
The Importance of Complement-Mediated Immune Signaling in Alzheimer’s Disease Pathogenesis

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Neuroinflammation and AD
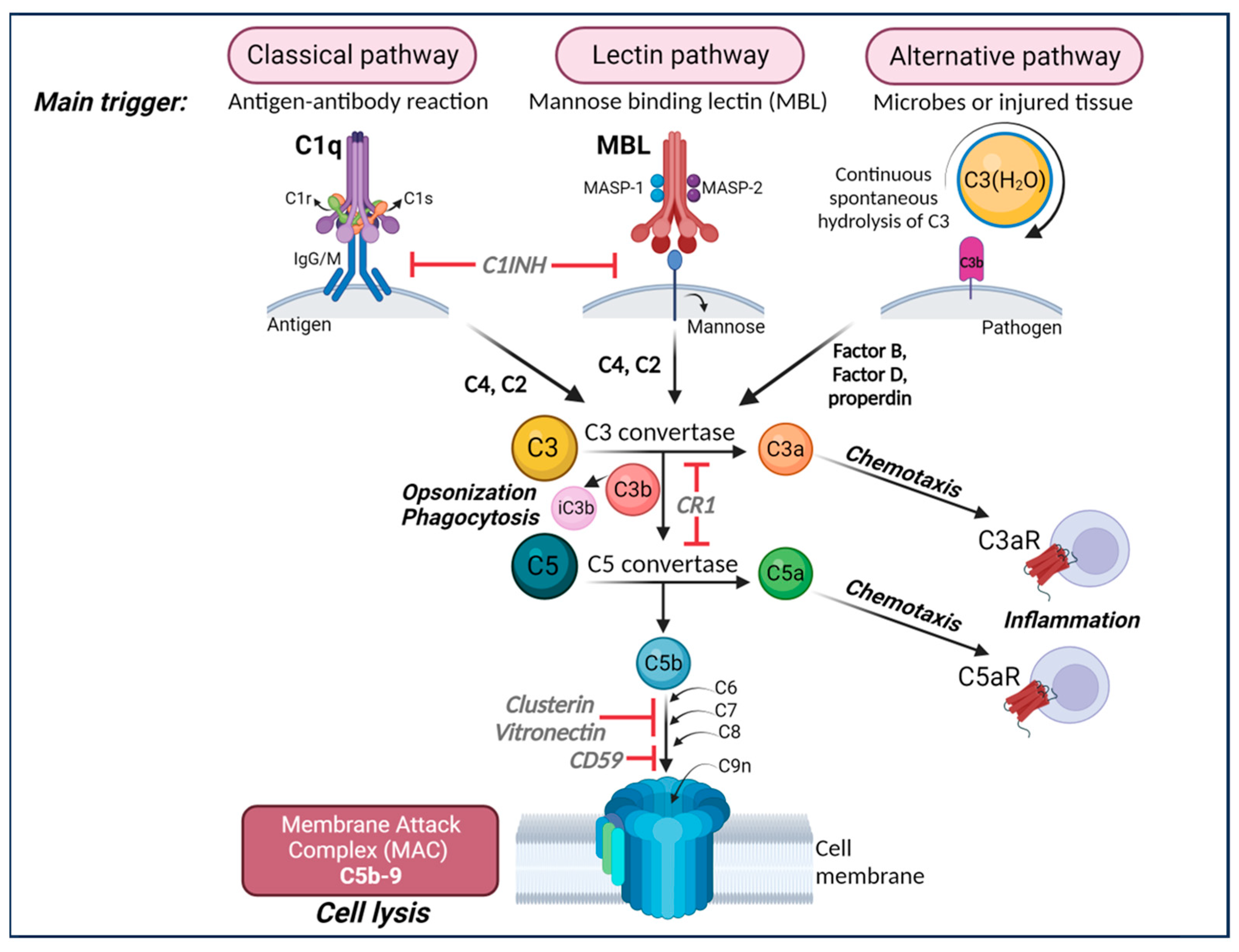
3. Complement System
4. Complement System in the Pathogenesis of AD
5. Complement Component C3 and AD
6. Driving Mechanisms of Complement-Mediated CNS Dysfunction
7. Complement Components as Therapeutical Targets in AD
8. Crosstalk between the Complement System and Other Inflammatory Players in AD
9. Complement and Tau Pathology
10. Future Directions
11. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Alzheimer’s Disease, International; Patterson, C. World Alzheimer Report 2018: The State of the Art of Dementia Research: New Frontiers; Alzheimer’s Disease International: London, UK, 2018. [Google Scholar]
- Alzheimer’s Association. 2023 Alzheimer’s disease facts and figures. Alzheimers Dement. 2023, 19, 1598–1695. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Available online: https://www.who.int/news-room/fact-sheets/details/dementia (accessed on 14 June 2023).
