1. Introduction
Damage to the central nervous system (CNS) due to injury or disease presents a multi-faceted pathophysiology [
1,
2]. However, demyelination commonly features and is associated with chronic pain [
3,
4], incontinence [
5,
6], cognitive decline [
7,
8], and paralysis [
9,
10]. Due to the highly complex nature of CNS pathologies, a combinatorial approach to treatment is likely to be needed. Cellular therapies that promote CNS re/myelination and repair are among the many promising strategies being explored in developing treatment regimens that restore function and quality of life to patients [
11,
12]. Among the candidate cell therapies being investigated, mesenchymal stromal cells (MSCs) are an attractive option [
13,
14,
15]. Their capacity for immunomodulation and tissue regeneration is well documented [
16,
17], and their culture requirements are extensively characterized [
18,
19]. Additionally, they do not necessarily require strict HLA-matching [
20,
21], with allogeneic MSCs considered preferable in some cases [
22,
23]. As a result, MSCs are highly amenable to use as cell therapy [
24]. Unlike human bone marrow-derived MSCs (hBM-MSCs), which require the invasive and painful aspiration of bone marrow to generate [
25], MSCs derived from human olfactory mucosa tissue (hOM-MSCs) can be established from biopsies taken during routine out-patient procedures [
26] and are obtained from an anatomical region that experiences neurogenesis throughout life [
27]. Transplantation of olfactory ensheathing cells in spinal cord injury patients in a phase I/IIa clinical trial demonstrated no adverse events over the 3-year study period, indicating that cells derived from olfactory mucosa are safe for use in vivo [
28]. Additionally, multiple trials of MSC transplantation in spinal cord injury have indicated that these cells are generally well tolerated and safe for treatment of the CNS [
13].
We have previously demonstrated that hOM-MSCs are potent mediators of CNS repair in vitro. In mixed cultures of dissociated rodent CNS cells, treatment with conditioned media (CM) from hOM-MSCs resulted in increased myelination levels compared with those seen with hBM-MSC CM [
29]. Multiplex analysis of the secretome of hOM-MSCs and hBM-MSCs indicated that while both secreted a wide array of cytokines and chemokines (as is typical of MSCs), CXCL12 was the only analyte found to be significantly higher in hOM-MSC-conditioned media (CM) compared with other cell groups [
30]. As well as secreting more CXCL12, hOM-MSCs secreted lower levels of several pro-inflammatory cytokines/chemokines (IL-6, CXCL8, and CCL2) compared to hBM-MSCs. MicroRNA expression analysis indicated that, while sharing 64% identity with hBM-MSCs, 24 were differentially expressed in hOM-MSCs [
30], with previous pathway analysis indicating that one of these (miRNA-140-5p) increased expression of CXCL12 [
31]. Direct CXCL12 treatment of in vitro myelinating cultures increased myelination, while use of antagomirs of miRNA-140-5p, neutralizing antibodies of CXCL12, or the CXCR4 antagonist AMD3100 all decreased myelination in these cultures (as well as those treated with hOM-MSC CM), confirming CXCL12 as a key secretory factor in this phenotype [
30]. hOM-MSCs also demonstrated the capacity to polarize microglia toward an anti-inflammatory phenotype in vitro [
30], further indicating potential reparative functions for these cells in the CNS.
We have also demonstrated the efficacy of hOM-MSCs in vivo. In the rat thoracic spinal cord contusion model, injection of hOM-MSCs into the injury site encouraged earlier coordinated gait recovery, with enhanced Schwann cell-mediated remyelination of spared nerve fibers surrounding the injury site [
32]. Moreover, in the experimental autoimmune encephalitis (EAE) model, intravenous (IV) injection of hOM-MSCs was seen to reduce disease severity and recovery time compared with control animals and those injected with hBM-MSCs. Furthermore, animals injected with hOM-MSCs saw reduced immune cell infiltrate and axonal abnormalities, as well as improved blood-brain barrier integrity [
33].
However, in these studies, hOM-MSCs were isolated and cultured using antibiotics and xeno-derived components. For advancement to clinical trials, these processes must be adapted to meet full GMP compliance. Herein, we describe a GMP-compatible method for the isolation and expansion of hOM-MSCs and evaluate the surface marker expression and secretory profile of the resultant cells. We also interrogate the effects of cryopreservation and different media formulations on cellular phenotypes. Finally, we evaluate these hOM-MSCs in the EAE model to determine efficacy in vivo.
3. Discussion
We describe in this study a standardized and GMP-compatible protocol for the isolation and expansion of hOM-MSCs. In the future, achieving a fully GMP-compliant cell product for use in humans would require the use of appropriate manufacturing facilities. However, by completely removing our reliance on xeno-derived components in the culture medium and refining our sample preparation and culture methods to eliminate microbial contamination (without relying on antibiotics), we are confident that we have developed a process for the establishment of hOM-MSCs that is readily translatable to full GMP compliance. These cells have been thoroughly characterized, conform to ISCT criteria for identification as MSCs [
34], demonstrate high expression of nestin and CXCL12 (a previously established trait of hOM-MSCs [
29,
30,
33]), and so we would propose these as key release criteria for any future cell product developed from this method. Additionally, in 3/3 samples tested in a GMP-compliant QC lab, endotoxin levels within spent culture media from these cells were below the limit of detection for the assay (
Appendix B). Given that the ultimate end goal for such a product would be the treatment of CNS injury and disease (an immune-privileged anatomical region), we would also suggest that an acceptable endotoxin release level of effectively zero be used for future release criteria.
