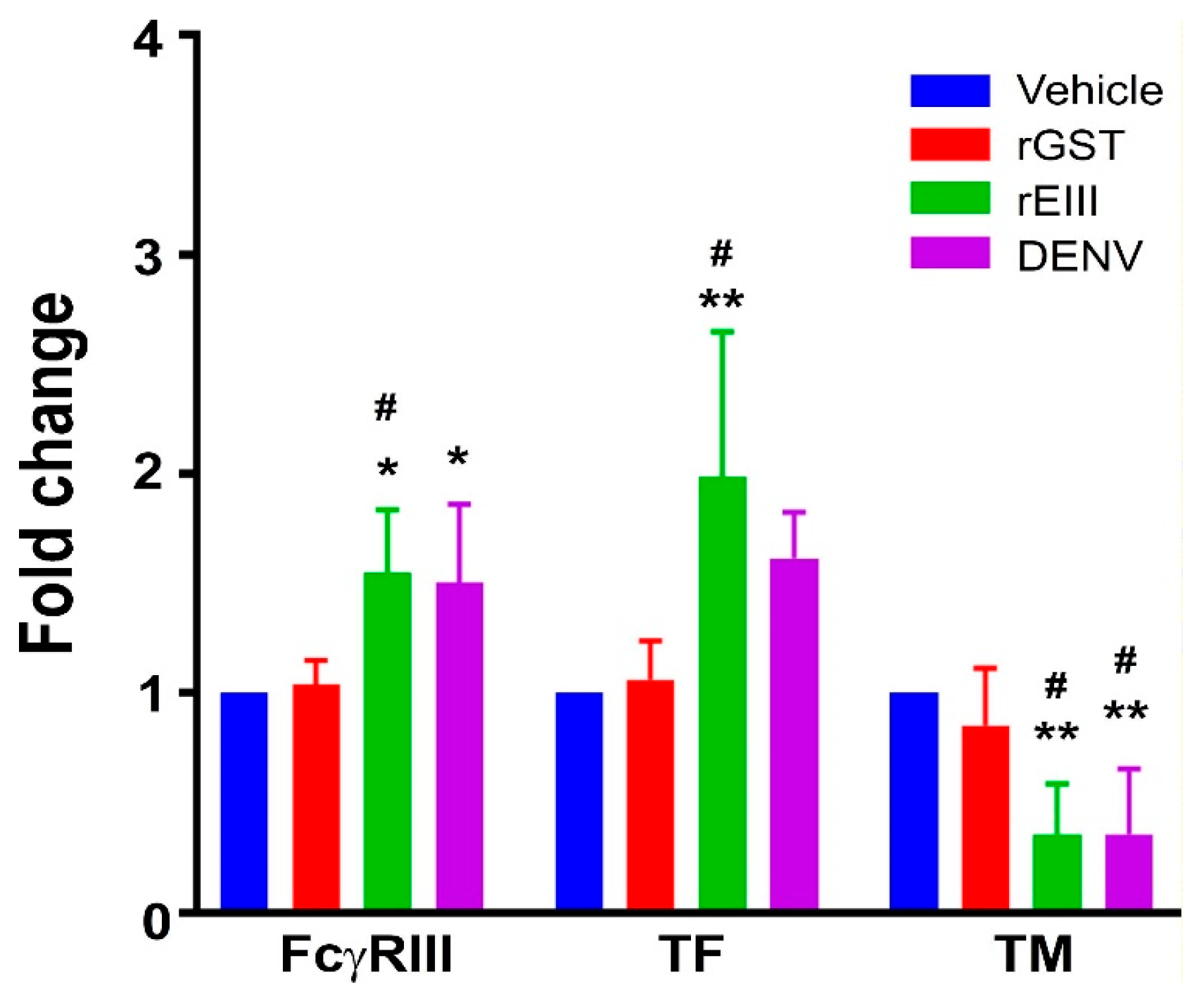
2.1. Treatments of Recombinant EIII Protein and Virion of DENV Markedly Induced Elevation of Endothelial Inflammation and Damage Markers in HMEC-1 Endothelial Cells
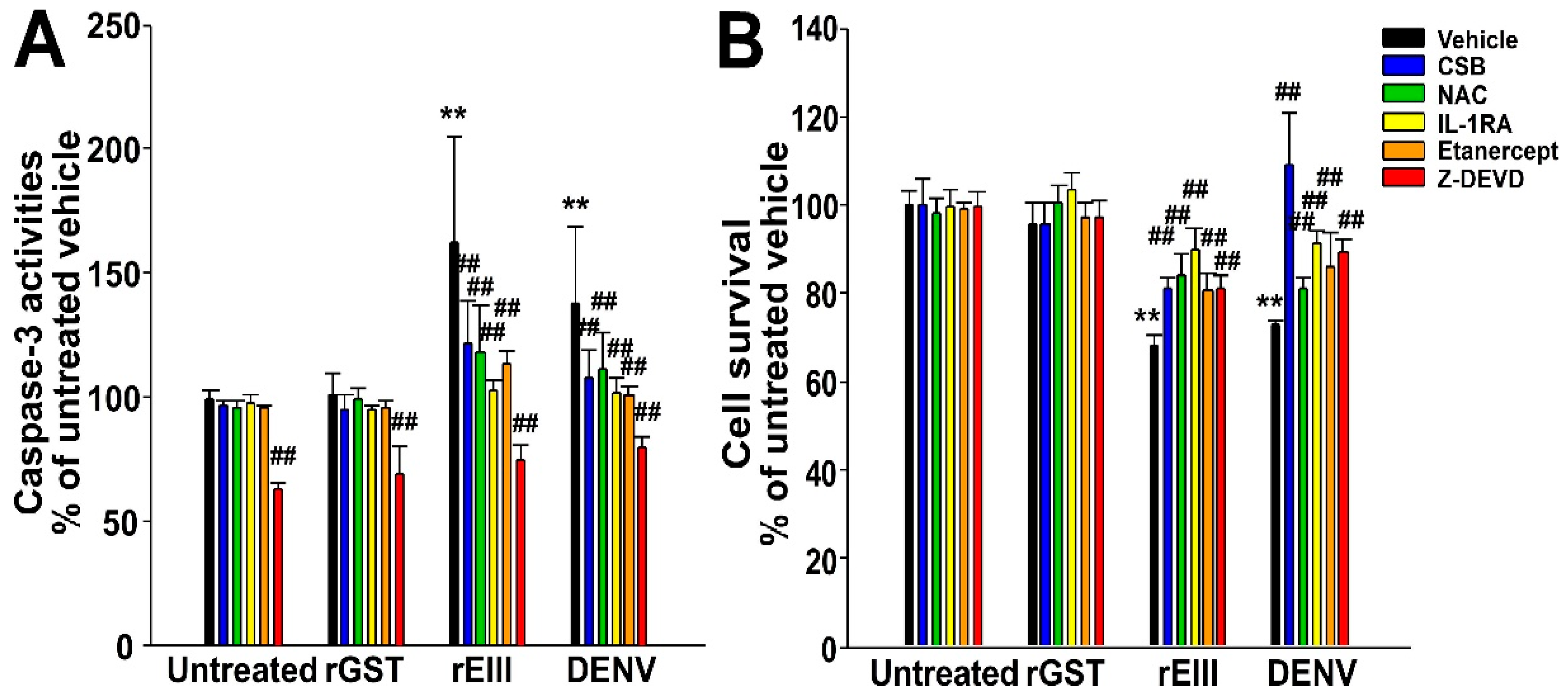
2.2. Treatments of Recombinant EIII Protein and Virion of DENV Markedly Induced Caspase-3-Associated Cell Death in HMEC-1 Endothelial Cells
2.3. Treatment with Recombinant EIII Protein and Anti-Platelet Ig in a Mouse Model Markedly Induced Caspase-3-Associated Damage in the Vascular Endothelial Cells of Hemorrhage Lesions
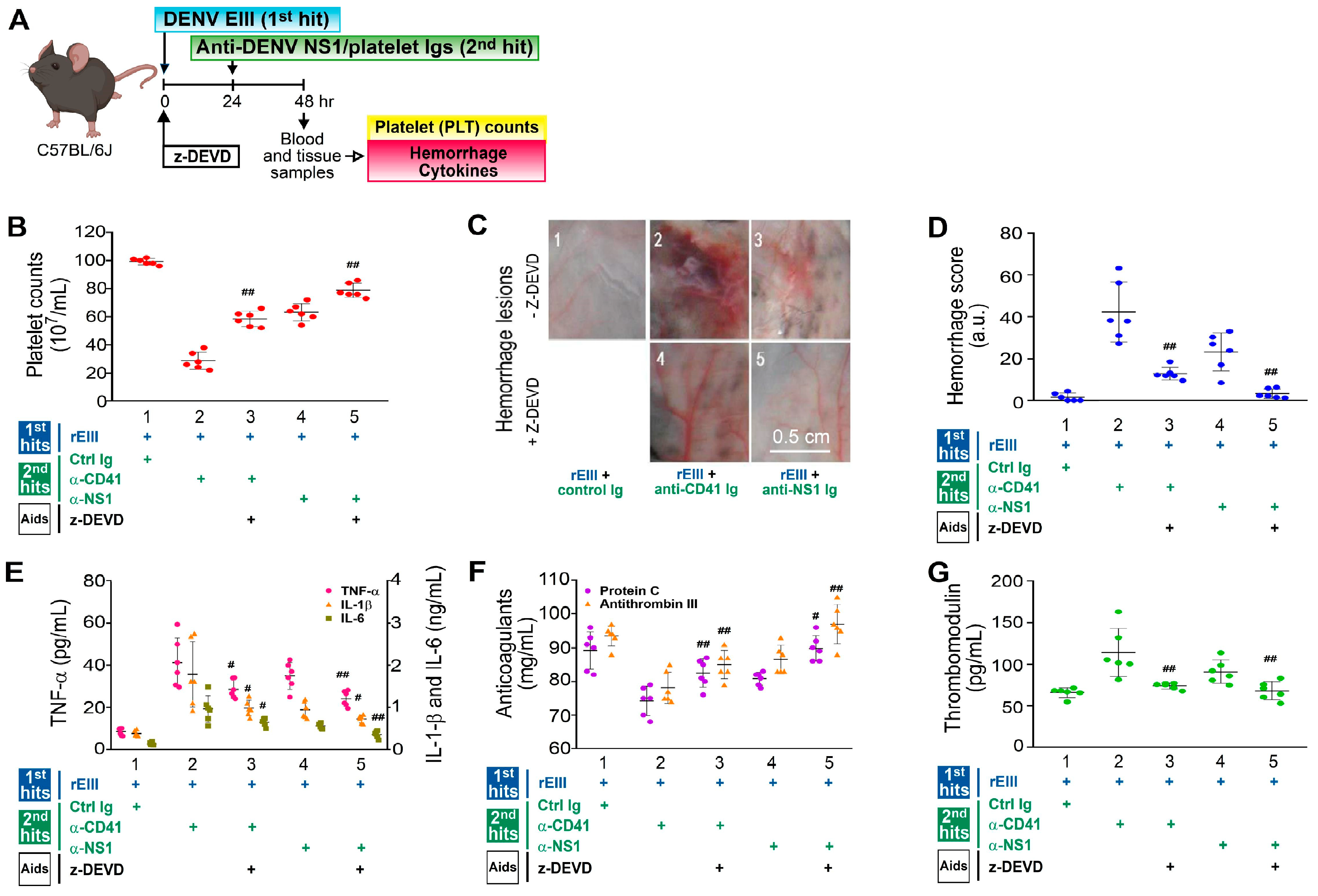
2.4. Treatment with Caspase-3 Inhibitor z-DEVD Markedly Mitigated Hemorrhage-Associated Pathogenesis Induced by the Recombinant EIII Protein and Anti-Platelet Ig Challenges in Mice
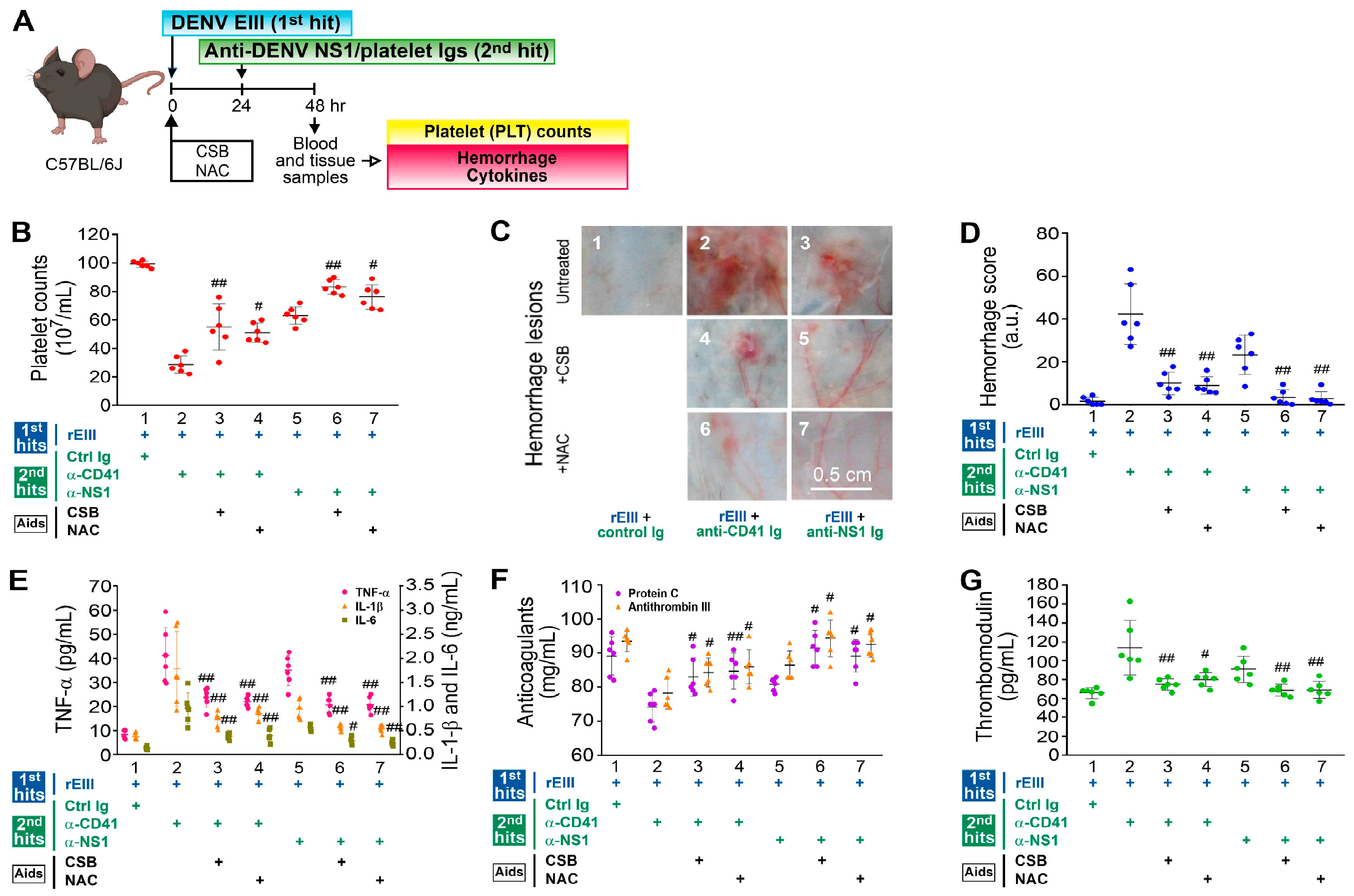
2.5. Treatment with CSB and NAC Markedly Rescued Hemorrhage-Associated Pathogenesis Induced by Recombinant EIII Protein and Anti-Platelet Ig Treatments in Mice
3. Discussion
4. Materials and Methods
4.1. DENV, Cell Lines, Recombinant Proteins, and Antibodies
4.2. HMEC-1 Endothelial Cell Experiments
4.3. Laboratory Mice
4.4. Induction of a Local Shwartzman Reaction-like Response with Dual Challenges Using rEIII and Anti-Platelet Ig
4.5. Quantification of Hemorrhage Severity Using Digitized Images
4.6. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pang, T.; Mak, T.K.; Gubler, D.J. Prevention and control of dengue-the light at the end of the tunnel. Lancet Infect. Dis. 2017, 17, e79–e87. [Google Scholar] [CrossRef] [PubMed]
- Gubler, D.J. Dengue and dengue hemorrhagic fever. Clin. Microbiol. Rev. 1998, 11, 480–496. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Bansal, D.; Patil, S.; Pandya, S.; Ilyas, Q.M.; Imran, S. A Retrospective Study of Climate Change Affecting Dengue: Evidences, Challenges and Future Directions. Front. Public Health 2022, 10, 884645. [Google Scholar] [CrossRef] [PubMed]
- Colon-Gonzalez, F.J.; Fezzi, C.; Lake, I.R.; Hunter, P.R. The effects of weather and climate change on dengue. PLoS Negl. Trop. Dis. 2013, 7, e2503. [Google Scholar] [CrossRef]
- Jansen, C.C.; Beebe, N.W. The dengue vector Aedes aegypti: What comes next. Microbes Infect. 2010, 12, 272–279. [Google Scholar] [CrossRef]
- Lai, Y.H. The climatic factors affecting dengue fever outbreaks in southern Taiwan: An application of symbolic data analysis. Biomed. Eng. Online 2018, 17, 148. [Google Scholar] [CrossRef]
