
Structural Analyses of Designed α-Helix and β-Sheet Peptide Nanofibers Using Solid-State Nuclear Magnetic Resonance and Cryo-Electron Microscopy and Introduction of Structure-Based Metal-Responsive Properties


 , and
, and
Abstract
1. Introduction
2. Results and Discussions
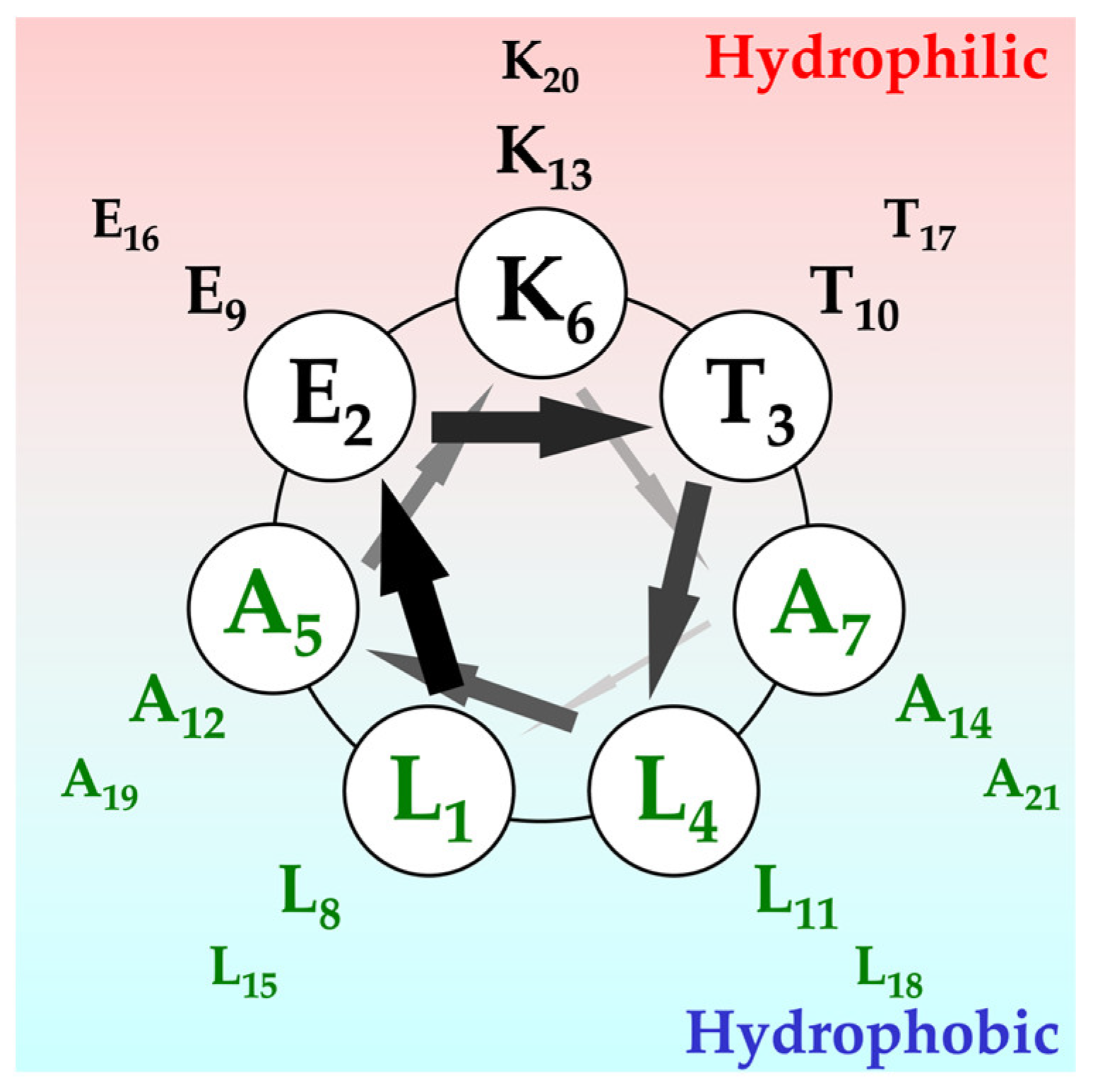
2.1. Secondary Structures of α3 and Their Orientation
2.2. Secondary Structures and Distances between 13C and 15N of α3 Peptide Revealed by 13C Solid-State NMR
2.3. Intermolecular Distance between α3 Molecules
2.4. Structure of α3 Peptide Nanofiber
2.5. Structural Analyses of β-Sheet-Type Peptide Nanofibers
2.6. Introducing Conformational Switching Capability upon Metal Ion Binding
2.7. Structure and Metal Ion Response of HDM1, -2, and -3
2.8. Identification of Ni-Binding Sites in HDM1
2.9. Conformational Switching Capability of HDM1 Fiber by Metal Ion
3. Materials and Methods
3.1. Synthesis of Peptides
3.2. Polarized IR Microscope
3.3. FT-IR Analysis
3.4. Solid-State NMR Measurement
3.5. Circular Dichroism Measurements
3.6. Atomic Force Microscopy
3.7. Cryo-Electron Microscopy and Image Processing
3.8. Chemical Modification of HDM1 and -2 via Diethylpyrocarbonate (DEPC)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lazarus, B.S.; Chadha, C.; Velasco-Hogan, A.; Barbosa, J.D.V.; Jasiuk, I.; Meyers, M.A. Engineering with keratin: A functional material and a source of bioinspiration. iScience 2021, 24, 102798. [Google Scholar] [CrossRef] [PubMed]
- Holmes, D.F.; Lu, Y.; Starborg, T.; Kadler, K.E. Collagen Fibril Assembly and Function. Curr. Top. Dev. Biol. 2018, 130, 107–142. [Google Scholar] [PubMed]
- Humenik, M.; Scheibel, T.; Smith, A. Spider silk: Understanding the structure-function relationship of a natural fiber. Prog. Mol. Biol. Transl. Sci. 2011, 103, 131–185. [Google Scholar] [PubMed]
- Burgess, N.C.; Sharp, T.H.; Thomas, F.; Wood, C.W.; Thomson, A.R.; Zaccai, N.R.; Brady, R.L.; Serpell, L.C.; Woolfson, D.N. Modular Design of Self-Assembling Peptide-Based Nanotubes. J. Am. Chem. Soc. 2015, 137, 10554–10562. [Google Scholar] [CrossRef]
- Wang, F.; Gnewou, O.; Wang, S.; Osinski, T.; Zuo, X.; Egelman, E.H.; Conticello, V.P. Deterministic chaos in the self-assembly of β sheet nanotubes from an amphipathic oligopeptide. Matter 2021, 4, 3217–3231. [Google Scholar] [CrossRef]
- Wu, Y.; Norberg, P.K.; Reap, E.A.; Congdon, K.L.; Fries, C.N.; Kelly, S.H.; Sampson, J.H.; Conticello, V.P.; Collier, J.H. A Supramolecular Vaccine Platform Based on α-Helical Peptide Nanofibers. ACS Biomater. Sci. Eng. 2017, 3, 3128–3132. [Google Scholar] [CrossRef]
- Najafi, H.; Jafari, M.; Farahavar, G.; Abolmaali, S.S.; Azarpira, N.; Borandeh, S.; Ravanfar, R. Recent advances in design and applications of biomimetic self-assembled peptide hydrogels for hard tissue regeneration. Biodes. Manuf. 2021, 4, 735–756. [Google Scholar] [CrossRef]