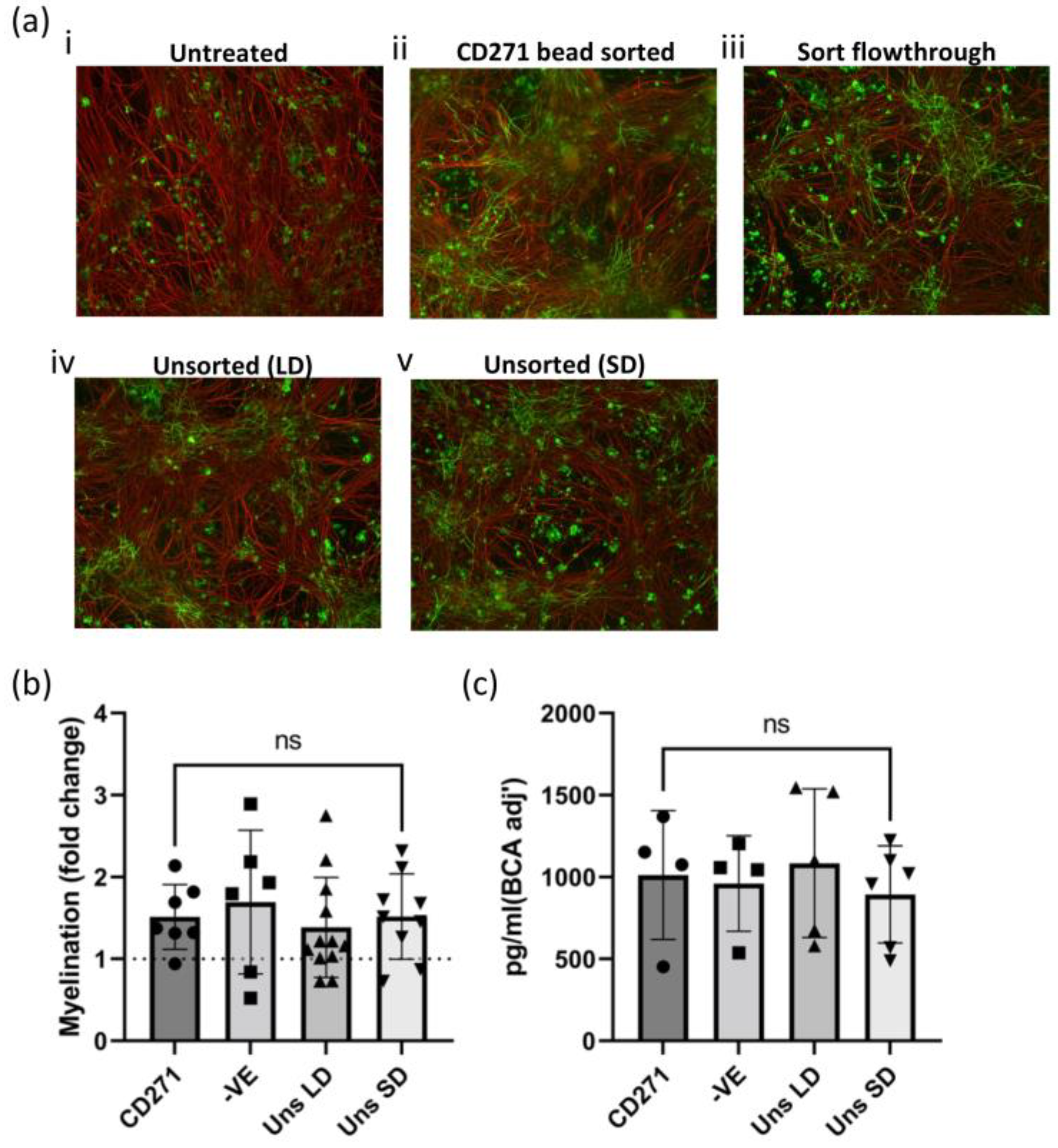
Previous methods of generating hOM-MSCs utilized non-GMP-compliant CD271 bead sorting [
29,
30]. We have demonstrated elsewhere that CD271-sorted cells have superior pro-myelinating and neuroprotective properties compared with cells grown from the negative sort fraction and hBM-MSCs [
29,
30,
32,
33]. However, MSCs isolated from other tissues often do not require sorting and instead are developed into a homogenous population by culture methods alone [
35,
36]. Given that hOM-MSC has previously demonstrated favorable growth kinetics, we reasoned that such an approach might work here. When analyzed by flow cytometry, pre-sort cells (P0) already demonstrated a predominance of MSC-like cells, with >90% being CD90, CD105, and nestin positive (near 100% for CD73). Interestingly, only a small percentage of P0 cells were positive for CD271 expression (~5%). Moreover, within this CD271+ population, only a smaller proportion (around 15% on average) expressed CD90, CD105, or nestin, with around 80% expressing CD73 (
Appendix A,
Figure A1), indicating not all outgrown CD271+ cells from nasal biopsies were MSCs. However, MSC marker expression was effectively uniform within one passage for both sorted and unsorted cell lines, with CD271 being undetectable from P1 onward (including in CD271-sorted cell lines). Additionally, unsorted cells were equivalent to sorted counterparts in their capacity to produce CXCL12 and promote myelination in vitro, further confirming that sorting is not required to generate cells with favorable characteristics. Collectively, these data would appear to indicate that CD271 positive selection may not be required to obtain a pure population of hOM-MSCs, and that cell lines established without sorting are phenotypically equivalent to those that are.
Enzymatic digestion was previously utilized in our tissue processing protocol [
29,
30], and was also recently used in the development of a GMP-compliant method for the isolation of olfactory ecto-mesenchymal stem cells (analogous to hOM-MSCs), where it was found to improve initial cell yield and reduce contamination [
37]. However, other investigations conflict with this, reporting that enzymatic digestion can negatively impact initial MSC recovery, viability, surface marker expression, and growth factor production and increase heterogeneity in the initial cell population [
38,
39,
40]. Considering this and our own flow cytometry results indicating > 90% MSC identity of initially outgrown cells, the decision was made to take a purely explant-based approach to tissue processing throughout this study.
