
The Dual Roles of Activating Transcription Factor 3 (ATF3) in Inflammation, Apoptosis, Ferroptosis, and Pathogen Infection Responses

Abstract
:1. Introduction
2. Literature Screening
3. Mechanisms of ATF3 Induction under Stressful Physiological Conditions
4. Implications of PAMPs and PRRs Activation on the Expression of ATF3
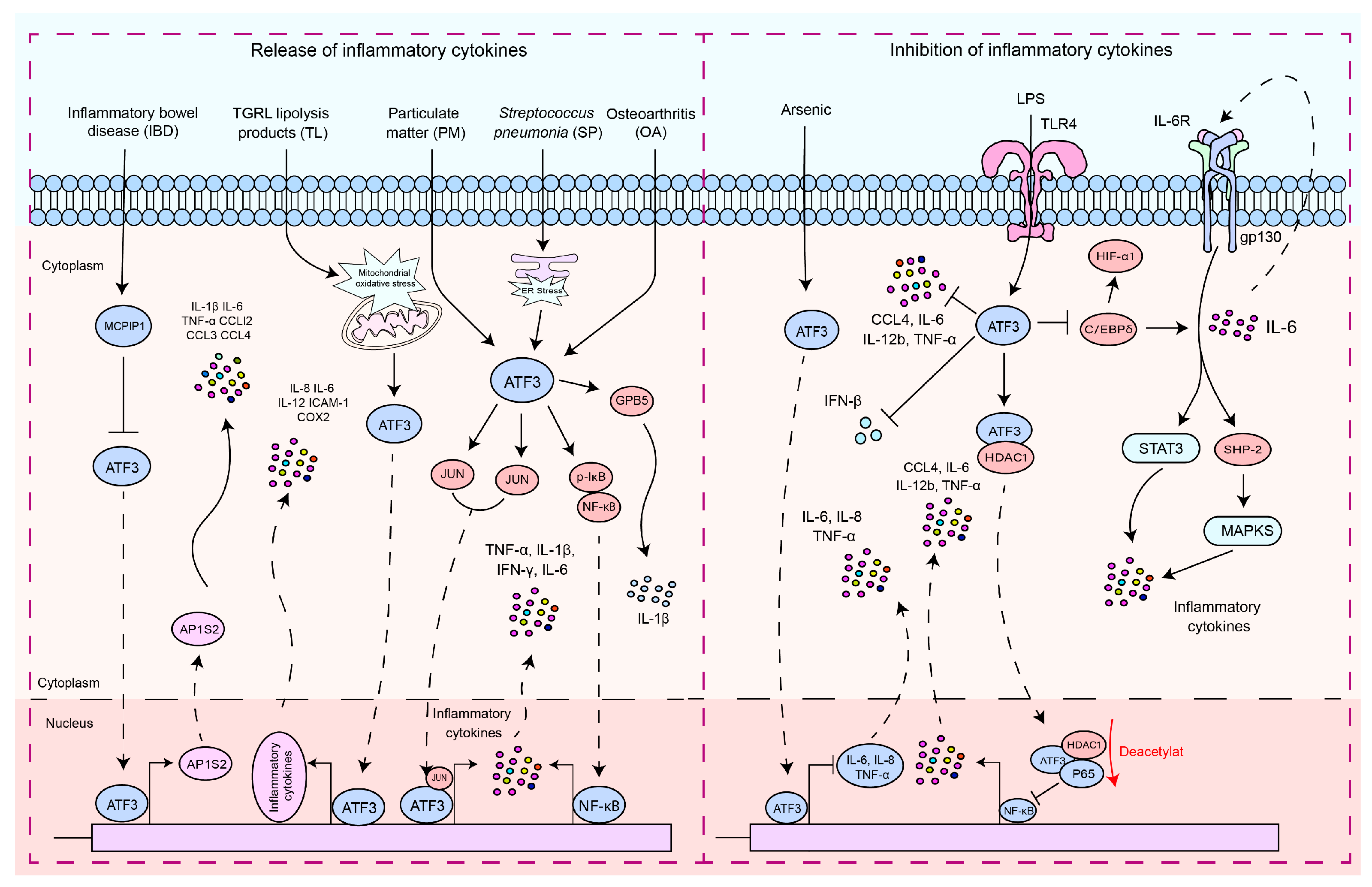
5. Modulatory Role of ATF3 in Inflammatory Cytokine
6. The Role of ATF3 in the Regulation of Cell Death
6.1. The Regulation of Apoptosis
6.2. The Regulation of Ferroptosis
7. The Functions of ATF3 in the Pathogenic Microbial Infection Process
7.1. The Functions of ATF3 in Viral Infection
7.1.1. DNA Virus

{kind=link}
{kind=link}
{kind=link}
| Microbial Name | Microbial Type | Functions of ATF3 | Research Models | References |
|---|---|---|---|---|
| Virus | ||||
| Hepatitis B Virus | DNA virus | ATF3 increases HBx mRNA degradation by regulating Ski2 expression. | HepG2, PXB, and AML12 cells. | [121,122] |
| Murine CytoMegalovirus | DNA virus | ATF3 regulates anti-MCMV responses by controlling the production of IFN-γ in NK cells. | C57BL/6, Rag1−/−, BALB/c, and ATF3−/− mice. | [126] |
| Herpes Simplex Virus 1 | DNA virus | ATF3 maintains the integrity of the neurons harboring latent virus. | HEp-2, Vero cell, HEK 293T, and CBA/J mice. | [132] |
| Human Papillomavirus | DNA virus | ATF3 plays a significant role in inducing apoptosis in HeLa cells. | HeLa cells. | [138,139] |
| Japanese Encephalitis Virus | RNA virus | ATF3 as a negative regulator of antiviral response and autophagy in mammalian cells during JEV infection. | Neuro2a, HEK, HeLa, and MEF cells. | [120] |
| Zika Virus | RNA virus | ATF3 acts to limit ZIKV infection by regulating autophagy and, thus, also ZIKV replication. | Wild-type and ATF3 knockout A549 cell lines. | [140] |
| Coxsackievirus B3 | RNA virus | -ATF3 regulates cell death induced by CVB3 infection. | HeLa cells. | [141] |
| Dengue Virus | RNA virus | Dengue virus degrades USP33-ATF3 axis via extracellular vesicles to activate microglial cells. | THP1 and HEK293T cells. | [142] |
| Human Immunodeficiency Virus | RNA virus | ATF3 orchestrates a recruitment of chromatin-modifying proteins. | Cervical carcinoma cell line C33A. | [143] |
| Bacteria | ||||
| Staphylococcus aureus | Gram-positive bacteria | ATF3 regulates antibacterial genes for antimicrobial processes. | Wild-type and ATF3 knockout mice. RAW 264.7 cell lines. | [14,15] |
| Streptococcus pneumoniae | Gram-positive bacteria | ATF3 promotes cytokine production (IL-17A TNF-α, IL-1β, and IFN-γ) in response to S. pneumoniae infection. | C57BL/6 WT and ATF3 KO mice. RAW 264.7 cells. | [4,13,17] |
| Listeria monocytogenes | Gram-positive bacteria | ATF3 provides protection from L. monocytogenes infections. | ATF3 knockout and wild-type mice. A549, HEp2, and RAW 264.7 cells. | [15] |
| Neisseria gonorrhoeae | Gram-negative bacteria | ATF3 negatively regulates IL-6 expression during N. gonorrhoeae infection. | T84 colorectal epithelial cells, End 1 endocervical cells, nasopharyngeal cells, and bronchial epithelial cell line 16HBE14. | [144] |