- Lopez, M.S.; Gomez, A.A.; Muller, G.V.; Walker, E.; Robert, M.A.; Estallo, E.L. Relationship between Climate Variables and Dengue Incidence in Argentina. Environ. Health Perspect. 2023, 131, 57008. [Google Scholar] [CrossRef]
- Susilawaty, A.; Ekasari, R.; Widiastuty, L.; Wijaya, D.R.; Arranury, Z.; Basri, S. Climate factors and dengue fever occurrence in Makassar during period of 2011–2017. Gac. Sanit. 2021, 35 (Suppl. 2), S408–S412. [Google Scholar] [CrossRef]
- Wong, J.M.; Rivera, A.; Volkman, H.R.; Torres-Velasquez, B.; Rodriguez, D.M.; Paz-Bailey, G.; Adams, L.E. Travel-Associated Dengue Cases—United States, 2010–2021. Mmwr. Morb. Mortal. Wkly. Rep. 2023, 72, 821–826. [Google Scholar] [CrossRef]
- Xu, C.; Xu, J.; Wang, L. Long-term effects of climate factors on dengue fever over a 40-year period. BMC Public Health 2024, 24, 1451. [Google Scholar] [CrossRef]
- Srikiatkhachorn, A.; Kelley, J.F. Endothelial cells in dengue hemorrhagic fever. Antivir. Res. 2014, 109, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Durbin, A.P. Dengue vascular leak syndrome: Insights into potentially new treatment modalities. J. Clin. Investig. 2019, 129, 4072–4073. [Google Scholar] [CrossRef] [PubMed]
- Spiropoulou, C.F.; Srikiatkhachorn, A. The role of endothelial activation in dengue hemorrhagic fever and hantavirus pulmonary syndrome. Virulence 2013, 4, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Srikiatkhachorn, A.; Ajariyakhajorn, C.; Endy, T.P.; Kalayanarooj, S.; Libraty, D.H.; Green, S.; Ennis, F.A.; Rothman, A.L. Virus-induced decline in soluble vascular endothelial growth receptor 2 is associated with plasma leakage in dengue hemorrhagic Fever. J. Virol. 2007, 81, 1592–1600. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Chaturvedi, U.C. Vascular endothelium: The battlefield of dengue viruses. FEMS Immunol. Med. Microbiol. 2008, 53, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Chunhakan, S.; Butthep, P.; Yoksan, S.; Tangnararatchakit, K.; Chuansumrit, A. Vascular leakage in dengue hemorrhagic Fever is associated with dengue infected monocytes, monocyte activation/exhaustion, and cytokines production. Int. J. Vasc. Med. 2015, 2015, 917143. [Google Scholar] [CrossRef]
- Liu, P.; Woda, M.; Ennis, F.A.; Libraty, D.H. Dengue virus infection differentially regulates endothelial barrier function over time through type I interferon effects. J. Infect. Dis. 2009, 200, 191–201. [Google Scholar] [CrossRef]