As well as substantially advancing hOM-MSCs toward GMP compliance, this explant and passaging method is also simpler, cheaper, and bypasses the need to develop GMP-compliant sorting methods (with expensive associated reagents). However, it does infer that, in previous studies, the sorting of hOM-MSCs mostly served to debulk the starting population to a far smaller number from which homogenous hOM-MSCs grew out. By starting with a comparatively sparse starting density of unsorted cells, we may, in essence, be replicating this effect. This would also be advantageous in expanding hOM-MSCs to clinically relevant quantities quickly, however, as a larger proportion (if not all) of the initial cellular outgrowth from a tissue sample could be used to establish a cell line rather than a relative few sorted cells.
The ability to be cryopreserved and recovered without significant loss of potency or viability greatly increases the utility of cell therapies, allowing for longer-term storage and the potential to establish cell banks for more rapid treatment [
41,
42]. Given that earlier intervention correlates with better recovery of function in conditions such as spinal cord injury [
13,
43], establishing that GMP-compatible hOM-MSCs are not negatively affected by cryopreservation and recovery is critical. We have demonstrated here that hOM-MSCs are highly amenable to cryopreservation, with no significant differences in phenotypic marker expression, CXCL12 production, or promotion of myelination compared with fresh cells. Therefore, GMP-compatible hOM-MSCs can be cryopreserved without altering their phenotype or compromising their favorable characteristics. In comparison, the reported effects of cryopreservation of hBM-MSCs show considerable variability, with a wide range of post-thaw viability described and some reports of attenuated paracrine effects [
44]. Consequently, hOM-MSCs may be more suitable for cryopreservation and ‘banking’ than hBM-MSCs.
Previous work utilized αMEM supplemented with 10% FCS to culture hOM-MSCs [
29,
30]. In this study, we have again used αMEM (as GMP-compliant variants are widely available and are identical in composition) but switched to a GMP-compliant human platelet lysate supplement (nLiven). While we have found hOM-MSCs thrive in this medium, changes in culture media have previously been observed to induce phenotypic changes in MSCs [
19,
45,
46]. However, when grown in parallel in either 10% FCS-supplemented or GMP-compatible media, hOM-MSCS demonstrated no significant changes in observed phenotype, indicating that the transition to GMP-compliant alternatives has not diminished their potential therapeutic value.
To evaluate GMP-compatible hOM-MSCs in vivo, the EAE model was selected, as hOM-MSCs have previously demonstrated potency in this model of immune-mediated demyelination [
33]. While treatment with hOM-MSCs did not cause any adverse events during the clinical course and demonstrated no enhanced lethality (
Appendix A,
Figure A2), it did not significantly improve disease outcomes. Differences between treated and control animals were minor and did not meet the threshold for statistical significance over several measures. This observation was surprising, given our in vitro data. However, this could be attributable to several factors. Firstly, disease severity was higher than anticipated in these experiments (particularly in animals immunized with full-length MOG protein), with a number of animals culled on welfare grounds. It is conceivable that in such a runaway inflammatory environment, hOM-MSCs are insufficient to curtail disease. Cells grown in 10% FCS did demonstrate some minor (but significant) changes in the expression of negative MSC markers compared with those grown in GMP-compliant media. However, critically, none approached the 5% threshold for these markers stipulated by the ISCT, so the biological significance of these differences is unclear. Additionally, cells grown in 10% FCS did trend toward slightly higher CXCL12 production than those grown in GMP-compliant media, although not significantly so, and so it is unclear if this would account for the lack of efficacy of GMP-compatible cells in vivo. Less overt phenotypic changes introduced by the change to GMP-compliant media cannot be detected in these assays and may require more in-depth transcriptomics to discern. It is also important to highlight that while we have evaluated these cells in both the historic and new media formulations, we did change the supplier of FCS from that used previously (due to issues of availability). Although unlikely to be a sole factor for observed differences from our prior findings, this, coupled with CD271 selection, may have introduced subtle phenotypic differences that were not observable in our in vitro characterization of these cells. Therefore, further pre-clinical work is warranted, likely in alternative models of demyelinating disease (such as the spinal cord contusion model).
Regardless, we demonstrate here that hOM-MSCs can be isolated and expanded using techniques and reagents that are readily translatable to full GMP compliance. These cells conform to ISCT minimal criteria for identification as MSCs, can be cryopreserved without phenotypic change, promote myelination in vitro, and are safe for intravenous delivery in vivo. As such, we would suggest further development of GMP-compatible hOM-MSCs as a potentially valuable asset in developing multi-faceted treatment strategies for demyelinating conditions in the CNS.