| Escherichia coli | Gram-negative bacteria | ATF3-mediated suppression of the innate cytokine storm abrogated the control of bacteria and causes high susceptibility to secondary infections. | C57BL/6 WT and ATF3 KO mice. A549, HEp2, and RAW 264.7 cells. | [15,53] |
| Pseudomonas aeruginosa | Gram-negative bacteria | ATF3 suppresses the progression of PA infection in hosts by inhibiting the activity of NF/κB. | AW264.7 and C57BL/6 ATF3 KO mice. | [145,146] |
| Mycobacterium tuberculosis | Other bacteria | ATF3 promotes cell autophagy and suppresses inflammatory response in Mycobacterium-tuberculosis-infected A549 cells. | A549 cells and RAW264.7 cells. BALB/c mice. | [12,147] |
| Mycoplasma pneumoniae | Other bacteria | ATF3 inhibits the expression and release of TNF-α, IL-1β, IL-6, and IL-18 induced by Mycoplasma pneumoniae in vitro and in vivo. | BALB/c mice, C57BL/6 mice, and RAW264.7 cells. | [148] |
| Fungi and Parasite | ||||
| Patulin | Fungal toxin | Patulin enhances ATF3 expression and promotes apoptosis in colorectal cancer cells. | HCT116 cells. | [149] |
| Deoxynivalenol | Fungal toxin | Deoxynivalenol induces G2/M cell cycle arrest in HepG2 cells by ATF3ΔZip2a/2b. | HepG2 cells. | [150] |
| Leishmania | Parasite | ATF3 promotes the survival of the Leishmania by regulating inflammatory response. | RAW 264.7 and BMDM cells. | [151,152] |
7.1.2. RNA Virus
7.2. The Functions of ATF3 in Bacterial Infection
7.2.1. Gram-Positive Bacteria
7.2.2. Gram-Negative Bacteria
7.2.3. Other Bacteria
7.3. The Functions of ATF3 in Fungal and Parasite Infections
8. Prospects for Clinical Applications
9. Conclusions and Future Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, W.; Chang, X.; Yao, W.; Wei, N.; Huo, N.; Wang, Y.; Wei, Q.; Liu, H.; Wang, X.; Zhang, S.; et al. Host CARD11 Inhibits Newcastle Disease Virus Replication by Suppressing Viral Polymerase Activity in Neurons. J. Virol. 2019, 93, e01499-19. [Google Scholar] [CrossRef] [PubMed]
- Caamano, J.; Hunter, C.A. NF-kappaB family of transcription factors: Central regulators of innate and adaptive immune functions. Clin. Microbiol. Rev. 2002, 15, 414–429. [Google Scholar] [CrossRef] [PubMed]
- Le Negrate, G. Subversion of innate immune responses by bacterial hindrance of NF-kappaB pathway. Cell Microbiol. 2012, 14, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kim, G.L.; Kim, N.Y.; Kim, S.J.; Ghosh, P.; Rhee, D.K. ATF3 Stimulates IL-17A by Regulating Intracellular Ca2+/ROS-Dependent IL-1beta Activation During Streptococcus pneumoniae Infection. Front. Immunol. 2018, 9, 1954. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Tian, M.; Ding, C.; Yu, S. The C/EBP Homologous Protein (CHOP) Transcription Factor Functions in Endoplasmic Reticulum Stress-Induced Apoptosis and Microbial Infection. Front. Immunol. 2018, 9, 3083. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Yang, B.; Shen, C.; Zhang, T.; Hao, Y.; Zhang, D.; Liu, H.; Shi, X.; Li, G.; Yang, J.; et al. MGF360-9L Is a Major Virulence Factor Associated with the African Swine Fever Virus by Antagonizing the JAK/STAT Signaling Pathway. mBio 2022, 13, e0233021. [Google Scholar] [CrossRef] [PubMed]