- Terry, R.D.; Masliah, E.; Salmon, D.P.; Butters, N.; DeTeresa, R.; Hill, R.; Hansen, L.A.; Katzman, R. Physical basis of cognitive alterations in Alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 1991, 30, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Hansen, L.; Albright, T.; Mallory, M.; Terry, R.D. Immunoelectron microscopic study of synaptic pathology in Alzheimer’s disease. Acta Neuropathol. 1991, 81, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Hammond, T.R.; Dufort, C.; Dissing-Olesen, L.; Giera, S.; Young, A.; Wysoker, A.; Walker, A.J.; Gergits, F.; Segel, M.; Nemesh, J.; et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 2019, 50, 253–271.e6. [Google Scholar] [CrossRef] [PubMed]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Stubbington, M.J.T.; Rozenblatt-Rosen, O.; Regev, A.; Teichmann, S.A. Single-cell transcriptomics to explore the immune system in health and disease. Science 2017, 358, 58–63. [Google Scholar] [CrossRef]
- Felsky, D.; Roostaei, T.; Nho, K.; Risacher, S.L.; Bradshaw, E.M.; Petyuk, V.; Schneider, J.A.; Saykin, A.; Bennett, D.A.; De Jager, P.L. Neuropathological correlates and genetic architecture of microglial activation in elderly human brain. Nat. Commun. 2019, 10, 409. [Google Scholar] [CrossRef]
- Jansen, I.E.; Savage, J.E.; Watanabe, K.; Bryois, J.; Williams, D.M.; Steinberg, S.; Sealock, J.; Karlsson, I.K.; Hagg, S.; Athanasiu, L.; et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 2019, 51, 404–413. [Google Scholar] [CrossRef]
- Ponath, G.; Lincoln, M.R.; Levine-Ritterman, M.; Park, C.; Dahlawi, S.; Mubarak, M.; Sumida, T.; Airas, L.; Zhang, S.; Isitan, C.; et al. Enhanced astrocyte responses are driven by a genetic risk allele associated with multiple sclerosis. Nat. Commun. 2018, 9, 5337. [Google Scholar] [CrossRef]
- Mathys, H.; Adaikkan, C.; Gao, F.; Young, J.Z.; Manet, E.; Hemberg, M.; De Jager, P.L.; Ransohoff, R.M.; Regev, A.; Tsai, L.H. Temporal Tracking of Microglia Activation in Neurodegeneration at Single-Cell Resolution. Cell Rep. 2017, 21, 366–380. [Google Scholar] [CrossRef] [PubMed]
- Walport, M.J. Complement. Second of two parts. N. Engl. J. Med. 2001, 344, 1140–1144. [Google Scholar] [CrossRef] [PubMed]
- Walport, M.J. Complement. First of two parts. N. Engl. J. Med. 2001, 344, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Stephan, A.H.; Barres, B.A.; Stevens, B. The complement system: An unexpected role in synaptic pruning during development and disease. Annu. Rev. Neurosci. 2012, 35, 369–389. [Google Scholar] [CrossRef] [PubMed]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef]
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B.; et al. The classical complement cascade mediates CNS synapse elimination. Cell 2007, 131, 1164–1178. [Google Scholar] [CrossRef]
- Gorelik, A.; Sapir, T.; Haffner-Krausz, R.; Olender, T.; Woodruff, T.M.; Reiner, O. Developmental activities of the complement pathway in migrating neurons. Nat. Commun. 2017, 8, 15096. [Google Scholar] [CrossRef]
- Carpanini, S.M.; Torvell, M.; Morgan, B.P. Therapeutic Inhibition of the Complement System in Diseases of the Central Nervous System. Front. Immunol. 2019, 10, 362. [Google Scholar] [CrossRef]
- Hammond, T.R.; Marsh, S.E.; Stevens, B. Immune Signaling in Neurodegeneration. Immunity 2019, 50, 955–974. [Google Scholar] [CrossRef]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef]
- Shi, Q.; Chowdhury, S.; Ma, R.; Le, K.X.; Hong, S.; Caldarone, B.J.; Stevens, B.; Lemere, C.A. Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci. Transl. Med. 2017, 9, eaaf6295. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Colodner, K.J.; Matousek, S.B.; Merry, K.; Hong, S.; Kenison, J.E.; Frost, J.L.; Le, K.X.; Li, S.; Dodart, J.C.; et al. Complement C3-Deficient Mice Fail to Display Age-Related Hippocampal Decline. J. Neurosci. 2015, 35, 13029–13042. [Google Scholar] [CrossRef] [PubMed]
- Krukowski, K.; Chou, A.; Feng, X.; Tiret, B.; Paladini, M.S.; Riparip, L.K.; Chaumeil, M.M.; Lemere, C.; Rosi, S. Traumatic Brain Injury in Aged Mice Induces Chronic Microglia Activation, Synapse Loss, and Complement-Dependent Memory Deficits. Int. J. Mol. Sci. 2018, 19, 3753. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Dejanovic, B.; Gandham, V.D.; Gogineni, A.; Edmonds, R.; Schauer, S.; Srinivasan, K.; Huntley, M.A.; Wang, Y.; Wang, T.M.; et al. Complement C3 Is Activated in Human AD Brain and Is Required for Neurodegeneration in Mouse Models of Amyloidosis and Tauopathy. Cell Rep. 2019, 28, 2111–2123.e6. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Rogers, J.; Cooper, N.R.; Webster, S.; Schultz, J.; McGeer, P.L.; Styren, S.D.; Civin, W.H.; Brachova, L.; Bradt, B.; Ward, P.; et al. Complement activation by beta-amyloid in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1992, 89, 10016–10020. [Google Scholar] [CrossRef]
- McGeer, P.L.; Akiyama, H.; Itagaki, S.; McGeer, E.G. Activation of the classical complement pathway in brain tissue of Alzheimer patients. Neurosci. Lett. 1989, 107, 341–346. [Google Scholar] [CrossRef]
- McGeer, P.L.; Itagaki, S.; Tago, H.; McGeer, E.G. Occurrence of HLA-DR reactive microglia in Alzheimer’s disease. Ann. N. Y. Acad. Sci. 1988, 540, 319–323. [Google Scholar] [CrossRef]
- McGeer, P.L.; Itagaki, S.; Tago, H.; McGeer, E.G. Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci. Lett. 1987, 79, 195–200. [Google Scholar] [CrossRef]
- Zotova, E.; Bharambe, V.; Cheaveau, M.; Morgan, W.; Holmes, C.; Harris, S.; Neal, J.W.; Love, S.; Nicoll, J.A.; Boche, D. Inflammatory components in human Alzheimer’s disease and after active amyloid-beta42 immunization. Brain 2013, 136, 2677–2696. [Google Scholar] [CrossRef]