4. Materials and Methods
4.1. Nasal Biopsy Processing and hOM-MSC Cell Line Generation
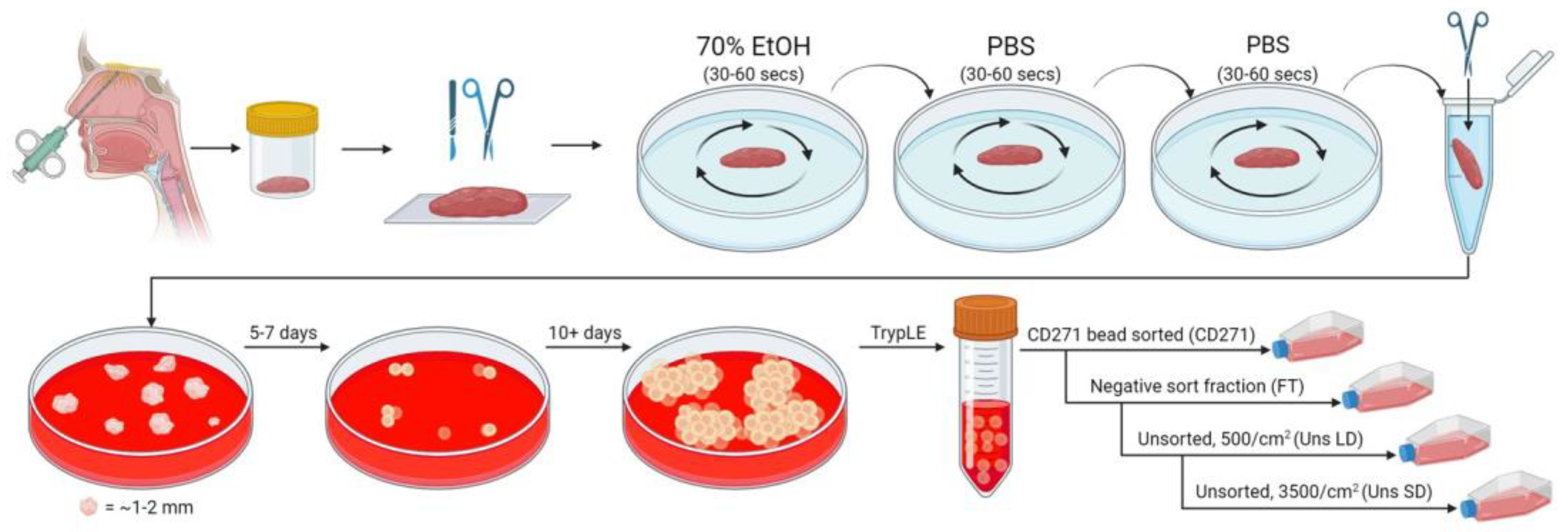
Biopsies were obtained with Central Office for Research Ethics Committees (COREC, REC reference 07/S0710/24) ethical approval and informed patient consent from six patients undergoing routine nasal septoplasty/polypectomy surgery (in accordance with the requirements of the Declaration of Helsinki). Tissue was taken from the nasal and sinus mucosa (primarily from ethmoid sinus mucosa), from which we have previously shown mesenchymal stromal cells (hOM-MSCs) that promote CNS repair can be derived [
29,
30]. Biopsies were collected on ice in Hanks balanced salt solution (HBSS) containing penicillin (100 units/mL), streptomycin (100 μg/mL), and fungizone (Amphotericin B, 1.25 μg/mL) (all Thermo Fisher Scientific, Waltham, MA, USA) for transport to our tissue culture facilities. In a biological safety cabinet (BSC) and under sterile conditions, tissue was placed in a Petri dish and extraneous bone and non-mucosa tissue removed. All subsequent steps were also conducted in a BSC under sterile conditions. The remaining tissue was ‘washed’ by swirling in a Petri containing 70% ethanol for 30–60 s, then ‘rinsed’ in the same manner in PBS (Thermo Fisher Scientific, Waltham, MA, USA, step performed twice) to remove residual alcohol. Tissue was then transferred to an Eppendorf containing 1 mL of culture media (αMEM with nucleosides and Glutamax (Thermo Fisher Scientific, Waltham, MA, USA), supplemented with 5% nLiven PR GMP-ready human platelet lysate (Sexton Biotechnologies, Indianapolis, IN, USA), henceforth referred to as ‘MSC media’ and used throughout this study unless otherwise stated). Using straight spring-bow surgical scissors, tissue was then minced until no pieces larger than 1–2 mm remained. The processed sample was transferred to a 10 cm TC-treated culture dish (Corning, New York, NY, USA) containing MSC media, which was then placed in a humidified incubator (set to 37 °C, 5% CO
2) and left undisturbed for 5–7 days. By this time, some tissue had adhered to the culture dish, and small clusters of outgrowing cells were apparent. Spent media and unadhered tissue were removed, and the adherent tissue and cells were washed gently with PBS. MSC media was replaced, the dish returned to the incubator, and the media was changed every 5 days. For endotoxin testing, the initial processed tissue was split between two dishes and grown in parallel in either MSC media or media further supplemented with antibiotics (1% penicillin/streptomycin solution, Sigma Aldrich, St. Louis, MO, USA). Spent media was reserved and sent for analysis at a clinical-grade QC laboratory for endotoxin testing. This testing indicated no detectable presence of endotoxin in any sample tested (
Appendix B).
Once large cell clusters were present and beginning to merge (~10 days), these were considered ready for further processing. Cell monolayers were washed twice with PBS, then incubated in TrypLE Select dissociation buffer (Thermo Fisher Scientific, Waltham, MA, USA), returned to the incubator, and agitated every few minutes until cells began to lift off. TrypLE was then neutralized with a 4× volume of culture media, and cell clumps were broken up by trituration. The cell suspension was passed through a 70 µm cell filter (Greiner, Kremsmünster, Austria), pelleted, and resuspended in PBS, with cell counts taken. Where possible, unsorted cells from this initial out-growth phase (considered passage 0/P0) were reserved for analysis by flow cytometry.