- Ariffianto, A.; Deng, L.; Harada, S.; Liang, Y.; Matsui, C.; Abe, T.; Shoji, I. Transcription Factor JunB Suppresses Hepatitis C Virus Replication. Kobe J. Med. Sci. 2023, 69, E86–E95. [Google Scholar]
- Li, D.; Jing, J.; Dong, X.; Zhang, C.; Wang, J.; Wan, X. Activating transcription factor 3: A potential therapeutic target for inflammatory pulmonary diseases. Immun. Inflamm. Dis. 2023, 11, e1028. [Google Scholar] [CrossRef]
- Rohini, M.; Haritha Menon, A.; Selvamurugan, N. Role of activating transcription factor 3 and its interacting proteins under physiological and pathological conditions. Int. J. Biol. Macromol. 2018, 120, 310–317. [Google Scholar] [CrossRef]
- Thompson, M.R.; Xu, D.; Williams, B.R. ATF3 transcription factor and its emerging roles in immunity and cancer. J. Mol. Med. 2009, 87, 1053–1060. [Google Scholar] [CrossRef]
- Zhou, H.; Li, N.; Yuan, Y.; Jin, Y.G.; Guo, H.; Deng, W.; Tang, Q.Z. Activating transcription factor 3 in cardiovascular diseases: A potential therapeutic target. Basic. Res. Cardiol. 2018, 113, 37. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Majumder, D.; Mal, S.; Chakraborty, S.; Gupta, P.; Jana, K.; Gupta, U.D.; Ghosh, Z.; Kundu, M.; Basu, J. Activating transcription factor 3 modulates the macrophage immune response to Mycobacterium tuberculosis infection via reciprocal regulation of inflammatory genes and lipid body formation. Cell Microbiol. 2020, 22, e13142. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.T.; Kim, E.H.; Luong, T.T.; Pyo, S.; Rhee, D.K. ATF3 confers resistance to pneumococcal infection through positive regulation of cytokine production. J. Infect. Dis. 2014, 210, 1745–1754. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Ma, Z.; Zheng, J.; Huang, S.; Yang, X.; Song, Y.; Dong, D.; Shi, L.; Xu, D. ATF3 Positively Regulates Antibacterial Immunity by Modulating Macrophage Killing and Migration Functions. Front. Immunol. 2022, 13, 839502. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.T.; Luong, T.T.; Lee, S.; Kim, G.L.; Pyo, S.; Rhee, D.K. ATF3 provides protection from Staphylococcus aureus and Listeria monocytogenes infections. FEMS Microbiol. Lett. 2016, 363, fnw062. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Takeuchi, H.; Nishioka, M.; Morimoto, N.; Kamioka, M.; Kumon, Y.; Sugiura, T. Relationship of IL-8 production and the CagA status in AGS cells infected with Helicobacter pylori exposed to low pH and activating transcription factor 3 (ATF3). Microbiol. Res. 2009, 164, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.T.; Kim, E.H.; Luong, T.T.; Pyo, S.; Rhee, D.K. TLR4 mediates pneumolysin-induced ATF3 expression through the JNK/p38 pathway in Streptococcus pneumoniae-infected RAW 264.7 cells. Mol. Cells 2015, 38, 58–64. [Google Scholar] [CrossRef]
- Zhao, J.; Li, X.; Guo, M.; Yu, J.; Yan, C. The common stress responsive transcription factor ATF3 binds genomic sites enriched with p300 and H3K27ac for transcriptional regulation. BMC Genom. 2016, 17, 335. [Google Scholar] [CrossRef]