- Eikelenboom, P.; Stam, F.C. Immunoglobulins and complement factors in senile plaques. An immunoperoxidase study. Acta Neuropathol. 1982, 57, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Kamphuis, W.; Mamber, C.; Moeton, M.; Kooijman, L.; Sluijs, J.A.; Jansen, A.H.; Verveer, M.; de Groot, L.R.; Smith, V.D.; Rangarajan, S.; et al. GFAP isoforms in adult mouse brain with a focus on neurogenic astrocytes and reactive astrogliosis in mouse models of Alzheimer disease. PLoS ONE 2012, 7, e42823. [Google Scholar] [CrossRef] [PubMed]
- Hanzel, D.K.; Trojanowski, J.Q.; Johnston, R.F.; Loring, J.F. High-throughput quantitative histological analysis of Alzheimer’s disease pathology using a confocal digital microscanner. Nat. Biotechnol. 1999, 17, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, J.; Nutma, E.; van der Valk, P.; Amor, S. Inflammation in CNS neurodegenerative diseases. Immunology 2018, 154, 204–219. [Google Scholar] [CrossRef]
- Brosseron, F.; Krauthausen, M.; Kummer, M.; Heneka, M.T. Body fluid cytokine levels in mild cognitive impairment and Alzheimer’s disease: A comparative overview. Mol. Neurobiol. 2014, 50, 534–544. [Google Scholar] [CrossRef]
- Brosseron, F.; Traschutz, A.; Widmann, C.N.; Kummer, M.P.; Tacik, P.; Santarelli, F.; Jessen, F.; Heneka, M.T. Characterization and clinical use of inflammatory cerebrospinal fluid protein markers in Alzheimer’s disease. Alzheimers Res. Ther. 2018, 10, 25. [Google Scholar] [CrossRef]
- Tian, Z.; Ji, X.; Liu, J. Neuroinflammation in Vascular Cognitive Impairment and Dementia: Current Evidence, Advances, and Prospects. Int. J. Mol. Sci. 2022, 23, 6224. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef]
- Sheng, J.; Ruedl, C.; Karjalainen, K. Most Tissue-Resident Macrophages Except Microglia Are Derived from Fetal Hematopoietic Stem Cells. Immunity 2015, 43, 382–393. [Google Scholar] [CrossRef]
- Lawson, L.J.; Perry, V.H.; Dri, P.; Gordon, S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 1990, 39, 151–170. [Google Scholar] [CrossRef]
- Hong, S.; Dissing-Olesen, L.; Stevens, B. New insights on the role of microglia in synaptic pruning in health and disease. Curr. Opin. Neurobiol. 2016, 36, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Sarlus, H.; Heneka, M.T. Microglia in Alzheimer’s disease. J. Clin. Investig. 2017, 127, 3240–3249. [Google Scholar] [CrossRef] [PubMed]
- Lian, H.; Litvinchuk, A.; Chiang, A.C.; Aithmitti, N.; Jankowsky, J.L.; Zheng, H. Astrocyte-Microglia Cross Talk through Complement Activation Modulates Amyloid Pathology in Mouse Models of Alzheimer’s Disease. J. Neurosci. 2016, 36, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Roy, E.R.; Wang, B.; Wan, Y.W.; Chiu, G.; Cole, A.; Yin, Z.; Propson, N.E.; Xu, Y.; Jankowsky, J.L.; Liu, Z.; et al. Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J. Clin. Investig. 2020, 130, 1912–1930. [Google Scholar] [CrossRef] [PubMed]
- Butovsky, O.; Weiner, H.L. Microglial signatures and their role in health and disease. Nat. Rev. Neurosci. 2018, 19, 622–635. [Google Scholar] [CrossRef] [PubMed]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef]
- Lambert, J.C.; Heath, S.; Even, G.; Campion, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Zelenika, D.; Bullido, M.J.; Tavernier, B.; et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1094–1099. [Google Scholar] [CrossRef]
- Schafer, D.P.; Lehrman, E.K.; Stevens, B. The “quad-partite” synapse: Microglia-synapse interactions in the developing and mature CNS. Glia 2013, 61, 24–36. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef]
- Filipello, F.; Morini, R.; Corradini, I.; Zerbi, V.; Canzi, A.; Michalski, B.; Erreni, M.; Markicevic, M.; Starvaggi-Cucuzza, C.; Otero, K.; et al. The Microglial Innate Immune Receptor TREM2 Is Required for Synapse Elimination and Normal Brain Connectivity. Immunity 2018, 48, 979–991.e8. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Gross, C.T. Microglia in development: Linking brain wiring to brain environment. Neuron Glia Biol. 2011, 7, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.T.; Morganti, J.M.; Bachstetter, A.D.; Hudson, C.E.; Peters, M.M.; Grimmig, B.A.; Weeber, E.J.; Bickford, P.C.; Gemma, C. CX3CR1 deficiency leads to impairment of hippocampal cognitive function and synaptic plasticity. J. Neurosci. 2011, 31, 16241–16250. [Google Scholar] [CrossRef]
- Bailey, C.H.; Kandel, E.R.; Harris, K.M. Structural Components of Synaptic Plasticity and Memory Consolidation. Cold Spring Harb. Perspect. Biol. 2015, 7, a021758. [Google Scholar] [CrossRef] [PubMed]
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P.; et al. Microglia-derived ASC specks cross-seed amyloid-beta in Alzheimer’s disease. Nature 2017, 552, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Arranz, A.M.; De Strooper, B. The role of astroglia in Alzheimer’s disease: Pathophysiology and clinical implications. Lancet Neurol. 2019, 18, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, R.; Hurtado, A.; Persaud, T.; Esham, K.; Pearse, D.D.; Oudega, M.; Bethea, J.R. Transgenic inhibition of astroglial NF-kappa B leads to increased axonal sparing and sprouting following spinal cord injury. J. Neurochem. 2009, 110, 765–778. [Google Scholar] [CrossRef] [PubMed]
- Diniz, L.P.; Tortelli, V.; Matias, I.; Morgado, J.; Bergamo Araujo, A.P.; Melo, H.M.; Seixas da Silva, G.S.; Alves-Leon, S.V.; de Souza, J.M.; Ferreira, S.T.; et al. Astrocyte Transforming Growth Factor Beta 1 Protects Synapses against Abeta Oligomers in Alzheimer’s Disease Model. J. Neurosci. 2017, 37, 6797–6809. [Google Scholar] [CrossRef]