Cell lines were established from outgrown cells in four different ways. Unsorted cells were used to establish cell lines by initially seeding them at either 500–750 cells/cm
2 (unsorted low density, Uns LD) or 3500 cells/cm
2 (unsorted standard density, Uns SD) in MSC media on TC-treated culture flasks (Corning, New York, NY, USA). Sorting of hOM-MSCs was also performed using a CD271 positive selection kit (StemCell Technologies, Vancouver, Canada) as described previously. In brief, cells were resuspended in 100 µL sorting buffer (PBS +0.5% bovine serum albumin (BSA, Sigma Aldrich, St. Louis, MO, USA) and 2 mM EDTA, Thermo Fisher Scientific, Waltham, MA, USA, henceforth referred to as PEB), and transferred to a 5 mL round bottom polystyrene tube (Corning, New York, NY, USA). Here, 2.5 µL FcR blocker and 5 µL antibody cocktail per sample were added to this, with the sample mixed and incubated for 15 min at room temperature. Then, 5 µL of RapidSpheres was added to each sample, mixed, incubated for a further 5 min, and then brought up to a volume of 2.5 mL with PEB. The tube was placed in the sorting magnet for 5 min, the supernatant was transferred to a clean tube, and cells were resuspended in 2.5 mL PEB. This step was repeated, and the remaining sorted cells were washed once in PBS and resuspended in 0.25 mL of MSC media. These were seeded in a single spot in a TC-treated flask and placed in the incubator for 2 h to allow cells to attach, after which the full volume of MSC media was added to the flask (CD271 sorted). Flowthrough cells from the sort (i.e., the negative fraction) were washed with PBS, resuspended in MSC media, cell counts taken, and seeded at 3500 cells/cm
2 in TC-treated flasks (–VE) (summarized in
Figure 1). Also, 75 cm
2 tissue culture flasks (Corning, New York, NY, USA) were used for maintaining cultures throughout. The only exception was with CD271-sorted cells, which were cultured initially in 25 cm
2 flasks (Corning, New York, NY, USA) at P0 due to low cell numbers and in 75 cm
2 for subsequent passages.
4.2. Cell Culture
Once established, all new cell lines were maintained in the same manner, and grown in MSC media on TC-treated plastic (Corning, New York, NY, USA). Once reaching 70–90% confluence, cell monolayers were washed twice with PBS, incubated with a volume of TrypLE sufficient to cover the cell layer, and returned to the incubator. Cells were occasionally agitated until lifted from the plastic, TrypLE was neutralized using 4× volume of MSC media, and a monocellular suspension was created by trituration by pipette. Cells were pelleted (300× g for 5 min), supernatant discarded, and cells resuspended in an appropriate volume of MSC media. Cell counts were performed, and new flasks were seeded at 3500 cells/cm2, with unused cells retained for further analysis. These were passaged a total of three times, with aliquots of cells at each passage cryopreserved wherever possible (see below). Where indicated, hOM-MSCs were also grown in αMEM supplemented with 10% FCS (Sigma Aldrich, St. Louis, MO, USA), as per their previous culture conditions.
4.3. Flow Cytometry and Antibody Panel
Single suspensions of hOM-MSCs were created as described above, resuspended in PBS, and cell counts taken. The preparation of the flow samples was performed as described in [
47]. In brief, aliquots of 2–3 × 10
5 cells per sample (consistent within each run) were transferred to a 96-well round bottom plate (Corning, New York, NY, USA), with residual cells pooled and used for setting up controls. From this point, all subsequent steps were performed while protected from the light, and at 4 °C unless specifically stated otherwise. Cells were washed in PBS, then stained with fixable viability stain (Thermo Fisher Scientific, Waltham, MA, USA, 1:1000 in PBS) for 15 min. Cells were washed in PEB, then incubated with a human FcR blocking agent (Miltenyi Biotec, Bergisch Gladbach, Germany, 1 µL per sample in 200 µL PEB) for 10 min. Samples were then resuspended in 50 µL of the full surface marker antibody staining cocktail or a fluorescence minus one control (where a single antibody is excluded as a negative control for that stain) in Brilliant Stain buffer (BD Biosciences, Franklin Lakes, NJ, USA, antibody cocktail detailed in
Table 1) for 20 min. Antibodies were obtained for each marker from the manufacturer indicated in
Table 1 (either Biolegend, San Diego, CA, USA or BD Biosciences, Franklin Lakes, NJ, USA). Samples were then washed with PEB and prepared and stained for intracellular markers (nestin; see
Table 1) using the eBiosciences Intracellular Fixation and Permeabilization kit (Thermo Fisher Scientific, Waltham, MA, USA), according to kit instructions. Samples were analyzed using a BD LSR Fortessa analyzer (BD Biosciences, Franklin Lakes, NJ, USA, based in the School of Immunity and Inflammation’s Flow Cytometry Core Facility), and the resulting data were analyzed using FlowJo v10 software (BD Biosciences, Franklin Lakes, NJ, USA). Flow cytometry data is expressed as the percentage (%) of live cells, unless otherwise stated. Gating strategies are described in
Appendix A,
Figure A3.