- Kojima, S.; Kuriki, Y.; Sato, Y.; Arisaka, F.; Kumagai, I.; Takahashi, S.; Miura, K. Synthesis of alpha-helix-forming peptides by gene engineering methods and their characterization by circular dichroism spectra measurements. Biochim. Biophys. Acta 1996, 1294, 129–137. [Google Scholar] [CrossRef]
- Kojima, S.; Kuriki, Y.; Yoshida, T.; Yazaki, K.; Miura, K. Fibril Formation by an Amphipathic α-Helix-Forming Polypeptide Produced by Gene Engineering. Proc. Jpn. Acad. Ser. B Phys. 1997, 73, 7–11. [Google Scholar] [CrossRef]
- Kojima, S.; Kuriki, Y.; Yazaki, K.; Miura, K. Stabilization of the fibrous structure of an alpha-helix-forming peptide by sequence reversal. Biochem. Biophys. Res. Commun. 2005, 331, 577–582. [Google Scholar] [CrossRef]
- Takei, T.; Okonogi, A.; Tateno, K.; Kimura, A.; Kojima, S.; Yazaki, K.; Miura, K. The effects of the side chains of hydrophobic aliphatic amino acid residues in an amphipathic polypeptide on the formation of alpha helix and its association. J. Biochem. 2006, 139, 271–278. [Google Scholar] [CrossRef]
- Gordon, D.J.; Meredith, S.C. Probing the role of backbone hydrogen bonding in beta-amyloid fibrils with inhibitor peptides containing ester bonds at alternate positions. Biochemistry 2003, 42, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Chatani, E.; Yuzu, K.; Ohhashi, Y.; Goto, Y. Current Understanding of the Structure, Stability and Dynamic Properties of Amyloid Fibrils. Int. J. Mol. Sci. 2021, 22, 4349. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.L.; Hulett, M.D.; Parish, C.R. Histidine-rich glycoprotein: A novel adaptor protein in plasma that modulates the immune, vascular and coagulation systems. Immunol. Cell Biol. 2005, 83, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Susi, H. Infrared spectroscopy—Conformation. Methods Enzymol. 1972, 26, 455–472. [Google Scholar] [PubMed]
- Dong, A.; Caughey, W.S. Infrared methods for study of hemoglobin reactions and structures. Methods. Enzymol. 1994, 232, 139–175. [Google Scholar]
- Shapovalov, M.; Vucetic, S.; Dunbrack, R.L., Jr. A new clustering and nomenclature for beta turns derived from high-resolution protein structures. PLoS Comput. Biol. 2019, 15, e1006844. [Google Scholar] [CrossRef]
- Sakajiri, K.; Saeki, S.; Kawaguchi, S.; Watanabe, J. Conformational Analysis by IR and Birefringence Measurements for Poly(p-phenylpropyl L-aspartate) Exhibiting a Helical Sense Inversion in the Solid State. Polym. J. 2000, 32, 803–806. [Google Scholar] [CrossRef]
- Wishart, D.S. Interpreting protein chemical shift data. Prog. Nucl. Magn. Reson. Spectrosc. 2011, 58, 62–87. [Google Scholar] [CrossRef]
- Saito, H.; Ando, I.; Naito, A. NMR Constrains for Determination of Secondary Structure. In Solid State NMR Spectroscopy for Biopolymers: Principles and Application, 1st ed.; Springer: Dordrecht, The Netherlands, 2006; pp. 127–199. [Google Scholar]