- Yacoub, S.; Wertheim, H.; Simmons, C.P.; Screaton, G.; Wills, B. Cardiovascular manifestations of the emerging dengue pandemic. Nat. Rev. Cardiol. 2014, 11, 335–345. [Google Scholar] [CrossRef]
- Chuansumrit, A.; Chaiyaratana, W. Hemostatic derangement in dengue hemorrhagic fever. Thromb. Res. 2014, 133, 10–16. [Google Scholar] [CrossRef]
- Avirutnan, P.; Punyadee, N.; Noisakran, S.; Komoltri, C.; Thiemmeca, S.; Auethavornanan, K.; Jairungsri, A.; Kanlaya, R.; Tangthawornchaikul, N.; Puttikhunt, C.; et al. Vascular leakage in severe dengue virus infections: A potential role for the nonstructural viral protein NS1 and complement. J. Infect. Dis. 2006, 193, 1078–1088. [Google Scholar] [CrossRef]
- Kelley, J.F.; Kaufusi, P.H.; Nerurkar, V.R. Dengue hemorrhagic fever-associated immunomediators induced via maturation of dengue virus nonstructural 4B protein in monocytes modulate endothelial cell adhesion molecules and human microvascular endothelial cells permeability. Virology 2012, 422, 326–337. [Google Scholar] [CrossRef] [PubMed]
- Malavige, G.N.; Ogg, G.S. Pathogenesis of vascular leak in dengue virus infection. Immunology 2017, 151, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Vervaeke, P.; Vermeire, K.; Liekens, S. Endothelial dysfunction in dengue virus pathology. Rev. Med. Virol. 2015, 25, 50–67. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Maguire, T.; Hileman, R.E.; Fromm, J.R.; Esko, J.D.; Linhardt, R.J.; Marks, R.M. Dengue virus infectivity depends on envelope protein binding to target cell heparan sulfate. Nat. Med. 1997, 3, 866–871. [Google Scholar] [CrossRef] [PubMed]
- Lien, T.S.; Sun, D.S.; Wu, C.Y.; Chang, H.H. Exposure to Dengue Envelope Protein Domain III Induces Nlrp3 Inflammasome-Dependent Endothelial Dysfunction and Hemorrhage in Mice. Front. Immunol. 2021, 12, 617251. [Google Scholar] [CrossRef]
- Lien, T.S.; Sun, D.S.; Hung, S.C.; Wu, W.S.; Chang, H.H. Dengue Virus Envelope Protein Domain III Induces Nlrp3 Inflammasome-Dependent NETosis-Mediated Inflammation in Mice. Front. Immunol. 2021, 12, 618577. [Google Scholar] [CrossRef]
- Lien, T.S.; Chan, H.; Sun, D.S.; Wu, J.C.; Lin, Y.Y.; Lin, G.L.; Chang, H.H. Exposure of Platelets to Dengue Virus and Envelope Protein Domain III Induces Nlrp3 Inflammasome-Dependent Platelet Cell Death and Thrombocytopenia in Mice. Front. Immunol. 2021, 12, 616394. [Google Scholar] [CrossRef]
- Vo, H.T.M.; Duong, V.; Ly, S.; Li, Q.Z.; Dussart, P.; Cantaert, T. Autoantibody Profiling in Plasma of Dengue Virus-Infected Individuals. Pathogens 2020, 9, 60. [Google Scholar] [CrossRef]
