
Cadmium Induces Kidney Iron Deficiency and Chronic Kidney Injury by Interfering with the Iron Metabolism in Rats




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
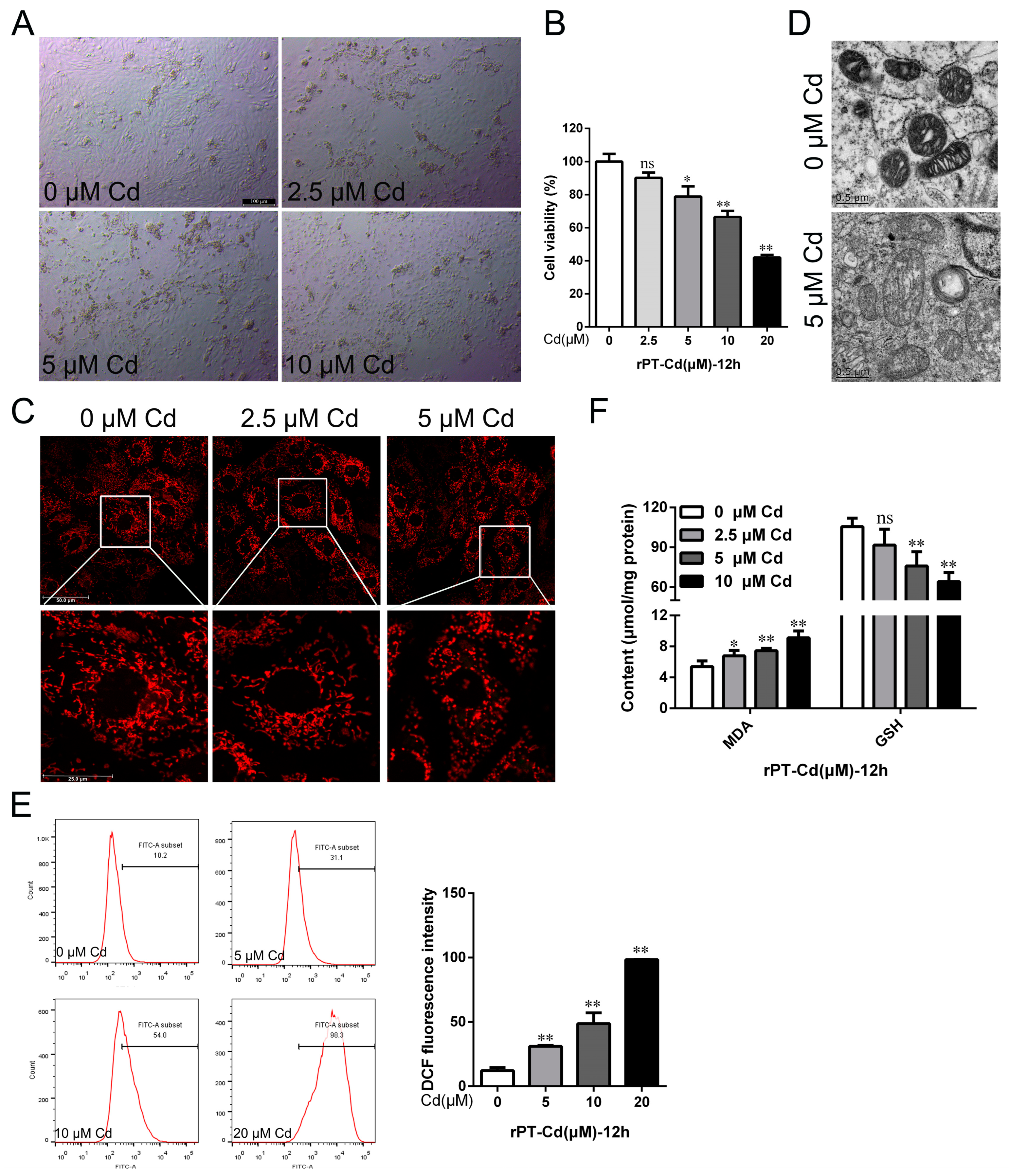
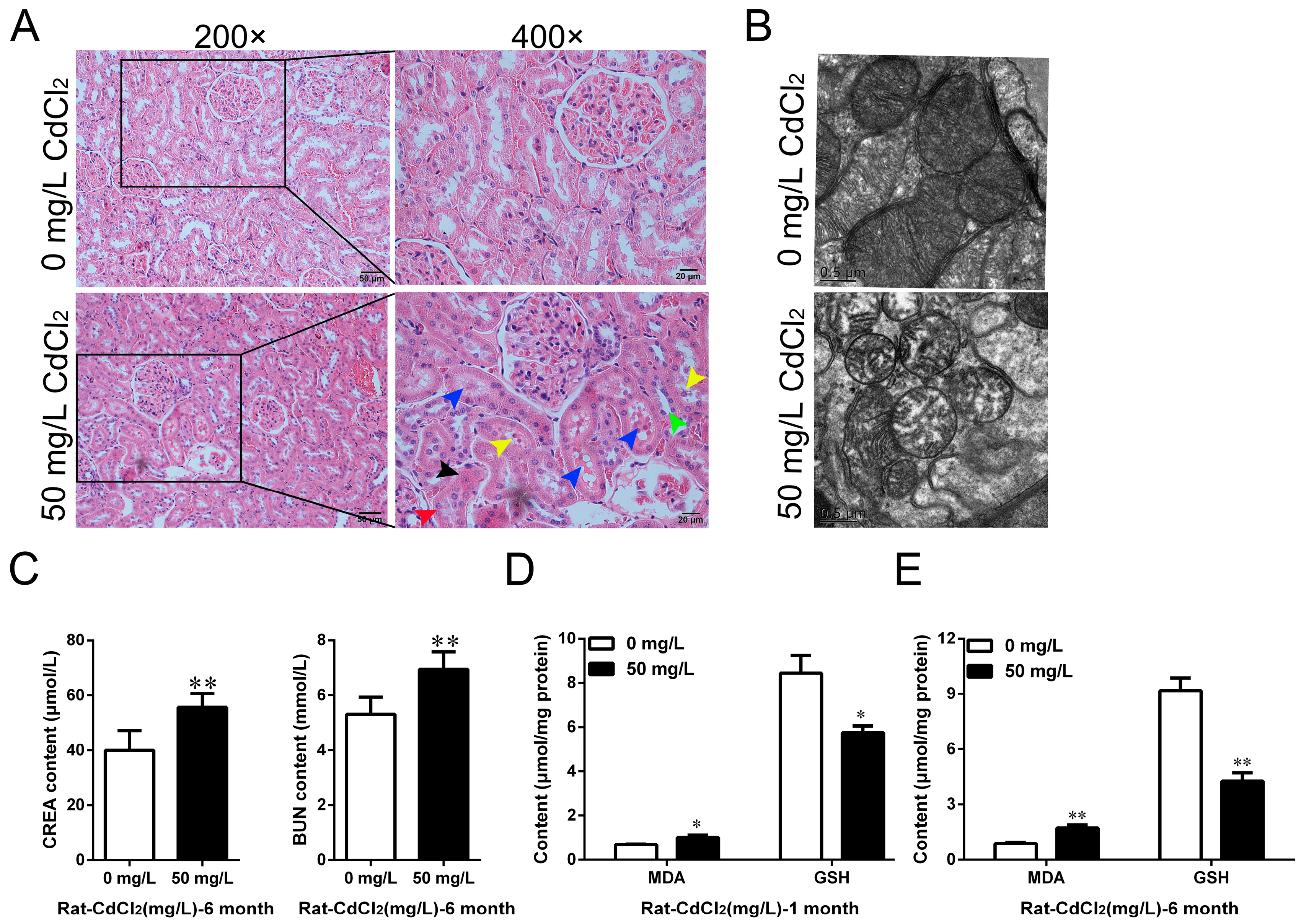
2.1. Cd Induced Oxidative Stress and Chronic Kidney Injury in Rats In Vivo and In Vitro
2.2. Cd Failed to Induce Typical Ferroptosis of Rat Kidneys In Vivo and In Vitro
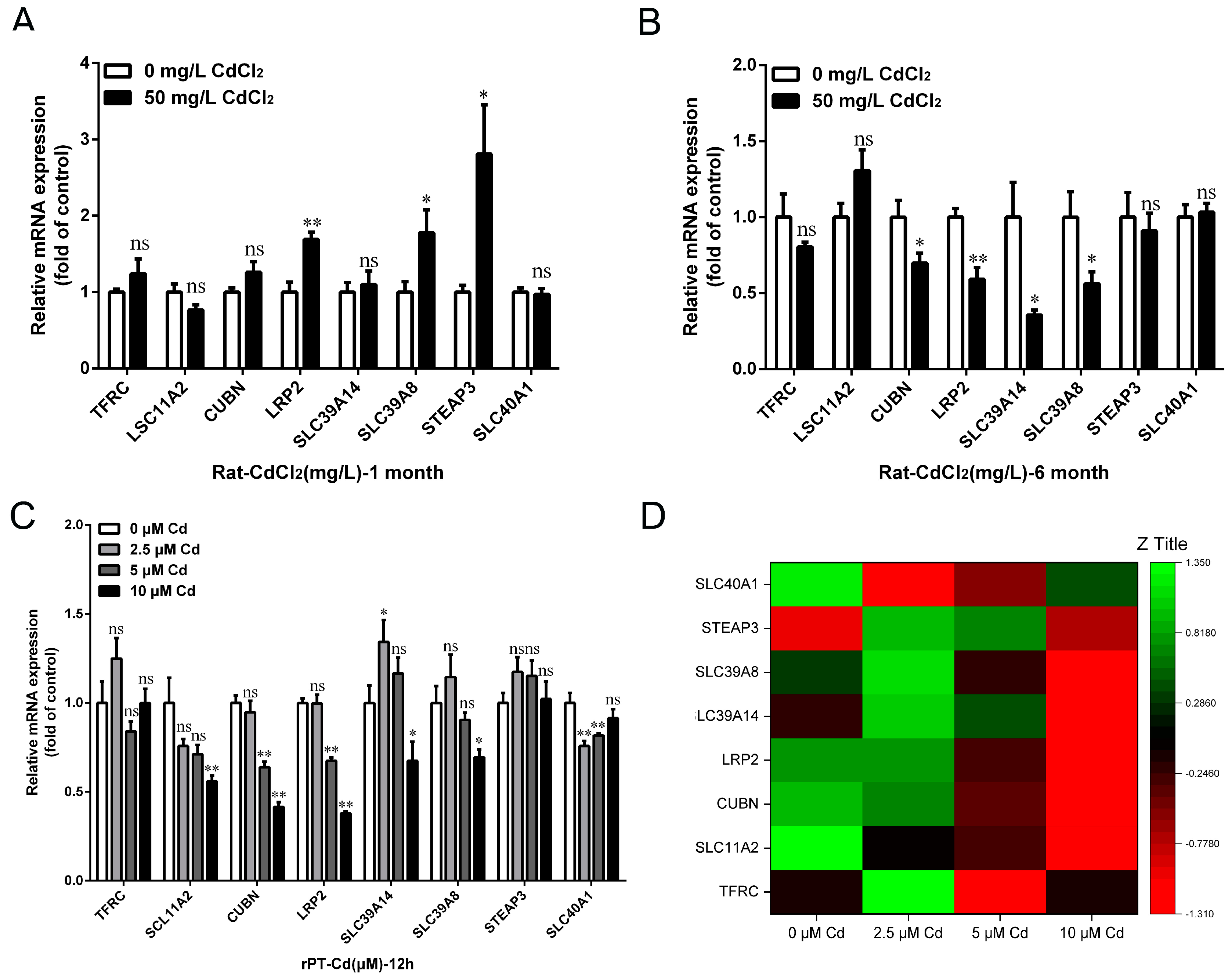
2.3. Cd Reduced Circulating Iron Content by Reducing Iron Absorption in the Small Intestine
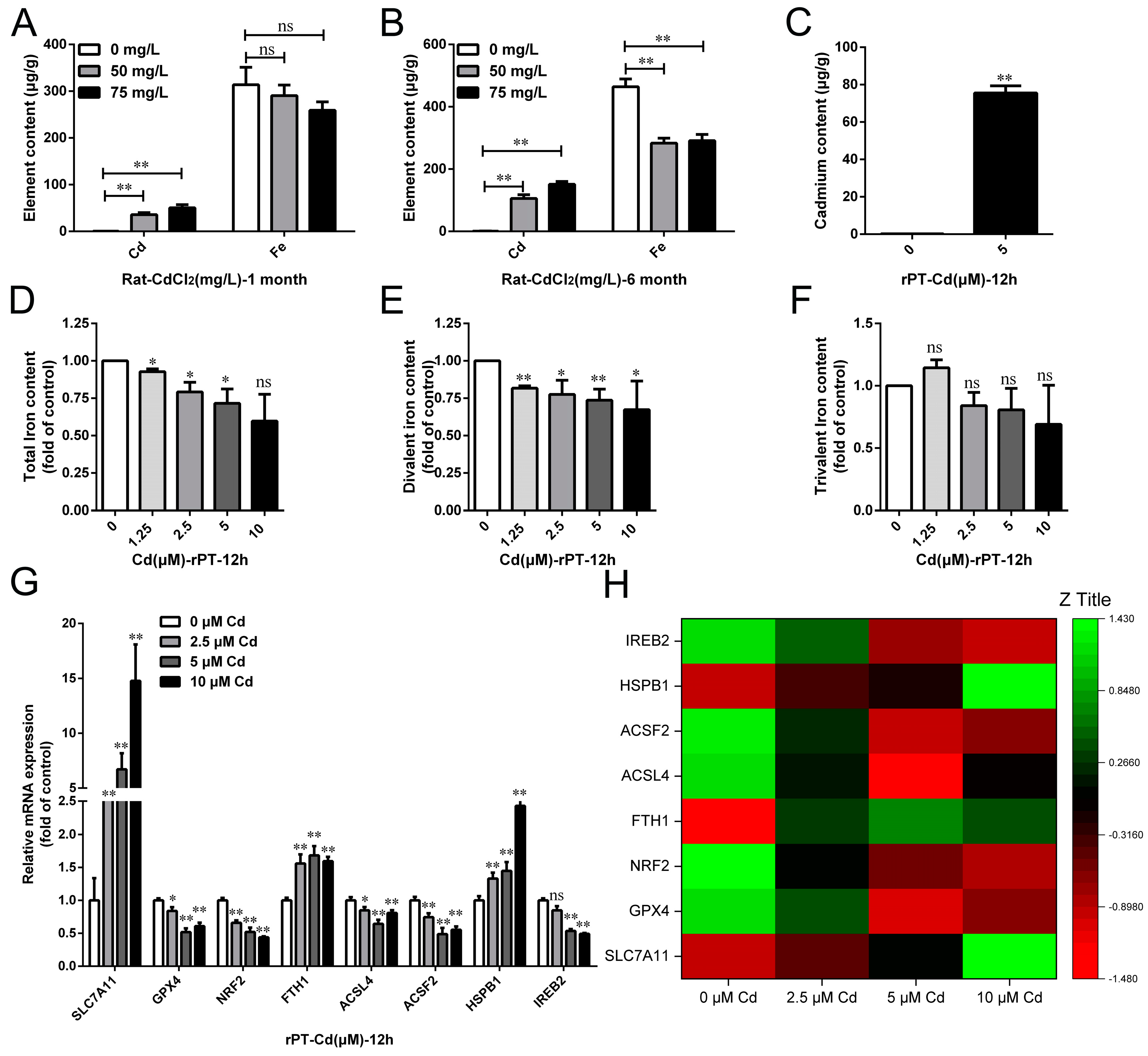
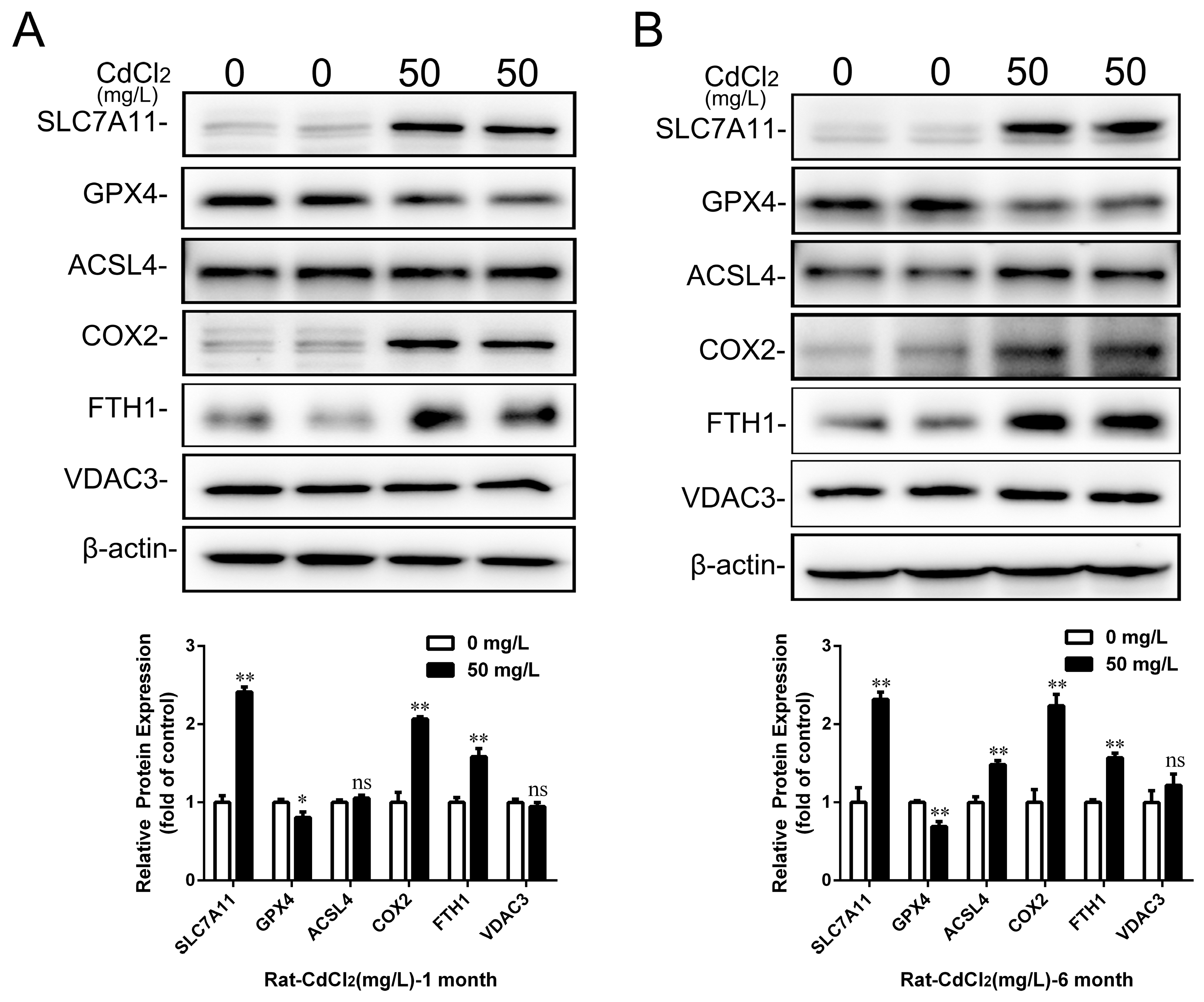
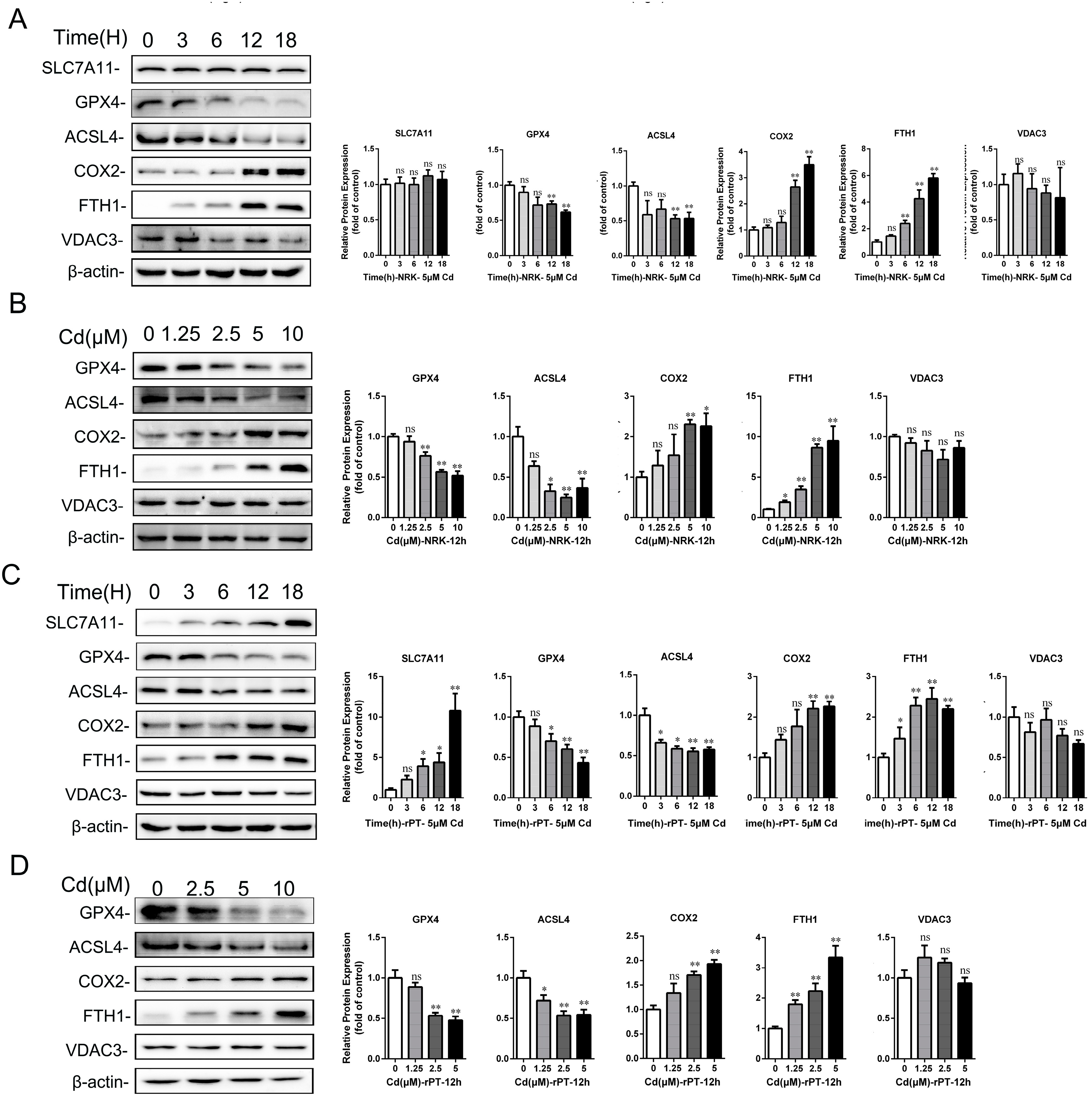
2.4. Cd Induced the Disorder of Iron Metabolism in Kidney and Proximal Tubular Cells of Rats
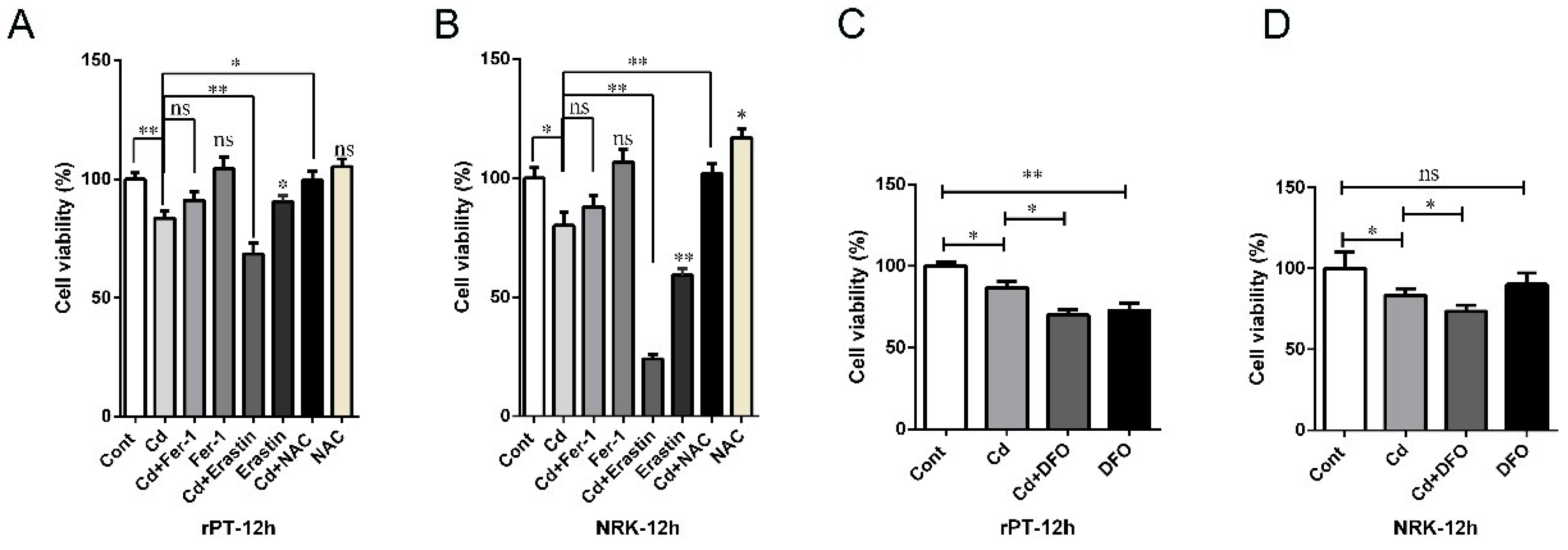
2.5. Cd-Induced Rat Proximal Tubular Cell Damage Was Alleviated by Fer-1 and NAC but Aggravated by Erastin and DFO

3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Animals and Treatments
4.3. Isolation and Culture of Renal Tubular Epithelial Cells
4.4. Detection of Cell Viability by CCK-8 Assay
4.5. Detection of Cadmium and Iron Contents
4.6. Detection of Blood Routine and Serum Biochemistry
4.7. Detection of MDA and GSH Contents
4.8. Detection of ROS Content by Flow Cytometry
4.9. Histopathological Observation
4.10. Observation of Mitochondrial Ultrastructure and Reticular Structure
4.11. Real-Time Quantitative Polymerase Chain Reaction
4.12. Western Blot Analysis
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Ren, S.; Song, C.; Ye, S.; Cheng, C.; Gao, P. The spatiotemporal variation in heavy metals in China’s farmland soil over the past 20 years: A meta-analysis. Sci. Total Environ. 2022, 806, 150322. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wang, L.; Wang, W.; Li, T.; He, Z.; Yang, X. Current status of agricultural soil pollution by heavy metals in China: A meta-analysis. Sci. Total Environ. 2019, 651, 3034–3042. [Google Scholar] [CrossRef] [PubMed]
- Lars, L. Hazards of heavy metal contamination. Br. Med. Bull. 2003, 68, 167–182. [Google Scholar] [CrossRef]