- Kawamura, I.; Norisada, K.; Naito, A. Structure Determination of Membrane Peptides and Proteins by Solid-State NMR. In Experimental Approaches of NMR Spectroscopy: Methodology and Application to Life Science and Materials Science, 1st ed.; Springer: Singapore, 2018; pp. 253–293. [Google Scholar]
- Pal, L.; Dasgupta, B.; Chakrabarti, P. 310-Helix adjoining α-helix and β-strand: Sequence and structural features and their conservation. Biopolymers 2005, 78, 147–162. [Google Scholar] [CrossRef]
- Pal, L.; Chakrabarti, P.; Basu, G. Sequence and Structure Patterns in Proteins from an Analysis of the Shortest Helices: Implications for Helix Nucleation. J. Mol. Biol. 2003, 326, 273–291. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, M.; Ohtsu, T.; Chatani, E.; Tamura, A. Hyper Thermostability and Liquid-Crystal-Like Properties of Designed α-Helical Peptide Nanofibers. J. Phys. Chem. B 2023, 127, 8331–8343. [Google Scholar] [CrossRef]
- Sadqi, M.; Hernández, F.; Pan, U.; Pérez, M.; Schaeberle, M.D.; Avila, J.; Muñoz, V. Alpha-helix structure in Alzheimer’s disease aggregates of tau-protein. Biochemistry 2002, 41, 7150–7155. [Google Scholar] [CrossRef]
- Bousset, L.; Thomson, N.H.; Radford, S.E.; Melki, R. The yeast prion Ure2p retains its native alpha-helical conformation upon assembly into protein fibrils in vitro. EMBO J. 2002, 21, 2903–2911. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.P.; Ranaghan, M.J.; Ganjei, A.Y.; Oprian, D.D. Crystal Structure of Recoverin with Calcium Ions Bound to Both Functional EF Hands. Biochemistry 2015, 54, 7222–7228. [Google Scholar] [CrossRef] [PubMed]
- Sasakawa, H.; Yoshinaga, S.; Kojima, S.; Tamura, A. Structure of POIA1, a homologous protein to the propeptide of subtilisin: Implication for protein foldability and the function as an intramolecular chaperone. J. Mol. Biol. 2002, 317, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Woody, W.R. Theory of Circular Dichroism of Proteins. In Circular Dichroism and the Conformational Analysis of Biomolecules; Plenum Press: New York, NY, USA; London, UK, 1996; pp. 25–67. [Google Scholar]
- Tilstra, L.; Mattice, W.L. The β Sheet Coil Transition of Polypeptides, as Determined by Circular Dichroism. In Circular Dichroism and the Conformational Analysis of Biomolecules; Plenum Press: New York, NY, USA; London, UK, 1996; pp. 261–283. [Google Scholar]
- Egelman, E.H.; Xu, C.; DiMaio, F.; Magnotti, E.; Modlin, C.; Yu, X.; Wright, E.; Baker, D.; Conticello, V.P. Structural Plasticity of Helical Nanotubes Based on Coiled-Coil Assemblies. Structure 2015, 23, 280–289. [Google Scholar] [CrossRef]
- Mendoza, V.L.; Vachet, R.W. Protein Surface Mapping Using Diethylpyrocarbonate with Mass Spectrometric Detection. Anal. Chem. 2008, 80, 2895–2904. [Google Scholar] [CrossRef]