- Wan, S.W.; Lin, C.F.; Yeh, T.M.; Liu, C.C.; Liu, H.S.; Wang, S.; Ling, P.; Anderson, R.; Lei, H.Y.; Lin, Y.S. Autoimmunity in dengue pathogenesis. J. Formos. Med. Assoc. 2013, 112, 3–11. [Google Scholar] [CrossRef]
- Ghorai, T.; Sarkar, A.; Roy, A.; Bhowmick, B.; Nayak, D.; Das, S. Role of auto-antibodies in the mechanisms of dengue pathogenesis and its progression: A comprehensive review. Arch. Microbiol. 2024, 206, 214. [Google Scholar] [CrossRef]
- Sun, D.S.; King, C.C.; Huang, H.S.; Shih, Y.L.; Lee, C.C.; Tsai, W.J.; Yu, C.C.; Chang, H.H. Antiplatelet autoantibodies elicited by dengue virus non-structural protein 1 cause thrombocytopenia and mortality in mice. J. Thromb. Haemost. 2007, 5, 2291–2299. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.F.; Lei, H.Y.; Liu, C.C.; Liu, H.S.; Yeh, T.M.; Wang, S.T.; Yang, T.I.; Sheu, F.C.; Kuo, C.F.; Lin, Y.S. Generation of IgM anti-platelet autoantibody in dengue patients. J. Med. Virol. 2001, 63, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Lien, T.S.; Sun, D.S.; Wu, W.S.; Chang, H.H. Simulation of Hemorrhage Pathogenesis in Mice through Dual Stimulation with Dengue Envelope Protein Domain III-Coated Nanoparticles and Antiplatelet Antibody. Int. J. Mol. Sci. 2023, 24, 9270. [Google Scholar] [CrossRef] [PubMed]
- Lei, H.Y.; Yeh, T.M.; Liu, H.S.; Lin, Y.S.; Chen, S.H.; Liu, C.C. Immunopathogenesis of dengue virus infection. J. Biomed. Sci. 2001, 8, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Lien, T.S.; Sun, D.S.; Chang, C.M.; Wu, C.Y.; Dai, M.S.; Chan, H.; Wu, W.S.; Su, S.H.; Lin, Y.Y.; Chang, H.H. Dengue virus and antiplatelet autoantibodies synergistically induce haemorrhage through Nlrp3-inflammasome and FcgammaRIII. Thromb. Haemost. 2015, 113, 1060–1070. [Google Scholar] [CrossRef]
- Lin, Y.S.; Yeh, T.M.; Lin, C.F.; Wan, S.W.; Chuang, Y.C.; Hsu, T.K.; Liu, H.S.; Liu, C.C.; Anderson, R.; Lei, H.Y. Molecular mimicry between virus and host and its implications for dengue disease pathogenesis. Exp. Biol. Med. 2011, 236, 515–523. [Google Scholar] [CrossRef]
- Sun, D.S.; Chang, Y.C.; Lien, T.S.; King, C.C.; Shih, Y.L.; Huang, H.S.; Wang, T.Y.; Li, C.R.; Lee, C.C.; Hsu, P.N.; et al. Endothelial Cell Sensitization by Death Receptor Fractions of an Anti-Dengue Nonstructural Protein 1 Antibody Induced Plasma Leakage, Coagulopathy, and Mortality in Mice. J. Immunol. 2015, 195, 2743–2753. [Google Scholar] [CrossRef]