- Gilchrist, M.; Thorsson, V.; Li, B.; Rust, A.G.; Korb, M.; Roach, J.C.; Kennedy, K.; Hai, T.; Bolouri, H.; Aderem, A. Systems biology approaches identify ATF3 as a negative regulator of Toll-like receptor 4. Nature 2006, 441, 173–178. [Google Scholar] [CrossRef]
- Hai, T.; Wolford, C.C.; Chang, Y.S. ATF3, a hub of the cellular adaptive-response network, in the pathogenesis of diseases: Is modulation of inflammation a unifying component? Gene Expr. 2010, 15, 1–11. [Google Scholar] [CrossRef]
- Hai, T.; Wolfgang, C.D.; Marsee, D.K.; Allen, A.E.; Sivaprasad, U. ATF3 and stress responses. Gene Expr. 1999, 7, 321–335. [Google Scholar] [PubMed]
- Chen, H.; Luo, S.; Chen, H.; Zhang, C. ATF3 regulates SPHK1 in cardiomyocyte injury via endoplasmic reticulum stress. Immun. Inflamm. Dis. 2023, 11, e998. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Shtil, A.A.; Tan, T.H.; Roninson, I.B.; Kong, A.N. Adriamycin activates c-jun N-terminal kinase in human leukemia cells: A relevance to apoptosis. Cancer Lett. 1996, 107, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Drysdale, B.E.; Howard, D.L.; Johnson, R.J. Identification of a lipopolysaccharide inducible transcription factor in murine macrophages. Mol. Immunol. 1996, 33, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Weir, E.; Chen, Q.; DeFrances, M.C.; Bell, A.; Taub, R.; Zarnegar, R. Rapid induction of mRNAs for liver regeneration factor and insulin-like growth factor binding protein-1 in primary cultures of rat hepatocytes by hepatocyte growth factor and epidermal growth factor. Hepatology 1994, 20, 955–960. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Jeong, J.Y.; Surh, Y.J.; Kim, K.W. Expression of stress-response ATF3 is mediated by Nrf2 in astrocytes. Nucleic Acids Res. 2010, 38, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Sun, L.; Liu, C.; Li, L. Naringin regulates endoplasmic reticulum stress and mitophagy through the ATF3/PINK1 signaling axis to alleviate pulmonary fibrosis. Naunyn Schmiedebergs Arch. Pharmacol. 2023, 396, 1155–1169. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, Y.; Du, T.; Yang, H.; Lei, L.; Guo, M.; Ding, H.F.; Zhang, J.; Wang, H.; Chen, X.; et al. ATF3 promotes erastin-induced ferroptosis by suppressing system Xc−. Cell Death Differ. 2020, 27, 662–675. [Google Scholar] [CrossRef]
- Fan, F.; Jin, S.; Amundson, S.A.; Tong, T.; Fan, W.; Zhao, H.; Zhu, X.; Mazzacurati, L.; Li, X.; Petrik, K.L.; et al. ATF3 induction following DNA damage is regulated by distinct signaling pathways and over-expression of ATF3 protein suppresses cells growth. Oncogene 2002, 21, 7488–7496. [Google Scholar] [CrossRef]
- Liang, G.; Wolfgang, C.D.; Chen, B.P.; Chen, T.H.; Hai, T. ATF3 gene. Genomic organization, promoter, and regulation. J. Biol. Chem. 1996, 271, 1695–1701. [Google Scholar] [CrossRef]
- American Association of Neurological Surgeons (AANS); American Society of Neuroradiology (ASNR); Cardiovascular and Interventional Radiology Society of Europe (CIRSE); Canadian Interventional Radiology Association (CIRA); Sacks, D.; Baxter, B.; Campbell, B.C.V.; Carpenter, J.S.; Cognard, C.; Dippel, D.; et al. Multisociety Consensus Quality Improvement Revised Consensus Statement for Endovascular Therapy of Acute Ischemic Stroke. Int. J. Stroke 2018, 13, 612–632. [Google Scholar] [CrossRef] [PubMed]