- Qin, K.; Yang, Y.; Mastrangelo, P.; Westaway, D. Mapping Cu(II) Binding Sites in Prion Proteins by Diethyl Pyrocarbonate Modification and Matrix-assisted Laser Desorption Ionization-Time of Flight (MALDI-TOF) Mass Spectrometric Footprinting. J. Biol. Chem. 2002, 277, 1981–1990. [Google Scholar] [CrossRef]
- Dong, A.; Huang, P.; Caughey, W.S. Protein secondary structures in water from second-derivative amide I infrared spectra. Biochemistry 1990, 29, 3303–3308. [Google Scholar] [CrossRef]
- Savitzky, A.; Golay, M.J.E. Smoothing and Differentiation of Data by Simplified Least Squares Procedures. Anal. Chem. 1964, 36, 1627–1639. [Google Scholar] [CrossRef]
- Susi, H.; Byler, D.M. Resolution-enhanced Fourier transform infrared spectroscopy of enzymes. Methods Enzymol. 1986, 130, 290–311. [Google Scholar] [PubMed]
- Kauppinen, J.K.; Moffatt, D.J.; Mantsch, H.H.; Cameron, D.G. Fourier transforms in the computation of self-deconvoluted and first-order derivative spectra of overlapped band contours. Anal. Chem. 1981, 53, 1454–1457. [Google Scholar] [CrossRef]
- Winningham, M.J.; Sogah, D.Y. A Modular Approach to Polymer Architecture Control via Catenation of Prefabricated Biomolecular Segments: Polymers Containing Parallel β-Sheets Templated by a Phenoxathiin-Based Reverse Turn Mimic. Macromolecules 1997, 30, 862–876. [Google Scholar] [CrossRef]
- Gullion, T.; Schaefer, J. Elimination of resonance offset effects in rotational-echo, double-resonance NMR. J. Magn. Reson. 1991, 92, 439–442. [Google Scholar] [CrossRef]
- Zheng, S.Q.; Palovcak, E.; Armache, J.P.; Verba, K.A.; Cheng, Y.; Agard, D.A. MotionCor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 2017, 14, 331–332. [Google Scholar] [CrossRef]
- Zhang, K. Gctf: Real-time CTF determination and correction. J. Struct. Biol. 2016, 193, 1–12. [Google Scholar] [CrossRef]
- Zivanov, J.; Nakane, T.; Forsberg, B.O.; Kimanius, D.; Hagen, W.J.; Lindahl, E.; Scheres, S.H. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 2018, 7, e42166. [Google Scholar] [CrossRef]
- Wagner, T.; Merino, F.; Stabrin, M.; Moriya, T.; Antoni, C.; Apelbaum, A.; Hagel, P.; Sitsel, O.; Raisch, T.; Prumbaum, D.; et al. SPHIRE-crYOLO is a fast and accurate fully automated particle picker for cryo-EM. Commun. Biol. 2019, 2, 218. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | N-Terminal | Sequence | C-Terminal | Number of Amino Acids |
|---|---|---|---|---|
| α3 | H3N+ | LETLAKALETLAKALETLAKA | COO− | 21 |
| CaRP2 | H3N+ | WDKDGNGTISFNE | CONH2 | 13 |
| βKE | H3N+ | KFVIFE | COO− | 7 |
| HDM1 | H3N+ | LETLAHALETLAHALETLAKA | COO− | 21 |
| HDM2 | H3N+ | LETLAKALHTLAHALETLAKA | COO− | 21 |
| HDM3 | H3N+ | LETLAKALEHLAHALETLAKA | COO− | 21 |
| Wave Number | Assignment | Area: Parallel | Area: Perpendicular | Dichroic Ratio | Angle (°) |
|---|---|---|---|---|---|
| 1648 cm−1 | random coil | 1.66 | 1.12 | 1.48 | 37.3 |
| 1654 cm−1 | α-helix | 3.28 | 1.41 | 2.33 | 0 |
| 1660 cm−1 | 310-helix or type III turn | 2.21 | 1.99 | 1.11 | 50.2 |