- Tsai, C.L.; Sun, D.S.; Su, M.T.; Lien, T.S.; Chen, Y.H.; Lin, C.Y.; Huang, C.H.; King, C.C.; Li, C.R.; Chen, T.H.; et al. Suppressed humoral immunity is associated with dengue nonstructural protein NS1-elicited anti-death receptor antibody fractions in mice. Sci. Rep. 2020, 10, 6294. [Google Scholar] [CrossRef]
- Halstead, S.B. Dengue. Lancet 2007, 370, 1644–1652. [Google Scholar] [CrossRef]
- Watanabe-Kusunoki, K.; Nakazawa, D.; Ishizu, A.; Atsumi, T. Thrombomodulin as a Physiological Modulator of Intravascular Injury. Front. Immunol. 2020, 11, 575890. [Google Scholar] [CrossRef]
- Remkova, A.; Kovacova, E.; Prikazska, M.; Kratochvil’ova, H. Thrombomodulin as a marker of endothelium damage in some clinical conditions. Eur. J. Intern. Med. 2000, 11, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, G.; Cirillo, P. Tissue factor: Newer concepts in thrombosis and its role beyond thrombosis and hemostasis. Cardiovasc. Diagn. Ther. 2018, 8, 581–593. [Google Scholar] [CrossRef]
- Pan, L.F.; Kreisle, R.A.; Shi, Y.D. Detection of Fcgamma receptors on human endothelial cells stimulated with cytokines tumour necrosis factor-alpha (TNF-alpha) and interferon-gamma (IFN-gamma). Clin. Exp. Immunol. 1998, 112, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Zille, M.; Ikhsan, M.; Jiang, Y.; Lampe, J.; Wenzel, J.; Schwaninger, M. The impact of endothelial cell death in the brain and its role after stroke: A systematic review. Cell Stress. 2019, 3, 330–347. [Google Scholar] [CrossRef] [PubMed]
- White, K.; Dempsie, Y.; Caruso, P.; Wallace, E.; McDonald, R.A.; Stevens, H.; Hatley, M.E.; Van Rooij, E.; Morrell, N.W.; MacLean, M.R.; et al. Endothelial apoptosis in pulmonary hypertension is controlled by a microRNA/programmed cell death 4/caspase-3 axis. Hypertension 2014, 64, 185–194. [Google Scholar] [CrossRef]
- Meuren, L.M.; Prestes, E.B.; Papa, M.P.; de Carvalho, L.R.P.; Mustafa, Y.M.; da Costa, L.S.; Da Poian, A.T.; Bozza, M.T.; Arruda, L.B. Infection of Endothelial Cells by Dengue Virus Induces ROS Production by Different Sources Affecting Virus Replication, Cellular Activation, Death and Vascular Permeability. Front. Immunol. 2022, 13, 810376. [Google Scholar] [CrossRef]
- Dhani, S.; Zhao, Y.; Zhivotovsky, B. A long way to go: Caspase inhibitors in clinical use. Cell Death Dis. 2021, 12, 949. [Google Scholar] [CrossRef]
- Shiri, R.; Yari, F.; Ahmadinejad, M.; Vaeli, S.; Tabatabaei, M.R. The caspase-3 inhibitor (peptide Z-DEVD-FMK) affects the survival and function of platelets in platelet concentrate during storage. Blood Res. 2014, 49, 49–53. [Google Scholar] [CrossRef]