- Hai, T.; Hartman, M.G. The molecular biology and nomenclature of the activating transcription factor/cAMP responsive element binding family of transcription factors: Activating transcription factor proteins and homeostasis. Gene 2001, 273, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Allen-Jennings, A.E.; Hartman, M.G.; Kociba, G.J.; Hai, T. The roles of ATF3 in glucose homeostasis. A transgenic mouse model with liver dysfunction and defects in endocrine pancreas. J. Biol. Chem. 2001, 276, 29507–29514. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.Y.; Shin, H.Y.; Kim, J.Y.; Kim, D.G.; Choi, Y.M.; Kwon, H.K.; Rhee, D.K.; Kim, Y.S.; Choi, S. ATF3 plays a key role in Kdo2-lipid A-induced TLR4-dependent gene expression via NF-kappaB activation. PLoS ONE 2010, 5, e14181. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.; Toftgard, R.; Bohm, S. Activated Ha-Ras but not TPA induces transcription through binding sites for activating transcription factor 3/Jun and a novel nuclear factor. J. Biol. Chem. 1995, 270, 12210–12218. [Google Scholar] [CrossRef] [PubMed]
- Nakagomi, S.; Suzuki, Y.; Namikawa, K.; Kiryu-Seo, S.; Kiyama, H. Expression of the activating transcription factor 3 prevents c-Jun N-terminal kinase-induced neuronal death by promoting heat shock protein 27 expression and Akt activation. J. Neurosci. 2003, 23, 5187–5196. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.P.; Liang, G.; Whelan, J.; Hai, T. ATF3 and ATF3 delta Zip. Transcriptional repression versus activation by alternatively spliced isoforms. J. Biol. Chem. 1994, 269, 15819–15826. [Google Scholar] [CrossRef]
- Shvedunova, M.; Akhtar, A. Modulation of cellular processes by histone and non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2022, 23, 329–349. [Google Scholar] [CrossRef]
- Furumatsu, T.; Tsuda, M.; Yoshida, K.; Taniguchi, N.; Ito, T.; Hashimoto, M.; Ito, T.; Asahara, H. Sox9 and p300 cooperatively regulate chromatin-mediated transcription. J. Biol. Chem. 2005, 280, 35203–35208. [Google Scholar] [CrossRef]
- Hua, B.; Tamamori-Adachi, M.; Luo, Y.; Tamura, K.; Morioka, M.; Fukuda, M.; Tanaka, Y.; Kitajima, S. A splice variant of stress response gene ATF3 counteracts NF-kappaB-dependent anti-apoptosis through inhibiting recruitment of CREB-binding protein/p300 coactivator. J. Biol. Chem. 2006, 281, 1620–1629. [Google Scholar] [CrossRef]
- Wolfgang, C.D.; Liang, G.; Okamoto, Y.; Allen, A.E.; Hai, T. Transcriptional autorepression of the stress-inducible gene ATF3. J. Biol. Chem. 2000, 275, 16865–16870. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. TLR signaling. Cell Death Differ. 2006, 13, 816–825. [Google Scholar] [CrossRef] [PubMed]
- Whitmore, M.M.; Iparraguirre, A.; Kubelka, L.; Weninger, W.; Hai, T.; Williams, B.R. Negative regulation of TLR-signaling pathways by activating transcription factor-3. J. Immunol. 2007, 179, 3622–3630. [Google Scholar] [CrossRef] [PubMed]