- Genchi, G.; Sinicropi, M.; Lauria, G.; Carocci, A.; Catalano, A. The Effects of Cadmium Toxicity. Int. J. Environ. Res. Public Health 2020, 17, 3782. [Google Scholar] [CrossRef] [PubMed]
- Bernhoft, R.A. Cadmium toxicity and treatment. Sci. World J. 2013, 2013, 394652. [Google Scholar] [CrossRef] [PubMed]
- Thévenod, F.; Lee, W.K. Toxicology of cadmium and its damage to mammalian organs. Met. Ions Life Sci. 2013, 11, 415–490. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, M.; Micali, A.; Marini, H.; Adamo, E.; Puzzolo, D.; Pisani, A.; Trichilo, V.; Altavilla, D.; Squadrito, F.; Minutoli, L. Cadmium, Organ Toxicity and Therapeutic Approaches: A Review on Brain, Kidney and Testis Damage. Curr. Med. Chem. 2017, 24, 3879–3893. [Google Scholar] [CrossRef]
- Ran, D.; Zhou, D.; Liu, G.; Ma, Y.; Ali, W.; Yu, R.; Wang, Q.; Zhao, H.; Zhu, J.; Zou, H.; et al. Reactive Oxygen Species Control Osteoblast Apoptosis through SIRT1/PGC-1α/P53 Signaling, Mediating the Onset of Cd-Induced Osteoporosis. J. Agric. Food Chem. 2023, 71, 5991–6002. [Google Scholar] [CrossRef]
- Satarug, S.; Vesey, D.A.; Gobe, G.C. Kidney Cadmium Toxicity, Diabetes and High Blood Pressure: The Perfect Storm. Tohoku J. Exp. Med. 2017, 241, 65–87. [Google Scholar] [CrossRef]
- Yang, H.; Shu, Y. Cadmium transporters in the kidney and cadmium-induced nephrotoxicity. Int. J. Mol. Sci. 2015, 16, 1484–1494. [Google Scholar] [CrossRef]
- Satarug, S. Dietary Cadmium Intake and Its Effects on Kidneys. Toxics 2018, 6, 15. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.C.; Tang, T.T.; Lv, L.L.; Lan, H.Y. Renal tubule injury: A driving force toward chronic kidney disease. Kidney Int. 2018, 93, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Gobe, G.; Crane, D. Mitochondria, reactive oxygen species and cadmium toxicity in the kidney. Toxicol. Lett. 2010, 198, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Yuan, Y.; Long, M.; Luo, T.; Bian, J.; Liu, X.; Gu, J.; Zou, H.; Song, R.; Wang, Y.; et al. Beclin-1-mediated Autophagy Protects Against Cadmium-activated Apoptosis via the Fas/FasL Pathway in Primary Rat Proximal Tubular Cell Culture. Sci. Rep. 2017, 7, 977. [Google Scholar] [CrossRef]