- Josic Dominovic, P.; Dobrivojevic Radmilovic, M.; Srakocic, S.; Miseric, I.; Skokic, S.; Gajovic, S. Validation and application of caged Z-DEVD-aminoluciferin bioluminescence for assessment of apoptosis of wild type and TLR2-deficient mice after ischemic stroke. J. Photochem. Photobiol. B 2024, 253, 112871. [Google Scholar] [CrossRef]
- Unnisa, A.; Greig, N.H.; Kamal, M.A. Inhibition of Caspase 3 and Caspase 9 Mediated Apoptosis: A Multimodal Therapeutic Target in Traumatic Brain Injury. Curr. Neuropharmacol. 2023, 21, 1001–1012. [Google Scholar] [CrossRef]
- Huang, H.S.; Sun, D.S.; Lien, T.S.; Chang, H.H. Dendritic cells modulate platelet activity in IVIg-mediated amelioration of ITP in mice. Blood 2010, 116, 5002–5009. [Google Scholar] [CrossRef] [PubMed]
- WHO. Dengue: Guidelines for Diagnosis, Treatment, Prevention and Control, New ed.; World Health Organization: Geneva, Switzerland, 2009; Chapters 1 and 2; pp. 11–28. [Google Scholar]
- WHO. Dengue Haemorrhagic Fever: Diagnosis, Treatment, Prevention and Control, 2nd ed.; World Health Organization: Geneva, Switzerland, 1997; Chapter 2; pp. 12–23. [Google Scholar]
- Iyngkaran, N.; Yadav, M.; Sinniah, M. Augmented inflammatory cytokines in primary dengue infection progressing to shock. Singap. Med. J. 1995, 36, 218–221. [Google Scholar]
- Coronel-Ruiz, C.; Gutierrez-Barbosa, H.; Medina-Moreno, S.; Velandia-Romero, M.L.; Chua, J.V.; Castellanos, J.E.; Zapata, J.C. Humanized Mice in Dengue Research: A Comparison with Other Mouse Models. Vaccines 2020, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Zellweger, R.M.; Shresta, S. Mouse models to study dengue virus immunology and pathogenesis. Front. Immunol. 2014, 5, 151. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L.; Verlaeten, O.; Cabie, A.; Kaidomar, S.; Moravie, V.; Martial, J.; Najioullah, F.; Plumelle, Y.; Fonteau, C.; Dussart, P.; et al. Influence of the dengue serotype, previous dengue infection, and plasma viral load on clinical presentation and outcome during a dengue-2 and dengue-4 co-epidemic. Am. J. Trop. Med. Hyg. 2008, 78, 990–998. [Google Scholar] [CrossRef]
- Brozna, J.P. Shwartzman reaction. Semin. Thromb. Hemost. 1990, 16, 326–332. [Google Scholar] [CrossRef]
- Chahin, A.B.; Opal, J.M.; Opal, S.M. Whatever happened to the Shwartzman phenomenon? Innate Immun. 2018, 24, 466–479. [Google Scholar] [CrossRef]