- Khuu, C.H.; Barrozo, R.M.; Hai, T.; Weinstein, S.L. Activating transcription factor 3 (ATF3) represses the expression of CCL4 in murine macrophages. Mol. Immunol. 2007, 44, 1598–1605. [Google Scholar] [CrossRef]
- Suganami, T.; Yuan, X.; Shimoda, Y.; Uchio-Yamada, K.; Nakagawa, N.; Shirakawa, I.; Usami, T.; Tsukahara, T.; Nakayama, K.; Miyamoto, Y.; et al. Activating transcription factor 3 constitutes a negative feedback mechanism that attenuates saturated Fatty acid/toll-like receptor 4 signaling and macrophage activation in obese adipose tissue. Circ. Res. 2009, 105, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Yu, L.; Li, J.; Xie, G.; Rong, T.; Zhang, L.; Chen, J.; Meng, Q.; Irving, A.T.; Wang, D.; et al. ATF3 suppresses metastasis of bladder cancer by regulating gelsolin-mediated remodeling of the actin cytoskeleton. Cancer Res. 2013, 73, 3625–3637. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Zhang, C.; Wu, W.; Chen, H.; Lin, R.; Sun, R.; Gao, X.; Li, G.; He, Q.; Gao, H.; et al. MCPIP1 restrains mucosal inflammation by orchestrating the intestinal monocyte to macrophage maturation via an ATF3-AP1S2 axis. Gut 2023, 72, 882–895. [Google Scholar] [CrossRef]
- Nyunt, T.; Britton, M.; Wanichthanarak, K.; Budamagunta, M.; Voss, J.C.; Wilson, D.W.; Rutledge, J.C.; Aung, H.H. Mitochondrial oxidative stress-induced transcript variants of ATF3 mediate lipotoxic brain microvascular injury. Free Radic. Biol. Med. 2019, 143, 25–46. [Google Scholar] [CrossRef]
- Dendorfer, U.; Oettgen, P.; Libermann, T.A. Multiple regulatory elements in the interleukin-6 gene mediate induction by prostaglandins, cyclic AMP, and lipopolysaccharide. Mol. Cell Biol. 1994, 14, 4443–4454. [Google Scholar] [CrossRef]
- de Haij, S.; Bakker, A.C.; van der Geest, R.N.; Haegeman, G.; Vanden Berghe, W.; Aarbiou, J.; Daha, M.R.; van Kooten, C. NF-kappaB mediated IL-6 production by renal epithelial cells is regulated by c-jun NH2-terminal kinase. J. Am. Soc. Nephrol. 2005, 16, 1603–1611. [Google Scholar] [CrossRef]
- Iezaki, T.; Ozaki, K.; Fukasawa, K.; Inoue, M.; Kitajima, S.; Muneta, T.; Takeda, S.; Fujita, H.; Onishi, Y.; Horie, T.; et al. ATF3 deficiency in chondrocytes alleviates osteoarthritis development. J. Pathol. 2016, 239, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.; Wu, Y.; Liu, H.; Wu, Y.; Shen, H.; Li, W. ATF3 is positively involved in particulate matter-induced airway inflammation in vitro and in vivo. Toxicol. Lett. 2018, 287, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Hoetzenecker, W.; Echtenacher, B.; Guenova, E.; Hoetzenecker, K.; Woelbing, F.; Bruck, J.; Teske, A.; Valtcheva, N.; Fuchs, K.; Kneilling, M.; et al. ROS-induced ATF3 causes susceptibility to secondary infections during sepsis-associated immunosuppression. Nat. Med. 2011, 18, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Balamurugan, K.; Mendoza-Villanueva, D.; Sharan, S.; Summers, G.H.; Dobrolecki, L.E.; Lewis, M.T.; Sterneck, E. C/EBPdelta links IL-6 and HIF-1 signaling to promote breast cancer stem cell-associated phenotypes. Oncogene 2019, 38, 3765–3780. [Google Scholar] [CrossRef] [PubMed]