| Classification and Location | Assigned Structure */13C-5N Distance ** | |
|---|---|---|
| 1L[13C=O], 18L[13C=O], or 19A[13C=O] | Leu or Ala, single label, and terminal region | mixture of α-helices and β-sheet |
| 4L[13C=O], 8L[13C=O], 11L[13C=O], or 15L[13C=O] | Leu, single label, and middle region | α-helix |
| 5A[13C=O], 7A[13C=O], 12A[13C=O], or 14A[13C=O] | Ala, single label, and middle region | α-helix |
| 1L[13C=O] and A5[15N] | double label, N-terminal, and vicinity | 4.6 (±0.1) Å |
| Positions of the isotope labels of α3 | double label, C-terminal, and vicinity | 4.5 (±0.1) Å |
| 1L[13C=O] and A21[15N] | double label, N-terminal, and C-terminal | 4.7 (±0.1) Å intermolecular |
| 1L[15N] and A21[13C=O] | double label, N-terminal, and C-terminal | 5.1 (±0.1) Å intermolecular |
| Peptide | Number of DEPC | Quantity | Assignments |
|---|---|---|---|
| HDM1 (Figure 10C) | 3 4 | major | His, His, N-terminal His, His, N-terminal,Lys |
| HDM1-Ni2+ (Figure 10D) | 0 1 2 3 | major | None * Lys His, His His, His, Lys |
| HDM2 (Figure 10E) | 3 4 | major | His, His, Lys His, His, Lys, Lys |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakagawa, S.; Kurokawa, M.; Kambara, O.; Takei, T.; Daidoji, K.; Naito, A.; Takita, M.; Kawamoto, A.; Hirose, M.; Tamura, A. Structural Analyses of Designed α-Helix and β-Sheet Peptide Nanofibers Using Solid-State Nuclear Magnetic Resonance and Cryo-Electron Microscopy and Introduction of Structure-Based Metal-Responsive Properties. Int. J. Mol. Sci. 2024, 25, 1111. https://doi.org/10.3390/ijms25021111
Nakagawa S, Kurokawa M, Kambara O, Takei T, Daidoji K, Naito A, Takita M, Kawamoto A, Hirose M, Tamura A. Structural Analyses of Designed α-Helix and β-Sheet Peptide Nanofibers Using Solid-State Nuclear Magnetic Resonance and Cryo-Electron Microscopy and Introduction of Structure-Based Metal-Responsive Properties. International Journal of Molecular Sciences. 2024; 25(2):1111. https://doi.org/10.3390/ijms25021111
Chicago/Turabian StyleNakagawa, Shota, Minami Kurokawa, Ohki Kambara, Toshiaki Takei, Kengo Daidoji, Akira Naito, Mao Takita, Akihiro Kawamoto, Mika Hirose, and Atsuo Tamura. 2024. "Structural Analyses of Designed α-Helix and β-Sheet Peptide Nanofibers Using Solid-State Nuclear Magnetic Resonance and Cryo-Electron Microscopy and Introduction of Structure-Based Metal-Responsive Properties" International Journal of Molecular Sciences 25, no. 2: 1111. https://doi.org/10.3390/ijms25021111
APA StyleNakagawa, S., Kurokawa, M., Kambara, O., Takei, T., Daidoji, K., Naito, A., Takita, M., Kawamoto, A., Hirose, M., & Tamura, A. (2024). Structural Analyses of Designed α-Helix and β-Sheet Peptide Nanofibers Using Solid-State Nuclear Magnetic Resonance and Cryo-Electron Microscopy and Introduction of Structure-Based Metal-Responsive Properties. International Journal of Molecular Sciences, 25(2), 1111. https://doi.org/10.3390/ijms25021111