- Aguillon, J.C.; Ferreira, V.; Nunez, E.; Paredes, L.; Molina, M.C.; Colombo, A.; Hermosilla, T.; Ferreira, A. Immunomodulation of LPS ability to induce the local Shwartzman reaction. Scand. J. Immunol. 1996, 44, 551–555. [Google Scholar] [CrossRef]
- Calderon-Pelaez, M.A.; Coronel-Ruiz, C.; Castellanos, J.E.; Velandia-Romero, M.L. Endothelial Dysfunction, HMGB1, and Dengue: An Enigma to Solve. Viruses 2022, 14, 1765. [Google Scholar] [CrossRef]
- Lin, C.F.; Lei, H.Y.; Shiau, A.L.; Liu, H.S.; Yeh, T.M.; Chen, S.H.; Liu, C.C.; Chiu, S.C.; Lin, Y.S. Endothelial cell apoptosis induced by antibodies against dengue virus nonstructural protein 1 via production of nitric oxide. J. Immunol. 2002, 169, 657–664. [Google Scholar] [CrossRef]
- Avirutnan, P.; Malasit, P.; Seliger, B.; Bhakdi, S.; Husmann, M. Dengue virus infection of human endothelial cells leads to chemokine production, complement activation, and apoptosis. J. Immunol. 1998, 161, 6338–6346. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.L.; Lin, C.F.; Wan, S.W.; Wei, L.S.; Chen, M.C.; Yeh, T.M.; Liu, H.S.; Anderson, R.; Lin, Y.S. Anti-dengue virus nonstructural protein 1 antibodies cause NO-mediated endothelial cell apoptosis via ceramide-regulated glycogen synthase kinase-3beta and NF-kappaB activation. J. Immunol. 2013, 191, 1744–1752. [Google Scholar] [CrossRef] [PubMed]
- Long, X.; Li, Y.; Qi, Y.; Xu, J.; Wang, Z.; Zhang, X.; Zhang, D.; Zhang, L.; Huang, J. XAF1 contributes to dengue virus-induced apoptosis in vascular endothelial cells. FASEB J. 2013, 27, 1062–1073. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Li, Y.; Zhang, Y.; Zhang, L.; Wang, Z.; Zhang, X.; Gui, L.; Huang, J. IFI6 Inhibits Apoptosis via Mitochondrial-Dependent Pathway in Dengue Virus 2 Infected Vascular Endothelial Cells. PLoS ONE 2015, 10, e0132743. [Google Scholar] [CrossRef]
- Dalrymple, N.A.; Mackow, E.R. Roles for endothelial cells in dengue virus infection. Adv. Virol. 2012, 2012, 840654. [Google Scholar] [CrossRef]
- Limonta, D.; Capo, V.; Torres, G.; Perez, A.B.; Guzman, M.G. Apoptosis in tissues from fatal dengue shock syndrome. J. Clin. Virol. 2007, 40, 50–54. [Google Scholar] [CrossRef]
- Lee, S.H.; Kim, J.Y.; Yoo, S.Y.; Kwon, S.M. Cytoprotective effect of dieckol on human endothelial progenitor cells (hEPCs) from oxidative stress-induced apoptosis. Free Radic. Res. 2013, 47, 526–534. [Google Scholar] [CrossRef]
- Warren, M.C.; Bump, E.A.; Medeiros, D.; Braunhut, S.J. Oxidative stress-induced apoptosis of endothelial cells. Free Radic. Biol. Med. 2000, 29, 537–547. [Google Scholar] [CrossRef]