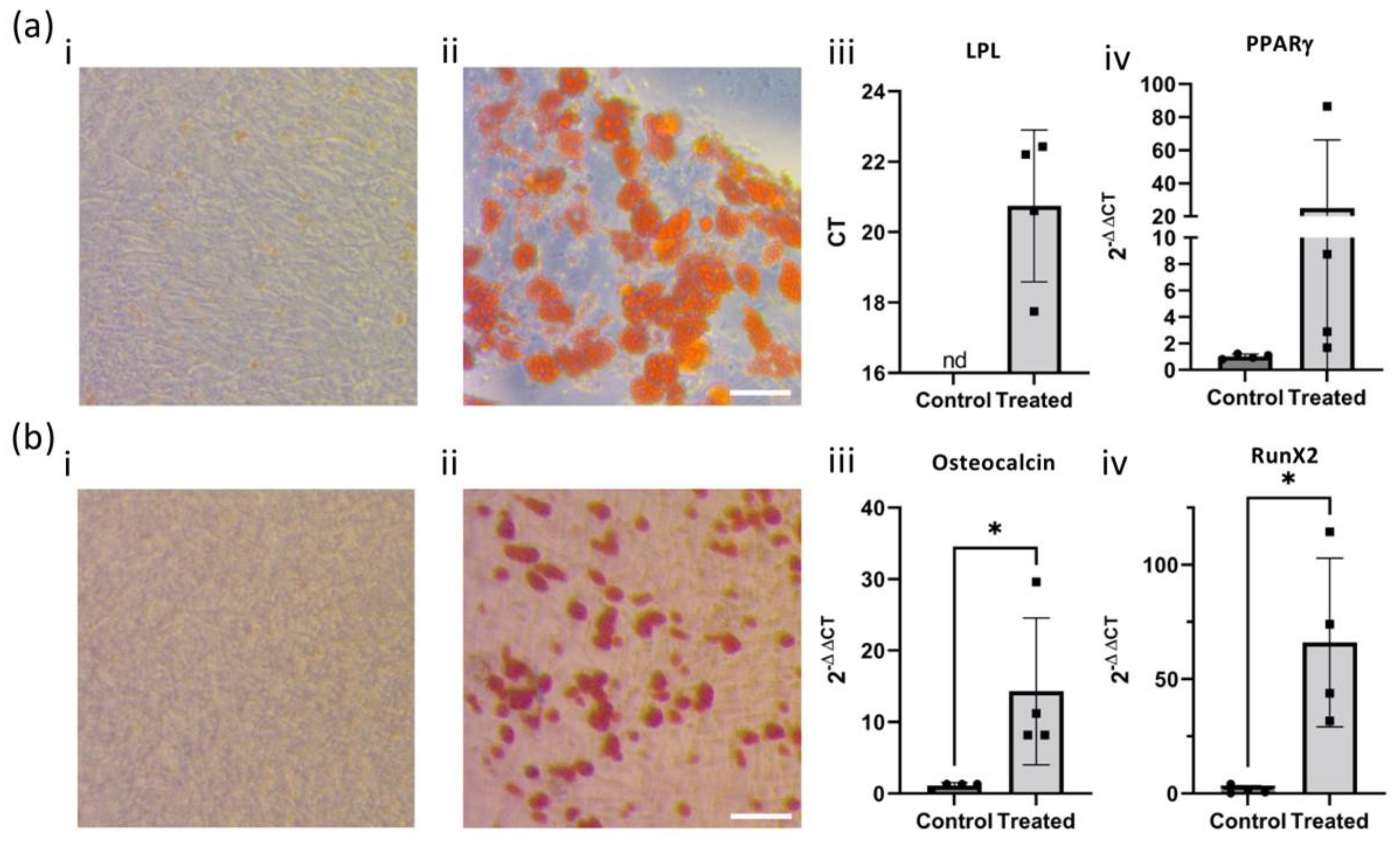
4.4. Adipogenic/Osteogenic Differentiation
A selection of hOM-MSCs (Uns SD, passage 3) were selected for use with either the StemPro Adipogenesis or Osteogenesis Differentiation Kits (Thermo Fisher Scientific, Waltham, MA, USA), in accordance with the manufacturer’s instructions with slight modifications. In brief, in both cases, hOM-MSCs were seeded into either 6-well (Corning, New York, NY, USA) or 48-well (StarLab, Milton Keynes, UK, seeded in triplicate) plates at a density of 1 × 104 cells/mL in MSC medium. Upon reaching 100% (adipogenesis) or 70% (osteogenesis) confluency, these were switched over to differentiation media, which was subsequently refreshed every 3–4 days. At either 28 (adipogenesis) or 42 (osteogenesis) days, cells were either stained for lipid/calcium deposition (48-well) or harvested for RNA (6-well).
4.5. Oil Red O/Alizarin Red Staining
Differentiated cultures were fixed by removing differentiation media, washing cell layers twice with PBS, and being fixed in 4% paraformaldehyde (PFA, Sigma Aldrich, St. Louis, MO, USA) in PBS for 20 min at room temperature. PFA was then removed, and cultures were washed twice with PBS. Oil Red O staining of lipid deposits was performed as previously described. In brief, PBS was removed, cells were washed ×1 with distilled water and ×1 with 60% isopropanol (Sigma Aldrich, St. Louis, MO), and they were incubated in a 0.3% Oil Red O (Sigma Aldrich, St. Louis, MO, USA) solution in 60% isopropanol for 15 min at room temperature. The staining solution was then removed, and cells were washed several times with 60% isopropanol until the non-bound stain could no longer be removed. Alizarin Red S staining for calcium deposition was also performed as described by others. In brief, PBS was removed, cells were washed twice with distilled water, and Alizarin Red S (Sigma Aldrich, St. Louis, MO, USA) staining solution pH 4.2 sufficient to cover the cell layers was added to each well and incubated at room temperature for 5 min, protected from light. Each well was then rinsed 3× with distilled water. In both cases, distilled water was added to cultures after final wash steps to prevent dehydration. These were imaged with a Zeiss Primovert light microscope at 400× magnification, with images captured using the AxioCam ERc5s camera using Zeiss Zen 2.3 (blue edition) software.
