
Targeting Alzheimer’s Disease: Evaluating the Efficacy of C-1 Functionalized N-Aryl-Tetrahydroisoquinolines as Cholinergic Enzyme Inhibitors and Promising Therapeutic Candidates
 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
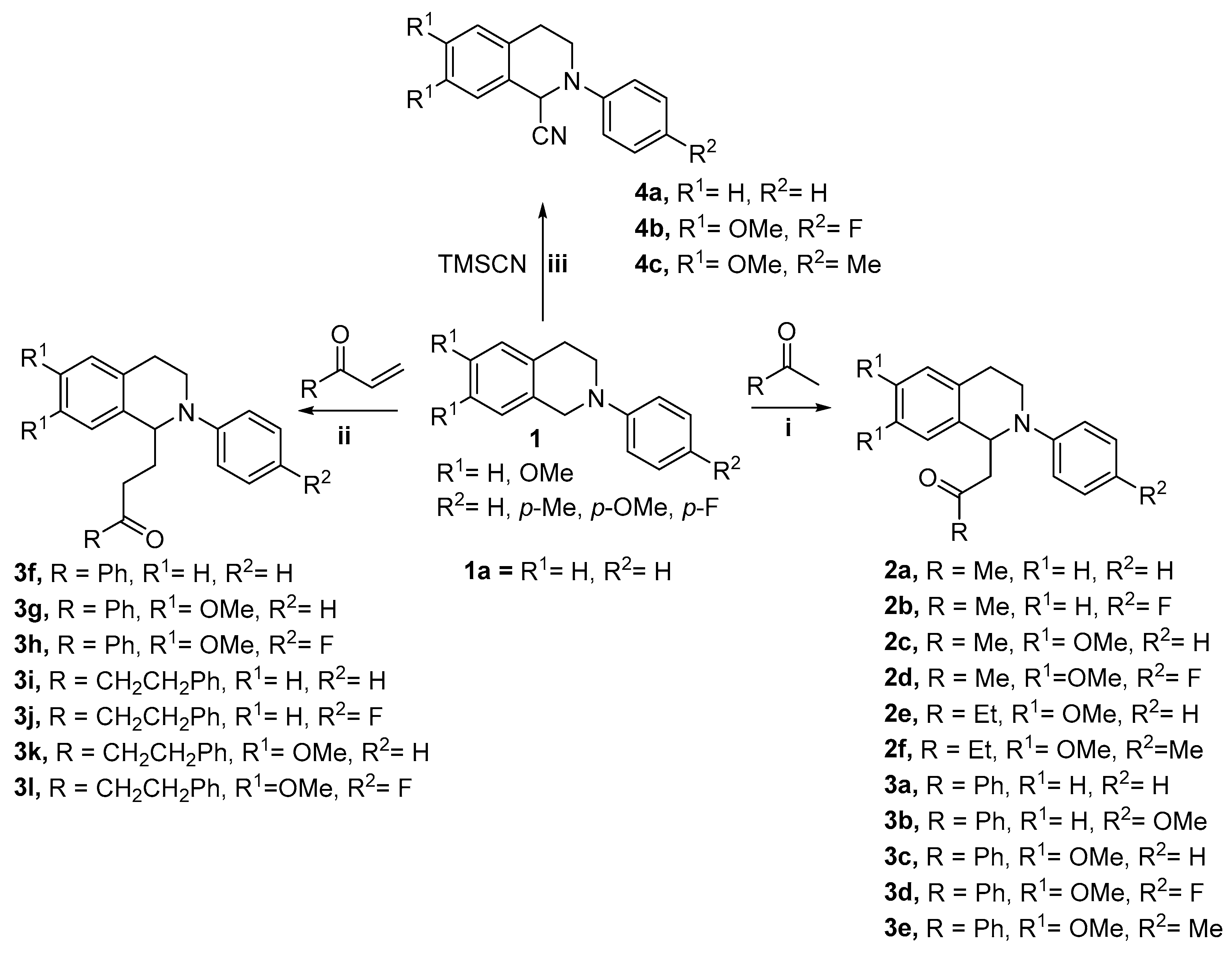
2.1. Chemistry—Synthesis of Compounds
2.2. Evaluation of the Cholinesterase’ Enzymes Inhibition Potency and SAR Discussion
2.3. Kinetic Studies of Enzymes Inhibition
2.4. Influence of Inhibitors Binding on the Trp−AChE/BuChE Fluorescence
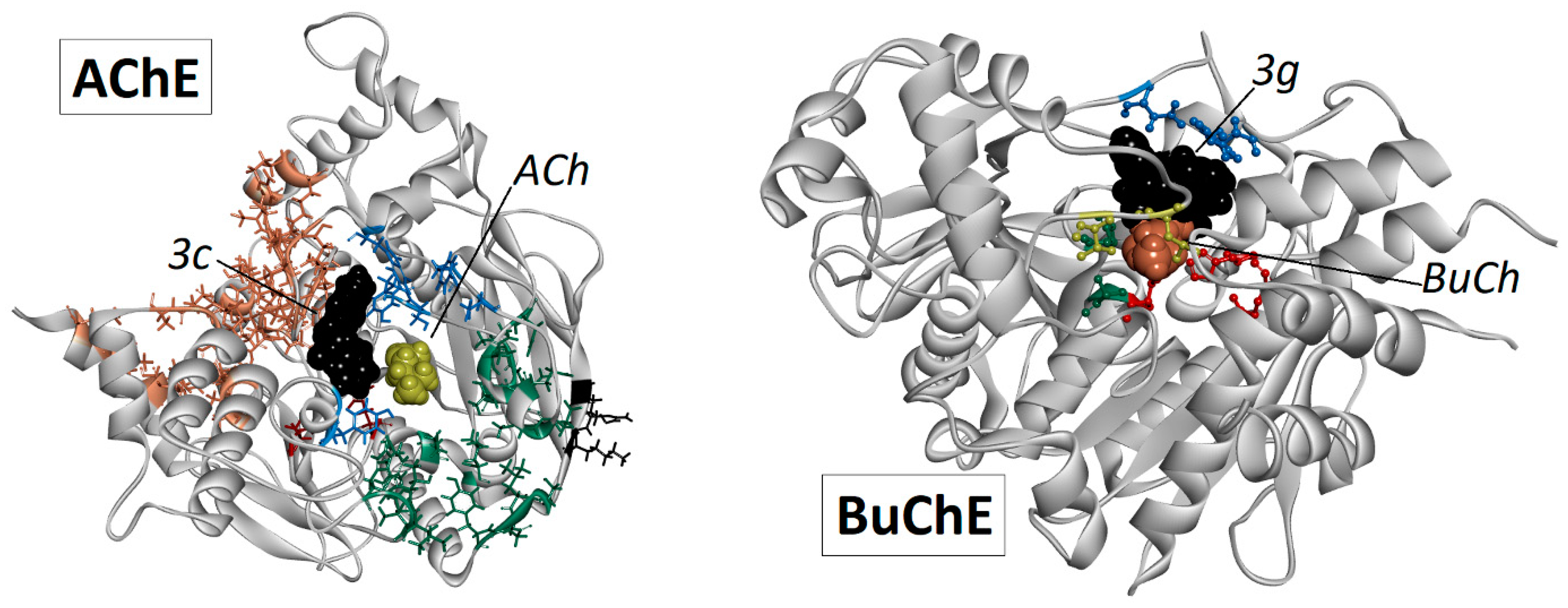
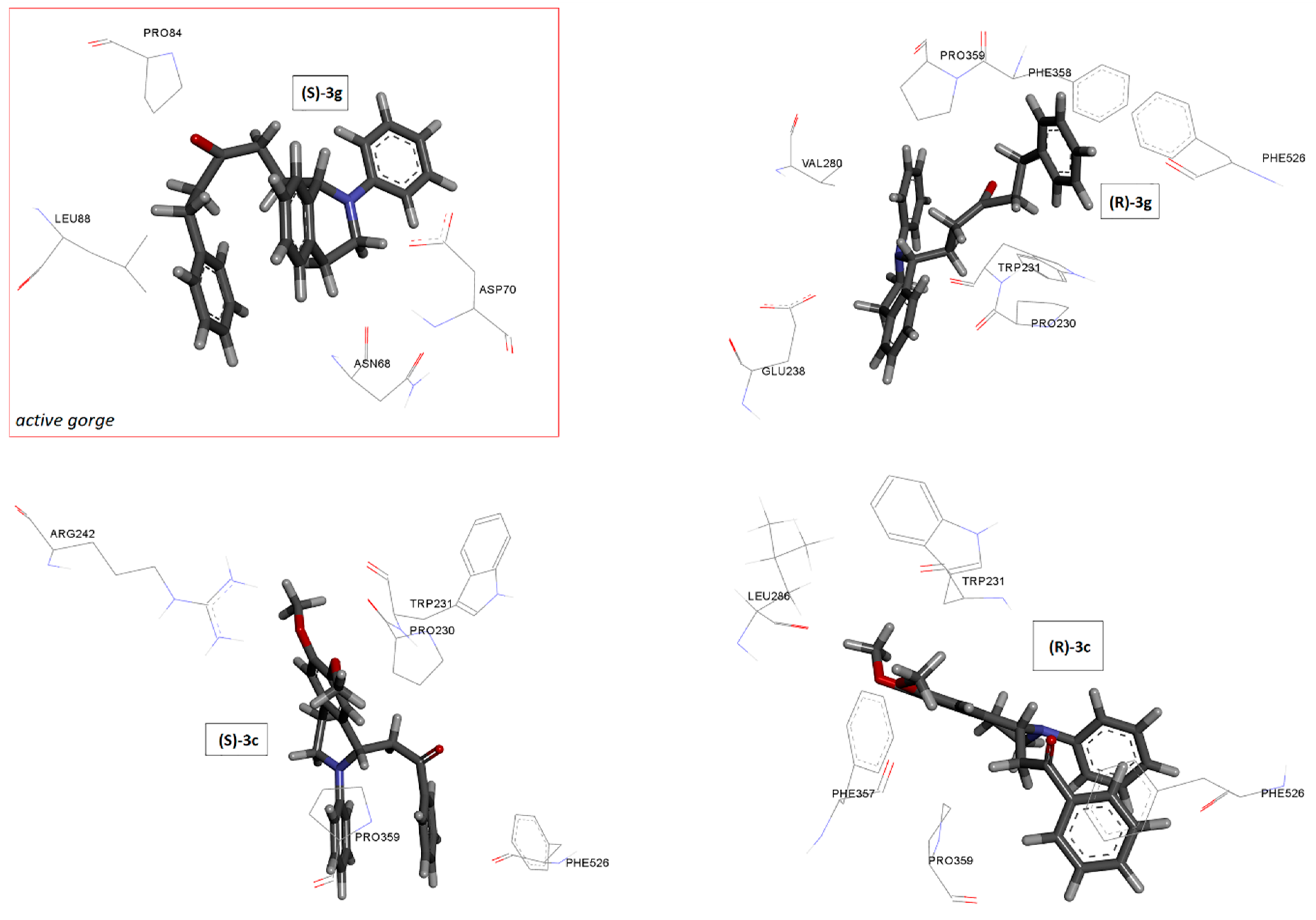
2.5. Docking Studies
2.6. ADME Prediction, log P, and the Blood−Brain Barrier Permeability Determination
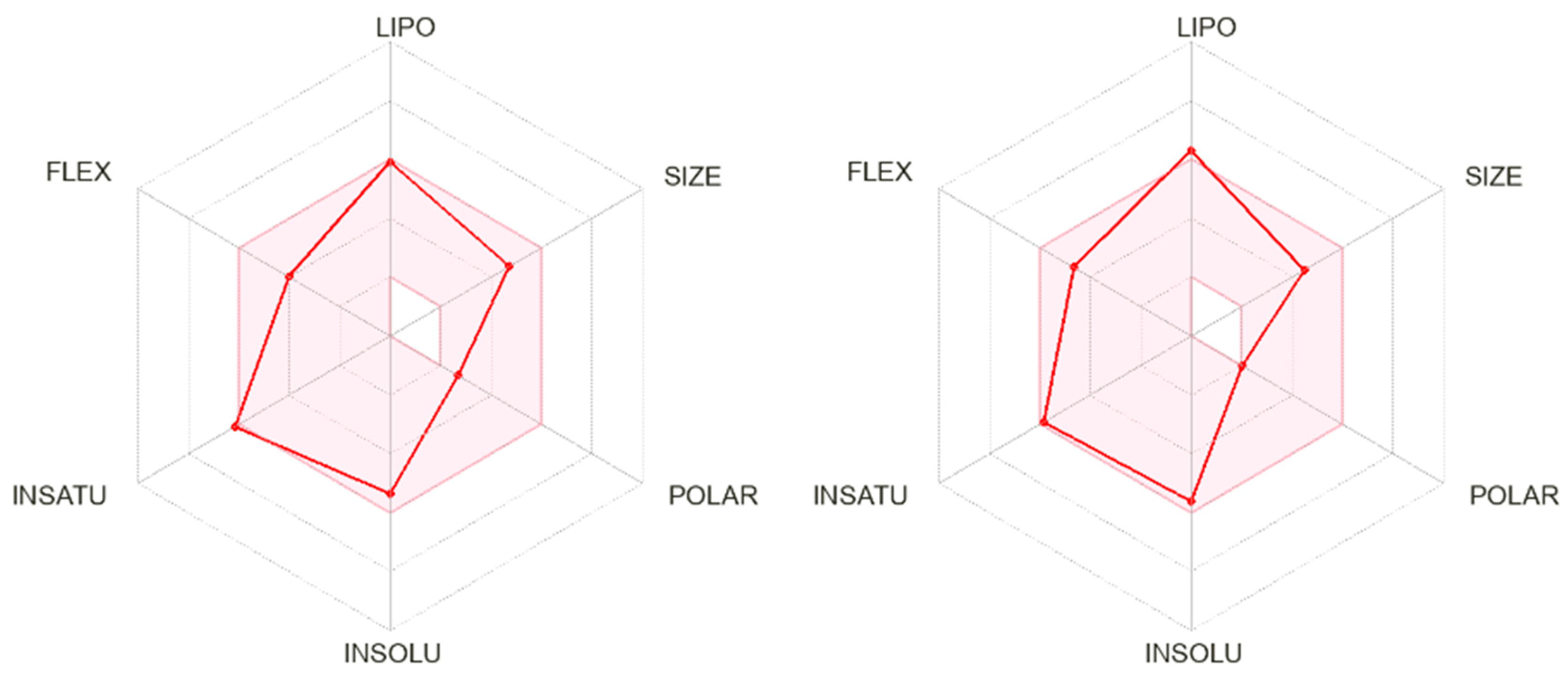
2.6.1. In Silico ADME Predictions for Selected THIQs
2.6.2. Ultra-High-Performance Chromatography log P Determination and the In Vitro BBB Permeability Study