- Chang, H.H.; Shyu, H.F.; Wang, Y.M.; Sun, D.S.; Shyu, R.H.; Tang, S.S.; Huang, Y.S. Facilitation of Cell Adhesion by Immobilized Dengue Viral Nonstructural Protein 1 (NS1): Arginine-Glycine-Aspartic Acid Structural Mimicry within the Dengue Viral NS1 Antigen. J. Infect. Dis. 2002, 186, 743–751. [Google Scholar] [CrossRef]
- Chang, H.H.; Kau, J.H.; Lo, S.J.; Sun, D.S. Cell-adhesion and morphological changes are not sufficient to support anchorage-dependent cell growth via non-integrin-mediated attachment. Cell Biol. Int. 2003, 27, 123–133. [Google Scholar] [CrossRef]
- Chuang, D.J.; Pethaperumal, S.; Siwakoti, B.; Chien, H.J.; Cheng, C.F.; Hung, S.C.; Lien, T.S.; Sun, D.S.; Chang, H.H. Activating Transcription Factor 3 Protects against Restraint Stress-Induced Gastrointestinal Injury in Mice. Cells 2021, 10, 3530. [Google Scholar] [CrossRef] [PubMed]
- Pethaperumal, S.; Hung, S.C.; Lien, T.S.; Sun, D.S.; Chang, H.H. P-Selectin is a Critical Factor for Platelet-Mediated Protection on Restraint Stress-Induced Gastrointestinal Injury in Mice. Int. J. Mol. Sci. 2022, 23, 11909. [Google Scholar] [CrossRef] [PubMed]
- Siwakoti, B.; Lien, T.S.; Lin, Y.Y.; Pethaperumal, S.; Hung, S.C.; Sun, D.S.; Cheng, C.F.; Chang, H.H. The Role of Activating Transcription Factor 3 in Metformin’s Alleviation of Gastrointestinal Injury Induced by Restraint Stress in Mice. Int. J. Mol. Sci. 2023, 24, 10995. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.S.; Lien, T.S.; Chang, H.H. Restraint stress-associated gastrointestinal injury and implications from the Evans blue-fed restraint stress mouse model. Tzu Chi Med. J. 2024, 36, 23–29. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lien, T.-S.; Sun, D.-S.; Wu, W.-S.; Chang, H.-H. Dengue Envelope Protein as a Cytotoxic Factor Inducing Hemorrhage and Endothelial Cell Death in Mice. Int. J. Mol. Sci. 2024, 25, 10858. https://doi.org/10.3390/ijms251910858
Lien T-S, Sun D-S, Wu W-S, Chang H-H. Dengue Envelope Protein as a Cytotoxic Factor Inducing Hemorrhage and Endothelial Cell Death in Mice. International Journal of Molecular Sciences. 2024; 25(19):10858. https://doi.org/10.3390/ijms251910858
Chicago/Turabian StyleLien, Te-Sheng, Der-Shan Sun, Wen-Sheng Wu, and Hsin-Hou Chang. 2024. "Dengue Envelope Protein as a Cytotoxic Factor Inducing Hemorrhage and Endothelial Cell Death in Mice" International Journal of Molecular Sciences 25, no. 19: 10858. https://doi.org/10.3390/ijms251910858