- Wang, X.; Yang, H.; Wang, M.; Yang, D.; Wang, Z.; Wang, L. Trehalose protects against cadmium-induced cytotoxicity in primary rat proximal tubular cells via inhibiting apoptosis and restoring autophagic flux. Cell Death Dis. 2017, 8, e3099. [Google Scholar] [CrossRef] [PubMed]
- Seyoum, Y.; Baye, K.; Humblot, C. Iron homeostasis in host and gut bacteria—A complex interrelationship. Gut Microbes 2021, 13, 1874855. [Google Scholar] [CrossRef] [PubMed]
- Rawee, P.; Kremer, D.; Nolte, I.; Leuvenink, H.; Touw, D.; De Borst, M.; Bakker, S.; Hanudel, M.; Eisenga, M. Iron Deficiency and Nephrotoxic Heavy Metals: A Dangerous Interplay? Int. J. Mol. Sci. 2023, 24, 5315. [Google Scholar] [CrossRef]
- van Swelm, R.; Wetzels, J.; Swinkels, D. The multifaceted role of iron in renal health and disease. Nat. Rev. Nephrol. 2020, 16, 77–98. [Google Scholar] [CrossRef]
- Thévenod, F.; Wolff, N. Iron transport in the kidney: Implications for physiology and cadmium nephrotoxicity. Met. Integr. Biometal Sci. 2016, 8, 17–42. [Google Scholar] [CrossRef]
- Lee, B.; Kim, S.; Kim, N.; Ham, J.; Kim, Y. Iron deficiency increases blood cadmium levels in adolescents surveyed in KNHANES 2010–2011. Biol. Trace Elem. Res. 2014, 159, 52–58. [Google Scholar] [CrossRef]
- Horiguchi, H.; Oguma, E.; Kayama, F. Cadmium induces anemia through interdependent progress of hemolysis, body iron accumulation, and insufficient erythropoietin production in rats. Toxicol. Sci. Off. J. Soc. Toxicol. 2011, 122, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, Y.; Lee, J.; Banno, H.; Imai, S.; Tokumoto, M.; Hasegawa, T.; Seko, Y.; Nagase, H.; Satoh, M. Cadmium induces iron deficiency anemia through the suppression of iron transport in the duodenum. Toxicol. Lett. 2020, 332, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Jurczuk, M.; Brzóska, M.; Rogalska, J.; Moniuszko-Jakoniuk, J. Iron body status of rats chronically exposed to cadmium and ethanol. Alcohol Alcohol. 