- Litvak, V.; Ramsey, S.A.; Rust, A.G.; Zak, D.E.; Kennedy, K.A.; Lampano, A.E.; Nykter, M.; Shmulevich, I.; Aderem, A. Function of C/EBPdelta in a regulatory circuit that discriminates between transient and persistent TLR4-induced signals. Nat. Immunol. 2009, 10, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wei, S.; Chen, M.; Li, J.; Wei, Y.; Zhang, J.; Dong, W. P2RY13 Exacerbates Intestinal Inflammation by Damaging the Intestinal Mucosal Barrier via Activating IL-6/STAT3 Pathway. Int. J. Biol. Sci. 2022, 18, 5056–5069. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef]
- Kwon, J.W.; Kwon, H.K.; Shin, H.J.; Choi, Y.M.; Anwar, M.A.; Choi, S. Activating transcription factor 3 represses inflammatory responses by binding to the p65 subunit of NF-kappaB. Sci. Rep. 2015, 5, 14470. [Google Scholar] [CrossRef]
- Labzin, L.I.; Schmidt, S.V.; Masters, S.L.; Beyer, M.; Krebs, W.; Klee, K.; Stahl, R.; Lutjohann, D.; Schultze, J.L.; Latz, E.; et al. ATF3 Is a Key Regulator of Macrophage IFN Responses. J. Immunol. 2015, 195, 4446–4455. [Google Scholar] [CrossRef]
- Ma, J.Q.; Li, Z.; Xie, W.R.; Liu, C.M.; Liu, S.S. Quercetin protects mouse liver against CCl4-induced inflammation by the TLR2/4 and MAPK/NF-kappaB pathway. Int. Immunopharmacol. 2015, 28, 531–539. [Google Scholar] [CrossRef]
- Shi, Q.; Hu, B.; Yang, C.; Deng, S.; Cheng, X.; Wu, J.; Qi, N. ATF3 inhibits arsenic-induced malignant transformation of human bronchial epithelial cells by attenuating inflammation. Toxicology 2021, 460, 152890. [Google Scholar] [CrossRef] [PubMed]
- De Nardo, D.; Labzin, L.I.; Kono, H.; Seki, R.; Schmidt, S.V.; Beyer, M.; Xu, D.; Zimmer, S.; Lahrmann, C.; Schildberg, F.A.; et al. High-density lipoprotein mediates anti-inflammatory reprogramming of macrophages via the transcriptional regulator ATF3. Nat. Immunol. 2014, 15, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Yue, J.; Lopez, J.M. Understanding MAPK Signaling Pathways in Apoptosis. Int. J. Mol. Sci. 2020, 21, 2346. [Google Scholar] [CrossRef] [PubMed]
- Morana, O.; Wood, W.; Gregory, C.D. The Apoptosis Paradox in Cancer. Int. J. Mol. Sci. 2022, 23, 1328. [Google Scholar] [CrossRef] [PubMed]
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Bertheloot, D.; Latz, E.; Franklin, B.S. Necroptosis, pyroptosis and apoptosis: An intricate game of cell death. Cell Mol. Immunol. 2021, 18, 1106–1121. [Google Scholar] [CrossRef] [PubMed]
- Kitakata, H.; Endo, J.; Ikura, H.; Moriyama, H.; Shirakawa, K.; Katsumata, Y.; Sano, M. Therapeutic Targets for DOX-Induced Cardiomyopathy: Role of Apoptosis vs. Ferroptosis. Int. J. Mol. Sci. 2022, 23, 1414. [Google Scholar] [CrossRef]
- Williams, G.T. Role of apoptosis in the immune system. Biochem. Cell Biol. 1994, 72, 447–450. [Google Scholar] [CrossRef]
- Nagata, S.; Tanaka, M. Programmed cell death and the immune system. Nat. Rev. Immunol. 2017, 17, 333–340. [Google Scholar] [CrossRef]
- Ekert, P.G.; Vaux, D.L. Apoptosis and the immune system. Br. Med. Bull. 1997, 53, 591–603. [Google Scholar] [CrossRef]