- Bellaver, B.; Povala, G.; Ferreira, P.C.L.; Ferrari-Souza, J.P.; Leffa, D.T.; Lussier, F.Z.; Benedet, A.L.; Ashton, N.J.; Triana-Baltzer, G.; Kolb, H.C.; et al. Astrocyte reactivity influences amyloid-beta effects on tau pathology in preclinical Alzheimer’s disease. Nat. Med. 2023, 29, 1775–1781. [Google Scholar] [CrossRef]
- Lian, H.; Zheng, H. Signaling pathways regulating neuron-glia interaction and their implications in Alzheimer’s disease. J. Neurochem. 2016, 136, 475–491. [Google Scholar] [CrossRef] [PubMed]
- Eikelenboom, P.; Hack, C.E.; Rozemuller, J.M.; Stam, F.C. Complement activation in amyloid plaques in Alzheimer’s dementia. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1989, 56, 259–262. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Haga, S. Immuno-electron-microscopic localization of complements in amyloid fibrils of senile plaques. Acta Neuropathol. 1984, 63, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Afagh, A.; Cummings, B.J.; Cribbs, D.H.; Cotman, C.W.; Tenner, A.J. Localization and cell association of C1q in Alzheimer’s disease brain. Exp. Neurol. 1996, 138, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Stoltzner, S.E.; Grenfell, T.J.; Mori, C.; Wisniewski, K.E.; Wisniewski, T.M.; Selkoe, D.J.; Lemere, C.A. Temporal accrual of complement proteins in amyloid plaques in Down’s syndrome with Alzheimer’s disease. Am. J. Pathol. 2000, 156, 489–499. [Google Scholar] [CrossRef]
- Reichwald, J.; Danner, S.; Wiederhold, K.H.; Staufenbiel, M. Expression of complement system components during aging and amyloid deposition in APP transgenic mice. J. Neuroinflamm. 2009, 6, 35. [Google Scholar] [CrossRef]
- Matsuoka, Y.; Picciano, M.; Malester, B.; LaFrancois, J.; Zehr, C.; Daeschner, J.M.; Olschowka, J.A.; Fonseca, M.I.; O’Banion, M.K.; Tenner, A.J.; et al. Inflammatory responses to amyloidosis in a transgenic mouse model of Alzheimer’s disease. Am. J. Pathol. 2001, 158, 1345–1354. [Google Scholar] [CrossRef]
- Nonaka, M.; Kimura, A. Genomic view of the evolution of the complement system. Immunogenetics 2006, 58, 701–713. [Google Scholar] [CrossRef]
- Bordet, J. Les leukocytes et les proprietes actives du serum chez les vaccines. Ann. Inst. Pasteur 1895, 9, 462–506. [Google Scholar]
- Hadding, U.; Muller-Eberhard, H.J. The ninth component of human complement: Isolation, description and mode of action. Immunology 1969, 16, 719–735. [Google Scholar]
- Nilsson, U. Separation and partial purification of the sixth, seventh and eighth components of human haemolytic complement. Acta Pathol. Microbiol. Scand. 1967, 70, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, U.R.; Mueller-Eberhard, H.J. Isolation of Beta If-Globulin from Human Serum and Its Characterization as the Fifth Component of Complement. J. Exp. Med. 1965, 122, 277–298. [Google Scholar] [CrossRef] [PubMed]
- Mueller-Eberhard, H.J.; Biro, C.E. Isolation and Description of the Fourth Component of Human Complement. J. Exp. Med. 1963, 118, 447–466. [Google Scholar] [CrossRef] [PubMed]
- Pillemer, L.; Ecker, E.E. The Terminology of the Components of Complement. Science 1941, 94, 437. [Google Scholar] [CrossRef] [PubMed]
- Merle, N.S.; Church, S.E.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part I—Molecular Mechanisms of Activation and Regulation. Front. Immunol. 2015, 6, 262. [Google Scholar] [CrossRef]
- Merle, N.S.; Noe, R.; Halbwachs-Mecarelli, L.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part II: Role in Immunity. Front. Immunol. 2015, 6, 257. [Google Scholar] [CrossRef]
- Coulthard, L.G.; Hawksworth, O.A.; Woodruff, T.M. Complement: The Emerging Architect of the Developing Brain. Trends Neurosci. 2018, 41, 373–384. [Google Scholar] [CrossRef]
- Bialas, A.R.; Stevens, B. TGF-beta signaling regulates neuronal C1q expression and developmental synaptic refinement. Nat. Neurosci. 2013, 16, 1773–1782. [Google Scholar] [CrossRef]
- Perez-Alcazar, M.; Daborg, J.; Stokowska, A.; Wasling, P.; Bjorefeldt, A.; Kalm, M.; Zetterberg, H.; Carlstrom, K.E.; Blomgren, K.; Ekdahl, C.T.; et al. Altered cognitive performance and synaptic function in the hippocampus of mice lacking C3. Exp. Neurol. 2014, 253, 154–164. [Google Scholar] [CrossRef]
- Fitzgerald, K.C.; Kim, K.; Smith, M.D.; Aston, S.A.; Fioravante, N.; Rothman, A.M.; Krieger, S.; Cofield, S.S.; Kimbrough, D.J.; Bhargava, P.; et al. Early complement genes are associated with visual system degeneration in multiple sclerosis. Brain 2019, 142, 2722–2736. [Google Scholar] [CrossRef]
- Werneburg, S.; Jung, J.; Kunjamma, R.B.; Ha, S.K.; Luciano, N.J.; Willis, C.M.; Gao, G.; Biscola, N.P.; Havton, L.A.; Crocker, S.J.; et al. Targeted Complement Inhibition at Synapses Prevents Microglial Synaptic Engulfment and Synapse Loss in Demyelinating Disease. Immunity 2020, 52, 167–182.e7. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Yin, Q.; Fang, R.; Yan, X.; Wang, Y.; Bezerianos, A.; Tang, H.; Miao, F.; Sun, J. Disrupted functional brain connectivity and its association to structural connectivity in amnestic mild cognitive impairment and Alzheimer’s disease. PLoS ONE 2014, 9, e96505. [Google Scholar] [CrossRef] [PubMed]
- Siegel, J.S.; Ramsey, L.E.; Snyder, A.Z.; Metcalf, N.V.; Chacko, R.V.; Weinberger, K.; Baldassarre, A.; Hacker, C.D.; Shulman, G.L.; Corbetta, M. Disruptions of network connectivity predict impairment in multiple behavioral domains after stroke. Proc. Natl. Acad. Sci. USA 2016, 113, E4367–E4376. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.S. Epidemiologic studies of exposure to prenatal infection and risk of schizophrenia and autism. Dev. Neurobiol. 2012, 72, 1272–1276. [Google Scholar] [CrossRef] [PubMed]