2003, 38, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Broadening horizons: The role of ferroptosis in cancer. Nat. Rev. Clin. Oncol. 2021, 18, 280–296. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis 2020, 11, 88. [Google Scholar] [CrossRef]
- Chen, X.; Li, J.; Kang, R.; Klionsky, D.; Tang, D. Ferroptosis: Machinery and regulation. Autophagy 2021, 17, 2054–2081. [Google Scholar] [CrossRef]
- Ge, J.; Zhang, C.; Sun, Y.; Zhang, Q.; Lv, M.; Guo, K.; Li, J. Cadmium exposure triggers mitochondrial dysfunction and oxidative stress in chicken (Gallus gallus) kidney via mitochondrial UPR inhibition and Nrf2-mediated antioxidant defense activation. Sci. Total Environ. 2019, 689, 1160–1171. [Google Scholar] [CrossRef]
- Aschner, M.; Skalny, A.; Martins, A.; Sinitskii, A.; Farina, M.; Lu, R.; Barbosa, F.; Gluhcheva, Y.; Santamaria, A.; Tinkov, A. Ferroptosis as a mechanism of non-ferrous metal toxicity. Arch. Toxicol. 2022, 96, 2391–2417. [Google Scholar] [CrossRef]
- Luan, P.; Sun, Y.; Zhu, Y.; Qiao, S.; Hu, G.; Liu, Q.; Zhang, Z. Cadmium exposure promotes activation of cerebrum and cerebellum ferroptosis and necrosis in swine. Ecotoxicol. Environ. Saf. 2021, 224, 112650. [Google Scholar] [CrossRef] [PubMed]
- Koye, D.; Magliano, D.; Nelson, R.; Pavkov, M. The Global Epidemiology of Diabetes and Kidney Disease. Adv. Chronic Kidney Dis. 2018, 25, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Zhang, L. Prevalence and Disease Burden of Chronic Kidney Disease. Adv. Exp. Med. Biol. 2019, 1165, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Lim, H.; Lim, J.; Son, J.; Lee, J.; Lee, B.; Chang, S.; Kim, H. Curcumin ameliorates cadmium-induced nephrotoxicity in Sprague-Dawley rats. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2018, 114, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Li, P.; Shen, Z.; Wang, J.; Diao, L. Protective effects of selenium yeast against cadmium-induced necroptosis through miR-26a-5p/PTEN/PI3K/AKT signaling pathway in chicken kidney. Ecotoxicol. Environ. Saf. 2021, 220, 112387. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Dong, W.; Li, J.; Gong, Z.; Liu, W.; He, S.; Zou, H.; Song, R.; Liu, G.; Liu, Z. Honokiol Antagonizes Cadmium-Induced Nephrotoxicity in Quail by Alleviating Autophagy Dysfunction, Apoptosis and Mitochondrial UPR Inhibition with Its Antioxidant Properties. Life 2022, 12, 1574. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Zou, H.; Luo, T.; Long, M.; Bian, J.; Liu, X.; Gu, J.; Yuan, Y.; Song, R.; Wang, Y.; et al. Caspase-Dependent and Caspase-Independent Pathways Are Involved in Cadmium-Induced Apoptosis in Primary Rat Proximal Tubular Cell Culture. PLoS ONE 2016, 11, e0166823. [Google Scholar] [CrossRef] [PubMed]
- Luo, T.; Yuan, Y.; Yu, Q.; Liu, G.; Long, M.; Zhang, K.; Bian, J.; Gu, J.; Zou, H.; Wang, Y.; et al. PARP-1 overexpression contributes to Cadmium-induced death in rat proximal tubular cells via parthanatos and the MAPK signalling pathway. Sci. Rep. 2017, 7, 4331. [Google Scholar] [CrossRef]
- Pavón, N.; Buelna-Chontal, M.; Macías-López, A.; Correa, F.; Uribe-Álvarez, C.; Hernández-Esquivel, L.; Chávez, E. On the oxidative damage by cadmium to kidney mitochondrial functions. Biochem. Cell Biol. = Biochim. Biol. Cell. 2019, 97, 187–192. [Google Scholar] [CrossRef]
- Nair, A.; Lee, W.; Smeets, K.; Swennen, Q.; Sanchez, A.; Thévenod, F.; Cuypers, A. Glutathione and mitochondria determine acute defense responses and adaptive processes in cadmium-induced oxidative stress and toxicity of the kidney. Arch. Toxicol. 2015, 89, 2273–2289. [Google Scholar] [CrossRef]
- He, Z.; Shen, P.; Feng, L.; Hao, H.; He, Y.; Fan, G.; Liu, Z.; Zhu, K.; Wang, Y.; Zhang, N.; et al. Cadmium induces liver dysfunction and ferroptosis through the endoplasmic stress-ferritinophagy axis. Ecotoxicol. Environ. Saf. 2022, 245, 114123. [Google Scholar] [CrossRef] [PubMed]