4.6. RNA Extractions, cDNA Conversions and qPCR Analysis
Primer sequences can be found in
Table 2. Genes of interest to confirm adipo/osteogenic differentiation were selected [
48] primer pairs were designed to span exon junctions using Primer3 [
49]. RNA extraction, cDNA conversion, and qPCR were performed as described [
50]. In brief, RNA was extracted from basal and differentiated hOM-MSCs (PureLink RNA Mini Kit, Thermo Fisher Scientific, Waltham, MA, USA), with on-column DNase digestion performed (RNase-Free DNase set, Qiagen, Hilden, Germany) to remove contaminating genomic DNA. RNA concentration was determined using the DeNovix DS-11+ spectrophotometer (DeNovix, Wilmington, DE, USA), and 0.5/1 μg of RNA was converted to cDNA (High-Capacity RNA-to-cDNA kit, Thermo Fisher Scientific, Waltham, MA, USA). The resulting cDNA was diluted in nuclease-free water (Qiagen, Hilden, Germany, diluted 1/5 or 1/10 depending on the starting RNA quantity) for use in qPCR reactions. qPCR reactions were prepared in 384-well plates using PerfeCTa SYBR Green FastMix ROX (Quantabio, Beverly, MA, USA), a final primer concentration of 0.75 μM, 1 μL cDNA template per reaction, and a total volume of 10 μL with nuclease-free water. Plates were run on the QuantStudio 7 Flex system (Thermo Fisher Scientific, Waltham, MA, USA), with results analyzed and exported to spreadsheets using the onboard software. Data were expressed relative to housekeeping and undifferentiated controls using the 2
−ΔΔCT method.
4.7. Conditioned Media and Myelinating Cultures
Conditioned media was prepared as follows: hOM-MSC at passage 3 was grown to 100% confluency in MSC media in 75 cm2 TC flasks (Corning, New York, NY, USA). At this point, spent media was aspirated, cells washed once with PBS, and media replaced with 11 mL DMEM-media (DMEM 4.5 g/L glucose, Invitrogen, 50 nM hydrocortisone, 0.5% N1 medium supplement, and 10 ng/mL biotin, all Sigma Aldrich, St. Louis, MO, USA). These cultures were left for 72 h, after which DMEM- was removed, filtered through a 0.2 µm syringe filter (Sigma Aldrich, St. Louis, MO, USA), and stored in a −80 °C freezer until required. When prepared for myelinating cultures, this was diluted 1:2–4 with fresh DMEM-.
Myelinating cultures were set up as described previously [
51]. Briefly, neurospheres generated using striata from 1-day-old Sprague-Dawley (SD) rats, triturated by pipette, were seeded onto poly-lysine-coated (using a 13 μg/mL solution, Sigma Aldrich, St. Louis, MO, USA) 13 mm coverslips (VWR), with 3 coverslips per 35 mm Petri dish (Corning, New York, NY, USA). These were incubated for 5–7 days in DMEM (1 g/mL glucose, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% FCS (Sigma Aldrich, St. Louis, MO, USA). Once complete monolayers of astrocytes were apparent, these were used as support cells to set up mixed-cell myelinating cultures. For these, spinal cords were dissected from E15.5 SD spinal cords and subsequently digested with a mixture of collagenase I (1.33%
w/
v in L-15 media, both Thermo Fisher Scientific, Waltham, MA, USA) and 0.25% Trypsin-EDTA (Sigma Aldrich, St. Louis, MO, USA) for 15 min at 37 °C. Reactions were quenched with a mix of 0.52 mg/mL soybean trypsin inhibitor, 3 μg/mL BSA fraction V, and 0.04 mg/mL DNase (all Sigma Aldrich, St. Louis, MO, USA), again prepared in L-15 media. The tissue mixture was pelleted (at 800 rpm for 5 min), and cells were resuspended in plating media (50%
v/
v DMEM, 1 g/mL glucose, 25%
v/
v horse serum, 25%
v/
v HBSS, and 2 mM L-glutamine, all Thermo Fisher Scientific, Waltham, MA, USA).
Cell counts were taken, volume adjusted to give a density of 1.5 × 106 cells/mL, and 100 μL of this suspension seeded directly on to the coverslips. These were placed in the incubator for 2 h to allow cells to adhere, after which they were supplemented with an additional 300 μL of plating media and 500 μL of DMEM+ (same formulation as DMEM-, with the addition of 0.5 mg/mL insulin, Sigma Aldrich, St. Louis, MO, USA). Media was replenished by removing 400 μL and adding 500 μL DMEM+ (to account for evaporation) every 2–3 days up to 12 days. From days 12–28, media was replenished with either basal DMEM- or the previously mentioned CM/DMEM- mix, again replenished every 2–3 days. After 28 days, media was removed, cultures washed twice with PBS, and fixed with 4% paraformaldehyde (PFA, Sigma Aldrich, St. Louis, MO, USA) for 20 min at room temperature. PFA was removed, cultures washed twice with PBS, and a volume of PBS sufficient to comfortably cover the cell layers was added for storage at 4 °C until required for staining and imaging (plates sealed with parafilm to prevent evaporation).
4.8. Immunocytochemistry
To immunolabel the cells, they were first permeabilized with 0.2% Triton X-100 (Sigma Aldrich, St. Louis, MO, USA) at room temperature (RT) for 15 min, and blocked with phosphate-buffered saline (PBS) with 0.2% porcine gelatin (blocking buffer, Sigma Aldrich, St. Louis, MO, USA) for 1 h at RT. The primary antibodies were diluted in a blocking buffer, and the cells were incubated for 1 h at RT. Mature myelin (proteolipid protein, PLP) was visualized using the AA3 antibody (1:100, anti-rat; hybridoma supernatant gift [
52]), and neurofilament was detected using SMI31 (mouse IgG1, 1:1500, Biolegend, San Diego, CA, USA). After washing, the cultures were incubated with the appropriate secondary antibodies for 45 min at RT and mounted under coverslips using Vectashield anti-fade (Vector Laboratories, Peterborough, UK).