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | logP | PAMPA-BBB Permeability Pe; 10−6 cm s−1 |
|---|---|---|
| 3c | 3.39 | 47.5 |
| 3f | 3.66 | High hydrophobicity; it retained in the membrane |
| 3i | 3.78 | High hydrophobicity; it retained in the membrane |
| Verapamil × HCl | 3.80 [32] | 15.8 |
| Caffeine | −0.07 [33] | 1.47 |
3. Conclusions
4. Experimental Section
4.1. Chemicals
4.2. In Vitro Inhibitory Evaluation on AChE/BChE
4.3. Kinetics Studies of ChE’s Inhibition
4.4. Fluorescence Measurements
4.5. In Silico Prediction of ADME
4.6. Determination of Partition Coefficient (logP) of Tested Compounds by UPLC Methods
4.7. PAMPA-BBB Permeability Assay
4.8. Quantum-Mechanical Calculations
4.9. Molecular Docking
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Walczak-Nowicka, Ł.J.; Herbet, M. Acetylcholinesterase Inhibitors in the Treatment of Neurodegenerative Diseases and the Role of Acetylcholinesterase in their Pathogenesis. Int. J. Mol. Sci. 2021, 22, 9290. [Google Scholar] [CrossRef]
- Čolović, M.B.; Krstić, D.Z.; Lazarević-Pašti, T.D.; Bondžić, A.M.; Vasić, V.M. Acetylcholinesterase inhibitors: Pharmacology and toxicology. Curr. Neuropharmacol. 2013, 11, 315–335. [Google Scholar] [CrossRef]
- FDA’s Decision to Approve New Treatment for Alzheimer’s Disease. Available online: https://www.fda.gov/drugs/our-perspective/fdas-decision-approve-new-treatment-alzheimers-disease (accessed on 23 December 2023).
- FDA Converts Novel Alzheimer’s Disease Treatment to Traditional Approval. Available online: https://www.fda.gov/news-events/press-announcements/fda-converts-novel-alzheimers-disease-treatment-traditional-approval (accessed on 23 December 2023).
- Söderberg, L.; Johannesson, M.; Nygren, P.; Laudon, H.; Eriksson, F.; Osswald, G.; Möller, C.; Lannfelt, L. Lecanemab, Aducanumab, and Gantenerumab—Binding Profiles to Different Forms of Amyloid-Beta Might Explain Efficacy and Side Effects in Clinical Trials for Alzheimer’s Disease. Neurotherapeutics 2023, 20, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Sims, J.R.; Zimmer, J.A.; Evans, C.D.; Lu, M.; Ardayfio, P.; Sparks, J.; Wessels, A.M.; Shcherbinin, S.; Wang, H.; Monkul Nery, E.S.; et al. Donanemab in Early Symptomatic Alzheimer Disease: The TRAILBLAZER-ALZ 2 Randomized Clinical Trial. JAMA 2023, 330, 512–527. [Google Scholar] [CrossRef] [PubMed]
- Van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2022, 388, 9–21. [Google Scholar] [CrossRef] [PubMed]
- National Library of Medicine. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Tacrine (accessed on 23 December 2023).