- Xiong, L.; Zhou, B.; Young, J.; Wintergerst, K.; Cai, L. Exposure to low-dose cadmium induces testicular ferroptosis. Ecotoxicol. Environ. Saf. 2022, 234, 113373. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Yu, D.; He, Z.; Bao, L.; Feng, L.; Chen, L.; Liu, Z.; Hu, X.; Zhang, N.; Wang, T.; et al. Endoplasmic reticulum stress-mediated autophagy activation is involved in cadmium-induced ferroptosis of renal tubular epithelial cells. Free Radic. Biol. Med. 2021, 175, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Kuwata, H.; Tomitsuka, Y.; Yoda, E.; Hara, S. Role of ACSL4 in the chemical-induced cell death in human proximal tubule epithelial HK-2 cells. Biosci. Rep. 2022, 42, BSR20212433. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J., 3rd; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Liu, Q.; Shan, X.; Gao, W.; Chen, Q. ATM orchestrates ferritinophagy and ferroptosis by phosphorylating NCOA4. Autophagy 2023, 19, 2062–2077. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Bai, Y.; Jia, Y.; Zhao, Y.; Kang, R.; Tang, D.; Dai, E. Ferroptosis is a lysosomal cell death process. Biochem. Biophys. Res. Commun. 2018, 503, 1550–1556. [Google Scholar] [CrossRef]
- Park, E.; Chung, S. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death Dis. 2019, 10, 822. [Google Scholar] [CrossRef]
- Song, X.; Liu, G.; Wang, Z.; Wang, L. Puerarin protects against cadmium-induced proximal tubular cell apoptosis by restoring mitochondrial function. Chem.-Biol. Interact. 2016, 260, 219–231. [Google Scholar] [CrossRef]
- Liu, F.; Wang, X.; Zhou, X.; Liu, Z.; Song, X.; Wang, Z.; Wang, L. Cadmium disrupts autophagic flux by inhibiting cytosolic Ca-dependent autophagosome-lysosome fusion in primary rat proximal tubular cells. Toxicology 2017, 383, 13–23. [Google Scholar] [CrossRef]
- Wang, L.; Fan, R.; Yang, D.; Zhang, D.; Wang, L. Puerarin reverses cadmium-induced lysosomal dysfunction in primary rat proximal tubular cells via inhibiting Nrf2 pathway. Biochem. Pharmacol. 