- Sekar, A.; Bialas, A.R.; de Rivera, H.; Davis, A.; Hammond, T.R.; Kamitaki, N.; Tooley, K.; Presumey, J.; Baum, M.; Van Doren, V.; et al. Schizophrenia risk from complex variation of complement component 4. Nature 2016, 530, 177–183. [Google Scholar] [CrossRef]
- Yilmaz, M.; Yalcin, E.; Presumey, J.; Aw, E.; Ma, M.; Whelan, C.W.; Stevens, B.; McCarroll, S.A.; Carroll, M.C. Overexpression of schizophrenia susceptibility factor human complement C4A promotes excessive synaptic loss and behavioral changes in mice. Nat. Neurosci. 2021, 24, 214–224. [Google Scholar] [CrossRef]
- Rahpeymai, Y.; Hietala, M.A.; Wilhelmsson, U.; Fotheringham, A.; Davies, I.; Nilsson, A.K.; Zwirner, J.; Wetsel, R.A.; Gerard, C.; Pekny, M.; et al. Complement: A novel factor in basal and ischemia-induced neurogenesis. EMBO J. 2006, 25, 1364–1374. [Google Scholar] [CrossRef]
- Westacott, L.J.; Haan, N.; Evison, C.; Marei, O.; Hall, J.; Hughes, T.R.; Zaben, M.; Morgan, B.P.; Humby, T.; Wilkinson, L.S.; et al. Dissociable effects of complement C3 and C3aR on survival and morphology of adult born hippocampal neurons, pattern separation, and cognitive flexibility in male mice. Brain Behav. Immun. 2021, 98, 136–150. [Google Scholar] [CrossRef]
- Figueiredo, C.P.; Barros-Aragao, F.G.Q.; Neris, R.L.S.; Frost, P.S.; Soares, C.; Souza, I.N.O.; Zeidler, J.D.; Zamberlan, D.C.; de Sousa, V.L.; Souza, A.S.; et al. Zika virus replicates in adult human brain tissue and impairs synapses and memory in mice. Nat. Commun. 2019, 10, 3890. [Google Scholar] [CrossRef]
- Jaffry, M.; Faiz, I.; Jaffry, K.; Souayah, N. Neurological Manifestations of SARS-CoV-2 Infection and the Role of Complement Activation. touchREVIEWS Neurol. 2022, 18, 86–92. [Google Scholar] [CrossRef]
- Lee, M.H.; Perl, D.P.; Steiner, J.; Pasternack, N.; Li, W.; Maric, D.; Safavi, F.; Horkayne-Szakaly, I.; Jones, R.; Stram, M.N.; et al. Neurovascular injury with complement activation and inflammation in COVID-19. Brain 2022, 145, 2555–2568. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, M.; Kobayashi, H.; Tatsumi, L.; Tomita, T. Mouse Models of Alzheimer’s Disease. Front. Mol. Neurosci. 2022, 15, 912995. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Fonseca, M.I.; Pisalyaput, K.; Tenner, A.J. Complement C3 and C4 expression in C1q sufficient and deficient mouse models of Alzheimer’s disease. J. Neurochem. 2008, 106, 2080–2092. [Google Scholar] [CrossRef] [PubMed]
- Maier, M.; Peng, Y.; Jiang, L.; Seabrook, T.J.; Carroll, M.C.; Lemere, C.A. Complement C3 deficiency leads to accelerated amyloid beta plaque deposition and neurodegeneration and modulation of the microglia/macrophage phenotype in amyloid precursor protein transgenic mice. J. Neurosci. 2008, 28, 6333–6341. [Google Scholar] [CrossRef] [PubMed]
- Thambisetty, M.; An, Y.; Nalls, M.; Sojkova, J.; Swaminathan, S.; Zhou, Y.; Singleton, A.B.; Wong, D.F.; Ferrucci, L.; Saykin, A.J.; et al. Effect of complement CR1 on brain amyloid burden during aging and its modification by APOE genotype. Biol. Psychiatry 2013, 73, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, M.X.; Jiang, S.; Cole, T.A.; Chu, S.H.; Fonseca, M.I.; Fang, M.J.; Hohsfield, L.A.; Torres, M.D.; Green, K.N.; Wetsel, R.A.; et al. Prevention of C5aR1 signaling delays microglial inflammatory polarization, favors clearance pathways and suppresses cognitive loss. Mol. Neurodegener. 2017, 12, 66. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Lue, L.; Yang, L.; Roher, A.; Kuo, Y.; Strohmeyer, R.; Goux, W.J.; Lee, V.; Johnson, G.V.; Webster, S.D.; et al. Complement activation by neurofibrillary tangles in Alzheimer’s disease. Neurosci. Lett. 2001, 305, 165–168. [Google Scholar] [CrossRef]
- Daborg, J.; Andreasson, U.; Pekna, M.; Lautner, R.; Hanse, E.; Minthon, L.; Blennow, K.; Hansson, O.; Zetterberg, H. Cerebrospinal fluid levels of complement proteins C3, C4 and CR1 in Alzheimer’s disease. J. Neural Transm. 2012, 119, 789–797. [Google Scholar] [CrossRef]
- Strohmeyer, R.; Shen, Y.; Rogers, J. Detection of complement alternative pathway mRNA and proteins in the Alzheimer’s disease brain. Brain Res. Mol. Brain Res. 2000, 81, 7–18. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Schwartz, J.B.; Abner, E.L.; Jicha, G.A.; Kapogiannis, D. High complement levels in astrocyte-derived exosomes of Alzheimer disease. Ann. Neurol. 2018, 83, 544–552. [Google Scholar] [CrossRef]
- Cribbs, D.H.; Berchtold, N.C.; Perreau, V.; Coleman, P.D.; Rogers, J.; Tenner, A.J.; Cotman, C.W. Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: A microarray study. J. Neuroinflamm. 2012, 9, 179. [Google Scholar] [CrossRef] [PubMed]
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1088–1093. [Google Scholar] [CrossRef] [PubMed]
- Bellenguez, C.; Kucukali, F.; Jansen, I.E.; Kleineidam, L.; Moreno-Grau, S.; Amin, N.; Naj, A.C.; Campos-Martin, R.; Grenier-Boley, B.; Andrade, V.; et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 2022, 54, 412–436. [Google Scholar] [CrossRef]
- Gatz, M.; Reynolds, C.A.; Fratiglioni, L.; Johansson, B.; Mortimer, J.A.; Berg, S.; Fiske, A.; Pedersen, N.L. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 2006, 63, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Crehan, H.; Holton, P.; Wray, S.; Pocock, J.; Guerreiro, R.; Hardy, J. Complement receptor 1 (CR1) and Alzheimer’s disease. Immunobiology 2012, 217, 244–250. [Google Scholar] [CrossRef]