- Marucci, G.; Buccioni, M.; Ben, D.D.; Lambertucci, C.; Volpini, R.; Amenta, F. Efficacy of acetylcholinesterase inhibitors in Alzheimer’s disease. Neuropharmacology 2021, 190, 108352. [Google Scholar] [CrossRef]
- Yang, X.; Dang, P.; Liu, W.; Ma, W.; Ge, X.; Zhu, K.; Wang, M.; Huang, X.; Ding, X.; Wang, X. The role of butyrylcholinesterase in the regulation of cognitive dysfunction in minimal hepatic encephalopathy: A potential blood marker of disease evolution. Front. Neurol. 2022, 13, 900997. [Google Scholar] [CrossRef]
- Mesulam, M.; Geula, C. Butyrylcholinesterase reactivity differentiates the amyloid plaques of aging from those of dementia. Ann. Neurol. 1994, 36, 722–727. [Google Scholar] [CrossRef]
- Soreq, H.; Seidman, S. Acetylcholinesterase—New roles for an old actor. Nat. Rev. Neurosci. 2001, 2, 294–302. [Google Scholar] [CrossRef]
- Scott, J.D.; Williams, R.M. Chemistry and biology of the tetrahydroisoquinoline antitumor antibiotics. Chem. Rev. 2002, 102, 1669–1730. [Google Scholar] [CrossRef]
- Faheem, M.; Karan Kumar, B.; Chandra Sekhar, K.V.G.; Chander, S.; Kunjiappan, S.; Murugesan, S. Medicinal chemistry perspectives of 1,2,3,4-tetrahydroisoquinoline analogs—biological activities and SAR studies. RSC Adv. 2021, 11, 12254–12287. [Google Scholar] [CrossRef] [PubMed]
- Zablotskaya, A.; Segal, I.; Geronikaki, A.; Eremkina, T.; Belyakov, S.; Petrova, M.; Shestakova, I.; Zvejniece, L.; Nikolajeva, V. Synthesis, physicochemical characterization, cytotoxicity, antimicrobial, anti-inflammatory and psychotropic activity of new N-[1,3-(benzo)thiazol-2-yl]-ω-[3,4-dihydroisoquinolin-2(1H)-yl]alkanamides. Eur. J. Med. Chem. 2013, 70, 846–856. [Google Scholar] [CrossRef]
- Zhu, P.; Ye, W.; Li, J.; Zhang, Y.; Huang, W.; Cheng, M.; Wang, Y.; Zhang, Y.; Liu, H.; Zuo, J. Design, synthesis, and biological evaluation of novel tetrahydroisoquinoline derivatives as potential antitumor candidate. Chem. Biol. Drug Des. 2017, 89, 443–455. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Su, C.; Wang, L.; Qin, J.; Wei, S.; Tang, H. Hybrids of oxoisoaporphine-tetrahydroisoquinoline: Novel multi-target inhibitors of inflammation and amyloid-β aggregation in Alzheimer’s disease. Mol. Divers. 2019, 23, 709–722. [Google Scholar] [CrossRef]
- Fang, Y.; Zhou, H.; Gu, Q.; Xu, J. Synthesis and evaluation of tetrahydroisoquinoline-benzimidazole hybrids as multifunctional agents for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2019, 167, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Ontoria, J.M.; Biancofiore, I.; Fezzardi, P.; Ferrigno, F.; Torrente, E.; Colarusso, S.; Bianchi, E.; Andreini, M.; Patsilinakos, A.; Kempf, G.; et al. Combined Peptide and Small-Molecule Approach toward Nonacidic THIQ Inhibitors of the KEAP1/NRF2 Interaction. ACS Med. Chem. Lett. 2020, 11, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Sang, Z.; Wang, K.; Han, X.; Cao, M.; Tan, Z.; Liu, W. Design, Synthesis, and Evaluation of Novel Ferulic Acid Derivatives as Multi-Target-Directed Ligands for the Treatment of Alzheimer’s Disease. ACS Chem. Neurosci. 2019, 10, 1008–1024. [Google Scholar] [CrossRef]
- Sukumarapillai, D.K.; Kooi-Yeong, K.; Kia, Y.; Murugaiyah, V.; Iyer, S.K. Design, synthesis and cholinesterase inhibitory evaluation study of fluorescent N-benzylpiperidine-4-one derivatives. Med. Chem. Res. 2016, 25, 1705–1715. [Google Scholar] [CrossRef]
- Sudhapriya, N.; Manikandan, A.; Kumar, M.R.; Perumal, P.T. Cu-mediated synthesis of differentially substituted diazepines as AChE inhibitors; validation through molecular docking and Lipinski’s filter to develop novel anti-neurodegenerative drugs. Bioorganic Med. Chem. Lett. 2019, 29, 1308–1312. [Google Scholar] [CrossRef]
- Ghamari, N.; Zarei, O.; Reiner, D.; Dastmalchi, S.; Stark, H.; Hamzeh-Mivehroud, M. Histamine H3 receptor ligands by hybrid virtual screening, docking, molecular dynamics simulations, and investigation of their biological effects. Chem. Biol. Drug Des. 2019, 93, 832–843. [Google Scholar] [CrossRef]
- Tofani, L.; Feis, A.; Snoke, R.E.; Berti, D.; Baglioni, P.; Smulevich, G. Spectroscopic and Interfacial Properties of Myoglobin/Surfactant Complexes. Biophys. J. 2004, 87, 1186–1195. [Google Scholar] [CrossRef] [PubMed]
- Shopova, M.; Genov, N. Protonated form of histidine 238 quenches the fluorescence of tryptophan 241 in subtilisin Novo. Int. J. Pept. Protein Res. 1983, 21, 475–478. [Google Scholar] [CrossRef]
- Masson, P.; Carletti, E.; Nachon, F. Structure, Activities and Biomedical Applications of Human Butyrylcholinesterase. Protein Pept. Lett. 2009, 16, 1215–1224. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- SwissADME. Available online: http://www.swissadme.ch (accessed on 4 January 2024).