2019, 162, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Zhou, J.; Wang, X.; Zhang, Y.; Wang, M.; Su, P. Cadmium attenuates testosterone synthesis by promoting ferroptosis and blocking autophagosome-lysosome fusion. Free Radic. Biol. Med. 2021, 176, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, J.; Zhang, K.; Liu, X.; Li, J. Effects of chronic cadmium poisoning on Zn, Cu, Fe, Ca, and metallothionein in liver and kidney of rats. Biol. Trace Elem. Res. 2012, 149, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, S.; Elsenhans, B. Iron retention and distribution in the cadmium-induced iron deficiency. Ecotoxicol. Environ. Saf. 1985, 10, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Hiratsuka, H.; Katsuta, O.; Toyota, N.; Tsuchitani, M.; Umemura, T.; Marumo, F. Chronic cadmium exposure-induced renal anemia in ovariectomized rats. Toxicol. Appl. Pharmacol. 1996, 137, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Park, S. Iron deficiency increases blood concentrations of neurotoxic metals in children. Korean J. Pediatr. 2014, 57, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Nordberg, G. Historical perspectives on cadmium toxicology. Toxicol. Appl. Pharmacol. 2009, 238, 192–200. [Google Scholar] [CrossRef]
- Chen, X.; Ren, S.; Zhu, G.; Wang, Z.; Wen, X. Emodin suppresses cadmium-induced osteoporosis by inhibiting osteoclast formation. Environ. Toxicol. Pharmacol. 2017, 54, 162–168. [Google Scholar] [CrossRef]
- Dabrowska, E.; Szynaka, B.; Kulikowska-Karpińska, E. Ultrastructural study of the submandibular gland of the rat after 6-month exposure to cadmium and zinc in drinking water. Adv. Med. Sci. 2006, 51, 245–249. [Google Scholar]
- Wang, L.; Wang, H.; Hu, M.; Cao, J.; Chen, D.; Liu, Z. Oxidative stress and apoptotic changes in primary cultures of rat proximal tubular cells exposed to lead. Arch. Toxicol. 2009, 83, 417–427. [Google Scholar] [CrossRef]
- Chen, S.; Liu, G.; Long, M.; Zou, H.; Cui, H. Alpha lipoic acid attenuates cadmium-induced nephrotoxicity via the mitochondrial apoptotic pathways in rat. J. Inorg. Biochem. 