- Kucukkilic, E.; Brookes, K.; Barber, I.; Guetta-Baranes, T.; Consortium, A.; Morgan, K.; Hollox, E.J. Complement receptor 1 gene (CR1) intragenic duplication and risk of Alzheimer’s disease. Hum. Genet. 2018, 137, 305–314. [Google Scholar] [CrossRef]
- Lian, H.; Yang, L.; Cole, A.; Sun, L.; Chiang, A.C.; Fowler, S.W.; Shim, D.J.; Rodriguez-Rivera, J.; Taglialatela, G.; Jankowsky, J.L.; et al. NFkappaB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer’s disease. Neuron 2015, 85, 101–115. [Google Scholar] [CrossRef]
- El Gaamouch, F.; Audrain, M.; Lin, W.J.; Beckmann, N.; Jiang, C.; Hariharan, S.; Heeger, P.S.; Schadt, E.E.; Gandy, S.; Ehrlich, M.E.; et al. VGF-derived peptide TLQP-21 modulates microglial function through C3aR1 signaling pathways and reduces neuropathology in 5xFAD mice. Mol. Neurodegener. 2020, 15, 4. [Google Scholar] [CrossRef]
- Xie, J.; Cools, L.; Van Imschoot, G.; Van Wonterghem, E.; Pauwels, M.J.; Vlaeminck, I.; De Witte, C.; El Andaloussi, S.; Wierda, K.; De Groef, L.; et al. Helicobacter pylori-derived outer membrane vesicles contribute to Alzheimer’s disease pathogenesis via C3-C3aR signalling. J. Extracell. Vesicles 2023, 12, e12306. [Google Scholar] [CrossRef]
- Fu, H.; Liu, B.; Frost, J.L.; Hong, S.; Jin, M.; Ostaszewski, B.; Shankar, G.M.; Costantino, I.M.; Carroll, M.C.; Mayadas, T.N.; et al. Complement component C3 and complement receptor type 3 contribute to the phagocytosis and clearance of fibrillar Abeta by microglia. Glia 2012, 60, 993–1003. [Google Scholar] [CrossRef]
- Tooyama, I.; Sato, H.; Yasuhara, O.; Kimura, H.; Konishi, Y.; Shen, Y.; Walker, D.G.; Beach, T.G.; Sue, L.I.; Rogers, J. Correlation of the expression level of C1q mRNA and the number of C1q-positive plaques in the Alzheimer Disease temporal cortex. analysis of C1q mrna and its protein using adjacent or nearby sections. Dement. Geriatr. Cogn. Disord. 2001, 12, 237–242. [Google Scholar] [CrossRef]
- Guttikonda, S.R.; Sikkema, L.; Tchieu, J.; Saurat, N.; Walsh, R.M.; Harschnitz, O.; Ciceri, G.; Sneeboer, M.; Mazutis, L.; Setty, M.; et al. Fully defined human pluripotent stem cell-derived microglia and tri-culture system model C3 production in Alzheimer’s disease. Nat. Neurosci. 2021, 24, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, M.; Ozdemir, S.; Kunadt, M.; Koel-Simmelink, M.; Boiten, W.; Piepkorn, L.; Pham, T.V.; Chiasserini, D.; Piersma, S.R.; Knol, J.C.; et al. C1q is increased in cerebrospinal fluid-derived extracellular vesicles in Alzheimer’s disease: A multi-cohort proteomics and immuno-assay validation study. Alzheimers Dement. 2023, 19, 4828–4840. [Google Scholar] [CrossRef] [PubMed]
- Stephan, A.H.; Madison, D.V.; Mateos, J.M.; Fraser, D.A.; Lovelett, E.A.; Coutellier, L.; Kim, L.; Tsai, H.H.; Huang, E.J.; Rowitch, D.H.; et al. A dramatic increase of C1q protein in the CNS during normal aging. J. Neurosci. 2013, 33, 13460–13474. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, M.I.; Zhou, J.; Botto, M.; Tenner, A.J. Absence of C1q leads to less neuropathology in transgenic mouse models of Alzheimer’s disease. J. Neurosci. 2004, 24, 6457–6465. [Google Scholar] [CrossRef] [PubMed]
- Benoit, M.E.; Hernandez, M.X.; Dinh, M.L.; Benavente, F.; Vasquez, O.; Tenner, A.J. C1q-induced LRP1B and GPR6 proteins expressed early in Alzheimer disease mouse models, are essential for the C1q-mediated protection against amyloid-beta neurotoxicity. J. Biol. Chem. 2013, 288, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Carpanini, S.M.; Torvell, M.; Bevan, R.J.; Byrne, R.A.J.; Daskoulidou, N.; Saito, T.; Saido, T.C.; Taylor, P.R.; Hughes, T.R.; Zelek, W.M.; et al. Terminal complement pathway activation drives synaptic loss in Alzheimer’s disease models. Acta Neuropathol. Commun. 2022, 10, 99. [Google Scholar] [CrossRef] [PubMed]
- Xin, Y.R.; Jiang, J.X.; Hu, Y.; Pan, J.P.; Mi, X.N.; Gao, Q.; Xiao, F.; Zhang, W.; Luo, H.M. The Immune System Drives Synapse Loss During Lipopolysaccharide-Induced Learning and Memory Impairment in Mice. Front. Aging Neurosci. 2019, 11, 279. [Google Scholar] [CrossRef]
- Propson, N.E.; Roy, E.R.; Litvinchuk, A.; Kohl, J.; Zheng, H. Endothelial C3a receptor mediates vascular inflammation and blood-brain barrier permeability during aging. J. Clin. Investig. 2021, 131, e140966. [Google Scholar] [CrossRef]
- Zhang, J.; Malik, A.; Choi, H.B.; Ko, R.W.; Dissing-Olesen, L.; MacVicar, B.A. Microglial CR3 activation triggers long-term synaptic depression in the hippocampus via NADPH oxidase. Neuron 2014, 82, 195–207. [Google Scholar] [CrossRef]
- Gedam, M.; Comerota, M.M.; Propson, N.E.; Chen, T.; Jin, F.; Wang, M.C.; Zheng, H. Complement C3aR depletion reverses HIF-1alpha-induced metabolic impairment and enhances microglial response to Abeta pathology. J. Clin. Investig. 2023, 133, e167501. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T.; Yan, F.; Lin, A.H.; Lambris, J.D.; Alexander, J.J.; Quigg, R.J.; Masliah, E. Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer’s mice. Proc. Natl. Acad. Sci. USA 2002, 99, 10837–10842. [Google Scholar] [CrossRef] [PubMed]