- Moriguchi, I.; Hirono, S.; Nakagome, I.; Hirano, H. Comparison of Reliability of log P Values for Drugs Calculated by Several Methods. Chem. Pharm. Bull. 1994, 42, 976–978. [Google Scholar] [CrossRef]
- Arnott, J.A.; Planey, S.L. The influence of lipophilicity in drug discovery and design. Expert Opin. Drug Discov. 2012, 7, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Di, L.; Kerns, E.H.; Fan, K.; McConnell, O.J.; Carter, G.T. High throughput artificial membrane permeability assay for blood-brain barrier. Eur. J. Med. Chem. 2003, 38, 223–232. [Google Scholar] [CrossRef]
- Yoshida, M.I.; Gomes, E.C.L.; Soares, C.D.V.; Cunha, A.F.; Oliveira, M.A. Thermal Analysis Applied to Verapamil Hydrochloride Characterization in Pharmaceutical Formulations. Molecules 2010, 15, 2439–2452. [Google Scholar] [CrossRef]
- Willson, C. The clinical toxicology of caffeine: A review and case study. Toxicol. Rep. 2018, 5, 1140–1152. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Valkó, K.; Snyder, L.R.; Glajch, J.L. Retention in reversed-phase liquid chromatography as a function of mobile-phase composition. J. Chromatogr. A 1993, 656, 501–520. [Google Scholar] [CrossRef]
- Cui, H.M.; Zhang, Q.Y.; Wang, J.L.; Chen, J.L.; Zhang, Y.L.; Tong, X.L. Poor permeability and absorption affect the activity of four alkaloids from Coptis. Mol. Med. Rep. 2015, 12, 7160–7168. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Sabus, C.; Carter, G.T.; Du, C.; Avdeef, A.; Tischler, M. In Vitro Permeability of Poorly Aqueous Soluble Compounds Using Different Solubilizers in the PAMPA Assay with Liquid Chromatography/Mass Spectrometry Detection. Pharm. Res. 2003, 20, 1820–1826. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B. Gaussian 09; Gaussian Inc: Wallingford, CT, USA, 2009. [Google Scholar]
- Bourne, Y.; Grassi, J.; Bougis, P.E.; Marchot, P. Conformational Flexibility of the Acetylcholinesterase Tetramer Suggested by X-ray Crystallography. J. Biol. Chem. 1999, 274, 30370–30376. [Google Scholar] [CrossRef]
- Bernstein, F.C.; Koetzle, T.F.; Williams, G.J.; Meyer, E.F., Jr.; Brice, M.D.; Rodgers, J.R.; Kennard, O.; Shimanouchi, T.; Tasumi, M. The Protein Data Bank: A computer-based archival file for macromolecular structures. J. Mol. Biol. 1977, 112, 535–542. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Dassault Systemes BIOVIA. Discovery Studio Modelling Environment (Release 2017); Dassault Systemes: San Diego, CA, USA, 2016. [Google Scholar]
- WebGRO for Macromolecular Simulations. Available online: https://simlab.uams.edu/ (accessed on 23 December 2023).