4.9. Microscopy and Image Analysis
Quantification of myelination was carried out by collecting images at 10× magnification using an Olympus BX51 (Olympus, Essex, UK). For each coverslip, 10 images were taken randomly, covering the entire coverslip. In each biological repeat, coverslips were stained in triplicate; thus, 30 images were taken per condition (at least n = 3 biological repeats). The analysis of treated cultures was carried out by comparing them to non-treated control cultures run in parallel. Analysis was carried out using Cell Profiler Image Analysis software (Broad Institute, v1.3) and is available to download at
https://github.com/muecs/cp (accessed 8 May 2022). This uses pattern recognition software to distinguish between linear myelinated internodes and oligodendrocyte cell bodies. In this manner, we track the co-expression of myelin sheaths (PLP) and axons (SMI31), which ignores immature oligodendrocytes lacking axonal contact, and therefore allows us to calculate the percentage of myelinated fibers. Once enumerated, samples were normalized to unconditioned media-treated controls, and values were expressed as fold changes from these controls.
4.10. Protein Extractions and ELISA Analysis
Whole-cell lysate (WCL) was prepared from cells used in the creation of CM. Cells were lifted with TrypLE as described previously, pelleted, and washed once in PBS, then resuspended in M-PER Mammalian Protein Extraction Reagent (Thermo Fisher Scientific, Waltham, MA, USA) and vortexed every 1–2 min for 15 min (with samples kept on ice when not being vortexed). Samples were then centrifuged (>10,000 RPM, 4 °C, 15 min) and supernatants transferred to clean Eppendorf’s and either kept on ice or stored at −20 °C for future analysis. Protein concentration was determined by a BCA assay (Thermo Fisher Scientific, Waltham, MA, USA) and used to normalize ELISA data. ELISA analysis for CXCL12 was conducted on the same CM used for myelinating cultures, using the Human SDF-1α (CXCL12) Mini ABTS ELISA Development Kit and associated ancillary reagents (Peprotech, now part of Thermo Fisher Scientific, Waltham, MA, USA). Plates were read on the Varioskan LUX plate reader (Thermo Fisher Scientific, Waltham, MA, USA, read at 405 nm absorbance with correction at 650 nm) over various timepoints. The concentration of CXCL12 in CM was calculated against the provided standard and expressed relative to 200/500 μg of WCL.
4.11. Cryopreservation and Recovery
‘Freezing media’ (FM) was prepared as follows: 90% v/v Knockout Serum Replacement (Thermo Fisher Scientific, Waltham, MA, USA), 10% v/v DMSO (Thermo Fisher Scientific, Waltham, MA, USA). Prior to cryopreservation, cells were prepared as a single-celled suspension as described already, resuspended in PBS, and cell counts taken. Cells were then resuspended at a density of 8 × 106 cells/mL in FM, and 2 × 106 cells were transferred to cryovials (Simport Sceintific, Saint-Mathieu-de-Beloeil, Canada). These were then slowly cooled (~−1 °C/minute) using a ‘Mr Frosty’ freezing container (Thermo Fisher Scientific, Waltham, MA, USA) in a −80 °C freezer before being transferred to liquid nitrogen tanks for long-term storage. For recovery, cells were thawed quickly in pre-warmed culture media, pelleted, resuspended in fresh culture media, and counted. They were then seeded in TC flasks at a density of 3500/cm2. The media was refreshed after 24 h to remove any residual DMSO that may have leached out of the cells and to remove any dead cells. From this point, these cells were then cultured as described previously.
4.12. EAE Induction
A total of 33 female (SL ADD) C57Bl/6 J mice were purchased from Harlan Laboratories (Loughborough, UK). All mice were housed under a 12 h light/dark cycle with ad libitum access to food and water in pathogen-free conditions. All experimental procedures were performed in accordance with the UK Animals (Scientific Procedures) Act 1986. All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. The research protocol was approved by the Ethical Committee for Animal Experimentation at the University of Glasgow, UK.
EAE was induced in female mice (7–8 weeks of age, weighing 18.5 ± 1.5 g) by subcutaneous injection at one site at the tail base with an emulsion (100 µL total) containing 200 µg recombinant rat myelin oligodendrocyte glycoprotein protein spanning amino acids 1-125 (MOG1-125) in complete Freund’s adjuvant (Sigma Aldrich, St. Louis, MO, USA) supplemented with 300 µg Mycobacterium tuberculosis (strain H37RA; BD Biosciences, Franklin Lakes, NJ, USA) [
34]. In some cases, mice were instead immunized with myelin oligodendrocyte glycoprotein peptide spanning amino acids 33–55 (MOG33-55) (SynPeptide, Shanghai, China) in complete Freund’s adjuvant (Sigma Aldrich, St. Louis, MO, USA) supplemented with 300 μg Mycobacterium tuberculosis (strain H37RA; BD Biosciences, Franklin Lakes, NJ, USA). Mice were then injected intraperitoneally with 200 ng pertussis toxin (Enzo Life Sciences, Farmingdale, NY, USA) in 100 µL of phosphate buffered saline solution (PBS, pH 7.6) immediately and 48 h after the immunization. The mice were scored daily for clinical manifestations of EAE on a half-point scale of 0–5. hOM-MSCs (1 × 10
6 cells in 100 μL PBS) or PBS (100 μL) were administered when animals showed signs of clinical disease (score of at least 2; hind limb paralysis) by intravenous (IV) injection of the tail vein. Mice were randomly distributed between groups on the day of IV administration and scored daily blind. This treatment strategy prevented the inclusion of asymptomatic animals.
4.13. Statistical Analysis
All statistical analysis performed in this study (and generation of graphical representations thereof) was carried out using the GraphPad Prism statistical software package (versions 9.3.1, GraphPad Software, Boston, MA, USA). The specific statistical tests employed are indicated in the relevant figure legends.

 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}