- Choucair-Jaafar, N.; Laporte, V.; Levy, R.; Poindron, P.; Lombard, Y.; Gies, J.P. Complement receptor 3 (CD11b/CD18) is implicated in the elimination of beta-amyloid peptides. Fundam. Clin. Pharmacol. 2011, 25, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Czirr, E.; Castello, N.A.; Mosher, K.I.; Castellano, J.M.; Hinkson, I.V.; Lucin, K.M.; Baeza-Raja, B.; Ryu, J.K.; Li, L.; Farina, S.N.; et al. Microglial complement receptor 3 regulates brain Abeta levels through secreted proteolytic activity. J. Exp. Med. 2017, 214, 1081–1092. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, M.I.; Ager, R.R.; Chu, S.H.; Yazan, O.; Sanderson, S.D.; LaFerla, F.M.; Taylor, S.M.; Woodruff, T.M.; Tenner, A.J. Treatment with a C5aR antagonist decreases pathology and enhances behavioral performance in murine models of Alzheimer’s disease. J. Immunol. 2009, 183, 1375–1383. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Arboledas, A.; Carvalho, K.; Balderrama-Gutierrez, G.; Chu, S.H.; Liang, H.Y.; Schartz, N.D.; Selvan, P.; Petrisko, T.J.; Pan, M.A.; Mortazavi, A.; et al. C5aR1 antagonism alters microglial polarization and mitigates disease progression in a mouse model of Alzheimer’s disease. Acta Neuropathol. Commun. 2022, 10, 116. [Google Scholar] [CrossRef] [PubMed]
- Panayiotou, E.; Fella, E.; Andreou, S.; Papacharalambous, R.; Gerasimou, P.; Costeas, P.; Angeli, S.; Kousiappa, I.; Papacostas, S.; Kyriakides, T. C5aR agonist enhances phagocytosis of fibrillar and non-fibrillar Abeta amyloid and preserves memory in a mouse model of familial Alzheimer’s disease. PLoS ONE 2019, 14, e0225417. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, K.; Schartz, N.D.; Balderrama-Gutierrez, G.; Liang, H.Y.; Chu, S.H.; Selvan, P.; Gomez-Arboledas, A.; Petrisko, T.J.; Fonseca, M.I.; Mortazavi, A.; et al. Modulation of C5a-C5aR1 signaling alters the dynamics of AD progression. J. Neuroinflamm. 2022, 19, 178. [Google Scholar] [CrossRef]
- Wang, C.; Yue, H.; Hu, Z.; Shen, Y.; Ma, J.; Li, J.; Wang, X.D.; Wang, L.; Sun, B.; Shi, P.; et al. Microglia mediate forgetting via complement-dependent synaptic elimination. Science 2020, 367, 688–694. [Google Scholar] [CrossRef]
- Kim, J.H.; Afridi, R.; Han, J.; Jung, H.G.; Kim, S.C.; Hwang, E.M.; Shim, H.S.; Ryu, H.; Choe, Y.; Hoe, H.S.; et al. Gamma subunit of complement component 8 is a neuroinflammation inhibitor. Brain 2021, 144, 528–552. [Google Scholar] [CrossRef]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef]
- Minter, M.R.; Moore, Z.; Zhang, M.; Brody, K.M.; Jones, N.C.; Shultz, S.R.; Taylor, J.M.; Crack, P.J. Deletion of the type-1 interferon receptor in APPSWE/PS1DeltaE9 mice preserves cognitive function and alters glial phenotype. Acta Neuropathol. Commun. 2016, 4, 72. [Google Scholar] [CrossRef] [PubMed]
- Roy, E.R.; Chiu, G.; Li, S.; Propson, N.E.; Kanchi, R.; Wang, B.; Coarfa, C.; Zheng, H.; Cao, W. Concerted type I interferon signaling in microglia and neural cells promotes memory impairment associated with amyloid beta plaques. Immunity 2022, 55, 879–894.e6. [Google Scholar] [CrossRef] [PubMed]
- Holtzman, D.M.; Herz, J.; Bu, G. Apolipoprotein E and apolipoprotein E receptors: Normal biology and roles in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006312. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef]
- Yin, C.; Ackermann, S.; Ma, Z.; Mohanta, S.K.; Zhang, C.; Li, Y.; Nietzsche, S.; Westermann, M.; Peng, L.; Hu, D.; et al. ApoE attenuates unresolvable inflammation by complex formation with activated C1q. Nat. Med. 2019, 25, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.; Wang, M.; Yin, Y.; Tang, Y. The Specific Mechanism of TREM2 Regulation of Synaptic Clearance in Alzheimer’s Disease. Front. Immunol. 2022, 13, 845897. [Google Scholar] [CrossRef]
- Vandendriessche, C.; Balusu, S.; Van Cauwenberghe, C.; Brkic, M.; Pauwels, M.; Plehiers, N.; Bruggeman, A.; Dujardin, P.; Van Imschoot, G.; Van Wonterghem, E.; et al. Importance of extracellular vesicle secretion at the blood-cerebrospinal fluid interface in the pathogenesis of Alzheimer’s disease. Acta Neuropathol. Commun. 2021, 9, 143. [Google Scholar] [CrossRef]
- Reiman, E.M.; Arboleda-Velasquez, J.F.; Quiroz, Y.T.; Huentelman, M.J.; Beach, T.G.; Caselli, R.J.; Chen, Y.; Su, Y.; Myers, A.J.; Hardy, J.; et al. Exceptionally low likelihood of Alzheimer’s dementia in APOE2 homozygotes from a 5,000-person neuropathological study. Nat. Commun. 2020, 11, 667. [Google Scholar] [CrossRef]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; van Duijn, C.M. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 1997, 278, 1349–1356. [Google Scholar] [CrossRef]
- Panitch, R.; Hu, J.; Chung, J.; Zhu, C.; Meng, G.; Xia, W.; Bennett, D.A.; Lunetta, K.L.; Ikezu, T.; Au, R.; et al. Integrative brain transcriptome analysis links complement component 4 and HSPA2 to the APOE epsilon2 protective effect in Alzheimer disease. Mol. Psychiatry 2021, 26, 6054–6064. [Google Scholar] [CrossRef] [PubMed]
- Ulland, T.K.; Song, W.M.; Huang, S.C.; Ulrich, J.D.; Sergushichev, A.; Beatty, W.L.; Loboda, A.A.; Zhou, Y.; Cairns, N.J.; Kambal, A.; et al. TREM2 Maintains Microglial Metabolic Fitness in Alzheimer’s Disease. Cell 2017, 170, 649–663.e13. [Google Scholar] [CrossRef] [PubMed]
- Pottier, C.; Wallon, D.; Rousseau, S.; Rovelet-Lecrux, A.; Richard, A.C.; Rollin-Sillaire, A.; Frebourg, T.; Campion, D.; Hannequin, D. TREM2 R47H variant as a risk factor for early-onset Alzheimer’s disease. J. Alzheimers Dis. 2013, 35, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Konishi, H.; Kiyama, H. Microglial TREM2/DAP12 Signaling: A Double-Edged Sword in Neural Diseases. Front. Cell Neurosci. 2018, 12, 206. [Google Scholar] [CrossRef] [PubMed]
- Qu, W.; Li, L. Loss of TREM2 Confers Resilience to Synaptic and Cognitive Impairment in Aged Mice. J. Neurosci. 2020, 40, 9552–9563. [Google Scholar] [CrossRef] [PubMed]
- Scott-Hewitt, N.; Perrucci, F.; Morini, R.; Erreni, M.; Mahoney, M.; Witkowska, A.; Carey, A.; Faggiani, E.; Schuetz, L.T.; Mason, S.; et al. Local externalization of phosphatidylserine mediates developmental synaptic pruning by microglia. EMBO J. 2020, 39, e105380. [Google Scholar] [CrossRef] [PubMed]