- Van Gunsteren, W.F.; Billeter, S.; Eising, A.; Hünenberger, P.; Krüger, P.; Mark, A.E.; Scott, W.R.P.; Tironi, I.G. Biomolecular Simulation: The GROMOS96 Manual and User Guide; Vdf Hochschulverlag AG an der ETH Zürich: Zürich, Switzerland, 1996; Volume 86, pp. 1–1044. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]




| Compound | Structure | Residual Activity at 1 × 10−5 M | IC50, μM | Selectivity Index for AChE a | ||
|---|---|---|---|---|---|---|
| AChE | BuChE | AChE | BuChE | |||
| 1a | 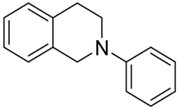 | n.a. | 11 | >10 | >10 | - |
| 2a |  | 30 | 43 | >10 | >10 | - |
| 2b | 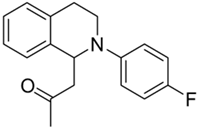 | 64 | 47 | 5.46 ± 0.58 | 8.90 ± 2.03 | 1.63 |
| 2c | 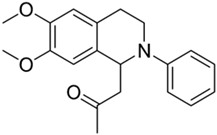 | 68 | 2 | 5.04 ± 0.28 | >10 | - |
| 2d | 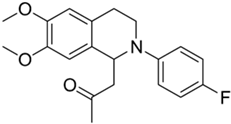 | 12 | 15 | >10 | >10 | - |
| 2e | 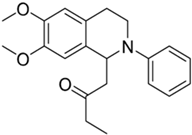 | 20 | n.a. | >10 | n.d. | - |
| 2f | 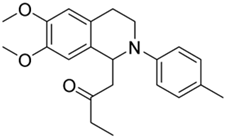 | n.a. | n.a. | n.d. | n.d. | - |
| 3a | 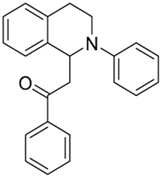 | 100 | 54 | 4.29 ± 0.09 | 8.26 ± 1.05 | 1.93 |
| 3b |  | 59 | 20 | 9.05 ± 0.81 | >10 | - |
| 3c |  | 100 | 37 | 1.95 ± 0.23 | >10 | >5 |
| 3d |  | 70 | 20 | 4.89 ± 0.15 | >10 | - |
| 3e |  | 45 | n.a. | >10 | n.d. | - |
| 3f | 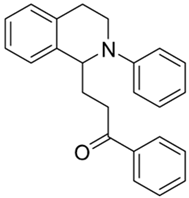 | 100 | 75 | 2.42 ± 0.09 | 6.28 ± 0.36 | 2.59 |
| 3g | 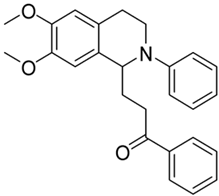 | 75 | 30 | 6.78 ± 0.22 | 8.01 ± 0.55 | 1.18 |
| 3h | 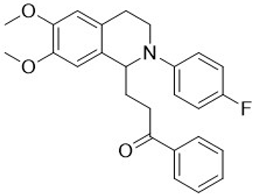 | 40 | 10 | >10 | >10 | - |
| 3i | 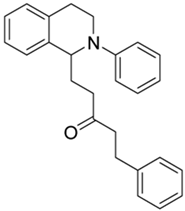 | 100 | 100 | 1.11 ± 0.04 | 2.72 ± 0.20 | 2.45 |
| 3j | 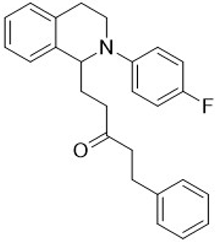 | 100 | 100 | 2.91 ± 0.09 | 4.52 ± 0.15 | 1.55 |
| 3k | 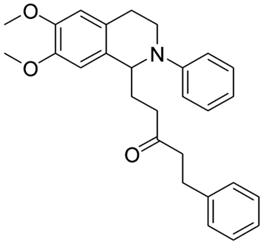 | 70 | 30 | 4.02 ± 0.19 | >10 | - |
| 3l | 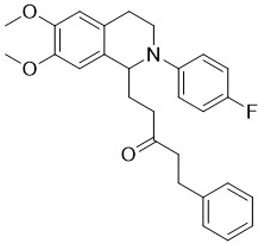 | 45 | 5 | >10 | >10 | - |
| 4a | 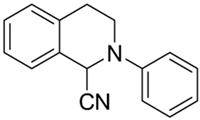 | 12 | 23 | >10 | >10 | - |
| 4b | 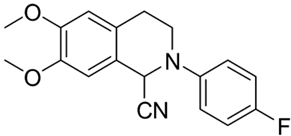 | 20 | 20 | >10 | >10 | - |
| 4c | 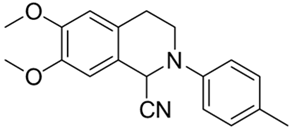 | 18 | 30 | >10 | >10 | - |
| Tacrine | - | - | - | 0.06 ± 0.001 | 0.008 ± 0.001 | 0.13 |
| Donepezil | - | - | - | 0.02 ± 0.001 | 3.67 ± 0.05 | 183 |
| Enzyme | Inhibitor | IC50, μM | Ki, μM | Type of Inhibition |
|---|---|---|---|---|
| AChE | 3c | 1.91 ± 0.11 | 1.78 ± 0.10 | Non-competitive |
| 3i | 1.11 ± 0.04 | 1.06 ± 0.01 | Non-competitive | |
| BuChE | 3c | n.a. | / | / |
| 3i | 2.71 ± 0.20 | 1.04 ± 0.1 | Mix non-competitive/uncompetitive |
| Ksv, M−1 | kq, M−1s−1 | Ka, M | n | |
|---|---|---|---|---|
| 3i | (6.96 ± 0.05) × 104 | (6.96 ± 0.05) × 1014 | (3.31 ± 0.05) × 104 | 0.94 ± 0.04 |
| Label of Compounds | AChE | BuChE | ||||
|---|---|---|---|---|---|---|
| Conf | ΔE | No | Conf | ΔE | No | |
| 3c | R | −9.2 | 1 | R | −7.9 | / |
| S | −8.9 | 1 | S | −7.5 | / | |
| 3i | R | −9.9 | 1 | R | −8.7 | 1 |
| S | −9.2 | 1 | S | −8.2 | 1 | |
| Compounds | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Parameters | 2b | 2c | 3a | 3b | 3c | 3d | 3f * | 3g | 3i * | 3j * | 3k | Tacrine | Donepezil |