2018, 184, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Curcic, M.; Buha, A.; Stankovic, S.; Milovanovic, V.; Bulat, Z.; Đukić-Ćosić, D.; Antonijević, E.; Vučinić, S.; Matović, V.; Antonijevic, B. Interactions between cadmium and decabrominated diphenyl ether on blood cells count in rats-Multiple factorial regression analysis. Toxicology 2017, 376, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Luo, T.; Liu, G.; Long, M.; Yang, J.; Song, R.; Wang, Y.; Yuan, Y.; Bian, J.; Liu, X.; Gu, J.; et al. Treatment of cadmium-induced renal oxidative damage in rats by administration of alpha-lipoic acid. Environ. Sci. Pollut. Res. Int. 2017, 24, 1832–1844. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Zhang, K.; Dong, W.; Tan, Y.; Long, M.; Zou, H.; Liu, Z. Puerarin restores the autophagic flux to alleviate cadmium-induced endoplasmic reticulum stress in NRK-52E cells. Mol. Med. Rep. 2020, 22, 2551–2563. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, K.; Long, M.; Dong, W.; Li, J.; Wang, X.; Liu, W.; Huang, Q.; Ping, Y.; Zou, H.; Song, R.; et al. Cadmium Induces Kidney Iron Deficiency and Chronic Kidney Injury by Interfering with the Iron Metabolism in Rats. Int. J. Mol. Sci. 2024, 25, 763. https://doi.org/10.3390/ijms25020763
Zhang K, Long M, Dong W, Li J, Wang X, Liu W, Huang Q, Ping Y, Zou H, Song R, et al. Cadmium Induces Kidney Iron Deficiency and Chronic Kidney Injury by Interfering with the Iron Metabolism in Rats. International Journal of Molecular Sciences. 2024; 25(2):763. https://doi.org/10.3390/ijms25020763
Chicago/Turabian StyleZhang, Kanglei, Mengfei Long, Wenxuan Dong, Jiahui Li, Xueru Wang, Wenjing Liu, Qing Huang, Yuyu Ping, Hui Zou, Ruilong Song, and et al. 2024. "Cadmium Induces Kidney Iron Deficiency and Chronic Kidney Injury by Interfering with the Iron Metabolism in Rats" International Journal of Molecular Sciences 25, no. 2: 763. https://doi.org/10.3390/ijms25020763
APA StyleZhang, K., Long, M., Dong, W., Li, J., Wang, X., Liu, W., Huang, Q., Ping, Y., Zou, H., Song, R., Liu, G., Ran, D., & Liu, Z. (2024). Cadmium Induces Kidney Iron Deficiency and Chronic Kidney Injury by Interfering with the Iron Metabolism in Rats. International Journal of Molecular Sciences, 25(2), 763. https://doi.org/10.3390/ijms25020763