- Fraser, D.A.; Pisalyaput, K.; Tenner, A.J. C1q enhances microglial clearance of apoptotic neurons and neuronal blebs, and modulates subsequent inflammatory cytokine production. J. Neurochem. 2010, 112, 733–743. [Google Scholar] [CrossRef]
- Zhang, B.; Gaiteri, C.; Bodea, L.G.; Wang, Z.; McElwee, J.; Podtelezhnikov, A.A.; Zhang, C.; Xie, T.; Tran, L.; Dobrin, R.; et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 2013, 153, 707–720. [Google Scholar] [CrossRef]
- Readhead, B.; Haure-Mirande, J.V.; Zhang, B.; Haroutunian, V.; Gandy, S.; Schadt, E.E.; Dudley, J.T.; Ehrlich, M.E. Molecular systems evaluation of oligomerogenic APP(E693Q) and fibrillogenic APP(KM670/671NL)/PSEN1(Deltaexon9) mouse models identifies shared features with human Alzheimer’s brain molecular pathology. Mol. Psychiatry 2016, 21, 1099–1111. [Google Scholar] [CrossRef]
- Haure-Mirande, J.V.; Audrain, M.; Fanutza, T.; Kim, S.H.; Klein, W.L.; Glabe, C.; Readhead, B.; Dudley, J.T.; Blitzer, R.D.; Wang, M.; et al. Deficiency of TYROBP, an adapter protein for TREM2 and CR3 receptors, is neuroprotective in a mouse model of early Alzheimer’s pathology. Acta Neuropathol. 2017, 134, 769–788. [Google Scholar] [CrossRef] [PubMed]
- Haure-Mirande, J.V.; Wang, M.; Audrain, M.; Fanutza, T.; Kim, S.H.; Heja, S.; Readhead, B.; Dudley, J.T.; Blitzer, R.D.; Schadt, E.E.; et al. Integrative approach to sporadic Alzheimer’s disease: Deficiency of TYROBP in cerebral Abeta amyloidosis mouse normalizes clinical phenotype and complement subnetwork molecular pathology without reducing Abeta burden. Mol. Psychiatry 2019, 24, 431–446. [Google Scholar] [CrossRef] [PubMed]
- Audrain, M.; Haure-Mirande, J.V.; Wang, M.; Kim, S.H.; Fanutza, T.; Chakrabarty, P.; Fraser, P.; St George-Hyslop, P.H.; Golde, T.E.; Blitzer, R.D.; et al. Integrative approach to sporadic Alzheimer’s disease: Deficiency of TYROBP in a tauopathy mouse model reduces C1q and normalizes clinical phenotype while increasing spread and state of phosphorylation of tau. Mol. Psychiatry 2019, 24, 1383–1397. [Google Scholar] [CrossRef] [PubMed]
- DeVos, S.L.; Corjuc, B.T.; Oakley, D.H.; Nobuhara, C.K.; Bannon, R.N.; Chase, A.; Commins, C.; Gonzalez, J.A.; Dooley, P.M.; Frosch, M.P.; et al. Synaptic Tau Seeding Precedes Tau Pathology in Human Alzheimer’s Disease Brain. Front. Neurosci. 2018, 12, 267. [Google Scholar] [CrossRef] [PubMed]
- Bejanin, A.; Schonhaut, D.R.; La Joie, R.; Kramer, J.H.; Baker, S.L.; Sosa, N.; Ayakta, N.; Cantwell, A.; Janabi, M.; Lauriola, M.; et al. Tau pathology and neurodegeneration contribute to cognitive impairment in Alzheimer’s disease. Brain 2017, 140, 3286–3300. [Google Scholar] [CrossRef]
- Britschgi, M.; Takeda-Uchimura, Y.; Rockenstein, E.; Johns, H.; Masliah, E.; Wyss-Coray, T. Deficiency of terminal complement pathway inhibitor promotes neuronal tau pathology and degeneration in mice. J. Neuroinflamm. 2012, 9, 220. [Google Scholar] [CrossRef] [PubMed]
- Dejanovic, B.; Huntley, M.A.; De Maziere, A.; Meilandt, W.J.; Wu, T.; Srinivasan, K.; Jiang, Z.; Gandham, V.; Friedman, B.A.; Ngu, H.; et al. Changes in the Synaptic Proteome in Tauopathy and Rescue of Tau-Induced Synapse Loss by C1q Antibodies. Neuron 2018, 100, 1322–1336.e7. [Google Scholar] [CrossRef]
- Litvinchuk, A.; Wan, Y.W.; Swartzlander, D.B.; Chen, F.; Cole, A.; Propson, N.E.; Wang, Q.; Zhang, B.; Liu, Z.; Zheng, H. Complement C3aR Inactivation Attenuates Tau Pathology and Reverses an Immune Network Deregulated in Tauopathy Models and Alzheimer’s Disease. Neuron 2018, 100, 1337–1353.e5. [Google Scholar] [CrossRef]
- Bonham, L.W.; Desikan, R.S.; Yokoyama, J.S.; Alzheimer’s Disease Neuroimaging Initiative. The relationship between complement factor C3, APOE epsilon4, amyloid and tau in Alzheimer’s disease. Acta Neuropathol. Commun. 2016, 4, 65. [Google Scholar] [CrossRef]
- Shi, Y.; Yamada, K.; Liddelow, S.A.; Smith, S.T.; Zhao, L.; Luo, W.; Tsai, R.M.; Spina, S.; Grinberg, L.T.; Rojas, J.C.; et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 2017, 549, 523–527. [Google Scholar] [CrossRef]
- Dejanovic, B.; Wu, T.; Tsai, M.C.; Graykowski, D.; Gandham, V.D.; Rose, C.M.; Bakalarski, C.E.; Ngu, H.; Wang, Y.; Pandey, S.; et al. Complement C1q-dependent excitatory and inhibitory synapse elimination by astrocytes and microglia in Alzheimer’s disease mouse models. Nat. Aging 2022, 2, 837–850. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Batista, A.F.; Khan, K.A.; Papavergi, M.-T.; Lemere, C.A. The Importance of Complement-Mediated Immune Signaling in Alzheimer’s Disease Pathogenesis. Int. J. Mol. Sci. 2024, 25, 817. https://doi.org/10.3390/ijms25020817
Batista AF, Khan KA, Papavergi M-T, Lemere CA. The Importance of Complement-Mediated Immune Signaling in Alzheimer’s Disease Pathogenesis. International Journal of Molecular Sciences. 2024; 25(2):817. https://doi.org/10.3390/ijms25020817
Chicago/Turabian StyleBatista, André F., Khyrul A. Khan, Maria-Tzousi Papavergi, and Cynthia A. Lemere. 2024. "The Importance of Complement-Mediated Immune Signaling in Alzheimer’s Disease Pathogenesis" International Journal of Molecular Sciences 25, no. 2: 817. https://doi.org/10.3390/ijms25020817
APA StyleBatista, A. F., Khan, K. A., Papavergi, M.-T., & Lemere, C. A. (2024). The Importance of Complement-Mediated Immune Signaling in Alzheimer’s Disease Pathogenesis. International Journal of Molecular Sciences, 25(2), 817. https://doi.org/10.3390/ijms25020817