| Mw, g/mol | 283.3 | 325.4 | 327.4 | 357.4 | 387.5 | 405.5 | 341.5 | 401.5 | 369.5 | 387.5 | 429.6 | 198.3 | 379.5 |
| TPSA, Å2 | 20.3 | 38.8 | 20.3 | 29.5 | 38.8 | 38.8 | 20.3 | 38.8 | 20.3 | 20.3 | 38.8 | 38.9 | 38.8 |
| ClogP | 3.45 | 3.09 | 4.25 | 4.21 | 4.16 | 4.48 | 4.57 | 4.46 | 5.00 | 5.26 | 4.90 | 2.59 | 4.00 |
| ilogP | 2.58 | 2.89 | 3.09 | 3.29 | 3.38 | 3.50 | 3.36 | 3.51 | 3.76 | 3.61 | 3.99 | 2.09 | 3.92 |
| MlogP | 3.58 | 2.46 | 4.08 | 3.68 | 3.31 | 3.68 | 4.30 | 3.51 | 4.71 | 5.08 | 3.91 | 2.33 | 3.06 |
| XlogP | 3.36 | 3.21 | 4.92 | 4.89 | 4.86 | 4.96 | 5.28 | 5.22 | 5.51 | 5.61 | 5.45 | 2.71 | 4.28 |
| WlogP | 3.62 | 3.08 | 4.36 | 4.37 | 4.38 | 4.93 | 4.75 | 4.77 | 5.07 | 5.63 | 5.08 | 2.70 | 3.83 |
| logS (ESOL) | −3.94 | −3.92 | −5.24 | −5.30 | −5.37 | −5.53 | −5.47 | −5.60 | −5.62 | −5.77 | −5.76 | −3.27 | −4.81 |
| No. H-bond acceptor | 2 | 3 | 1 | 2 | 3 | 4 | 1 | 3 | 1 | 2 | 3 | 1 | 4 |
| No. H-bond donor | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| No. rotable bonds | 3 | 5 | 4 | 5 | 6 | 6 | 5 | 7 | 7 | 7 | 9 | 0 | 6 |
| BBB permeation | yes | yes | yes | yes | yes | yes | yes | yes | yes | yes | yes | yes | yes |
| LogKP (skin permeation), cm/s | −5.64 | −6.01 | −4.80 | −5.01 | −5.21 | −5.25 | −4.63 | −5.04 | −4.64 | −4.68 | −5.05 | −5.59 | −5.58 |
| Lipinski’s rule violations | no | no | no | no | no | no | yes | no | yes | yes | no | no | no |
| Bioavailability score | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 |
| GI absorbtion | high | high | high | high | high | high | high | high | high | high | high | high | high |
| PAINS alerts | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| P-pg substrate | yes | yes | yes | yes | yes | yes | yes | yes | yes | yes | yes | yes | yes |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jovanović, D.; Filipović, A.; Janjić, G.; Lazarević-Pašti, T.; Džambaski, Z.; Bondžić, B.P.; Bondžić, A.M. Targeting Alzheimer’s Disease: Evaluating the Efficacy of C-1 Functionalized N-Aryl-Tetrahydroisoquinolines as Cholinergic Enzyme Inhibitors and Promising Therapeutic Candidates. Int. J. Mol. Sci. 2024, 25, 1033. https://doi.org/10.3390/ijms25021033
Jovanović D, Filipović A, Janjić G, Lazarević-Pašti T, Džambaski Z, Bondžić BP, Bondžić AM. Targeting Alzheimer’s Disease: Evaluating the Efficacy of C-1 Functionalized N-Aryl-Tetrahydroisoquinolines as Cholinergic Enzyme Inhibitors and Promising Therapeutic Candidates. International Journal of Molecular Sciences. 2024; 25(2):1033. https://doi.org/10.3390/ijms25021033
Chicago/Turabian StyleJovanović, Dunja, Ana Filipović, Goran Janjić, Tamara Lazarević-Pašti, Zdravko Džambaski, Bojan P. Bondžić, and Aleksandra M. Bondžić. 2024. "Targeting Alzheimer’s Disease: Evaluating the Efficacy of C-1 Functionalized N-Aryl-Tetrahydroisoquinolines as Cholinergic Enzyme Inhibitors and Promising Therapeutic Candidates" International Journal of Molecular Sciences 25, no. 2: 1033. https://doi.org/10.3390/ijms25021033