- Place, D.E.; Lee, S.; Kanneganti, T.D. PANoptosis in microbial infection. Curr. Opin. Microbiol. 2021, 59, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zychlinsky, A. Apoptosis induced by bacterial pathogens. Microb. Pathog. 1994, 17, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Hartman, M.G.; Lu, D.; Kim, M.L.; Kociba, G.J.; Shukri, T.; Buteau, J.; Wang, X.; Frankel, W.L.; Guttridge, D.; Prentki, M.; et al. Role for activating transcription factor 3 in stress-induced beta-cell apoptosis. Mol. Cell Biol. 2004, 24, 5721–5732. [Google Scholar] [CrossRef] [PubMed]
- Nobori, K.; Ito, H.; Tamamori-Adachi, M.; Adachi, S.; Ono, Y.; Kawauchi, J.; Kitajima, S.; Marumo, F.; Isobe, M. ATF3 inhibits doxorubicin-induced apoptosis in cardiac myocytes: A novel cardioprotective role of ATF3. J. Mol. Cell Cardiol. 2002, 34, 1387–1397. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.M.; Liu, X.M.; Wang, Y.; Chen, Z.Y. Activating transcription factor 3 (ATF3) regulates cell growth, apoptosis, invasion and collagen synthesis in keloid fibroblast through transforming growth factor beta (TGF-beta)/SMAD signaling pathway. Bioengineered 2021, 12, 117–126. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Deng, L.; Zou, K.; Tian, Y.; Tang, X. ATF3 Modulates the Proliferation, Migration, and Apoptosis of Synovial Fibroblasts after Arthroscopy by Promoting RGS1 Transcription. Curr. Mol. Med. 2023, 23, 981–990. [Google Scholar] [CrossRef]
- Lin, H.; Cheng, C.F. Activating transcription factor 3, an early cellular adaptive responder in ischemia/reperfusion-induced injury. Ci Ji Yi Xue Za Zhi 2018, 30, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.F.; Lin, H. Acute kidney injury and the potential for ATF3-regulated epigenetic therapy. Toxicol. Mech. Methods 2011, 21, 362–366. [Google Scholar] [CrossRef]
- Yoshida, T.; Sugiura, H.; Mitobe, M.; Tsuchiya, K.; Shirota, S.; Nishimura, S.; Shiohira, S.; Ito, H.; Nobori, K.; Gullans, S.R.; et al. ATF3 protects against renal ischemia-reperfusion injury. J. Am. Soc. Nephrol. 2008, 19, 217–224. [Google Scholar] [CrossRef]
- Thiel, D.A.; Reeder, M.K.; Pfaff, A.; Coleman, T.R.; Sells, M.A.; Chernoff, J. Cell cycle-regulated phosphorylation of p21-activated kinase 1. Curr. Biol. 2002, 12, 1227–1232. [Google Scholar] [CrossRef]
- Meulmeester, E.; Jochemsen, A.G. p53: A guide to apoptosis. Curr. Cancer Drug Targets 2008, 8, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Deng, S.; Lu, Y.; Zhang, Y.; Yang, L.; Guan, Y.; Jiang, H.; Li, H. Increased inflammation and brain injury after transient focal cerebral ischemia in activating transcription factor 3 knockout mice. Neuroscience 2012, 220, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Chen, J.J.; Wu, J.S.; Tsai, H.D.; Lin, H.; Yan, Y.T.; Hsu, C.Y.; Ho, Y.S.; Lin, T.N. Novel link of anti-apoptotic ATF3 with pro-apoptotic CTMP in the ischemic brain. Mol. Neurobiol. 2015, 51, 543–557. [Google Scholar] [CrossRef] [PubMed]
- Mashima, T.; Udagawa, S.; Tsuruo, T. Involvement of transcriptional repressor ATF3 in acceleration of caspase protease activation during DNA damaging agent-induced apoptosis. J. Cell Physiol. 2001, 188, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Pan, D.; Wang, X.; Huo, Z.; Wu, X.; Li, J.; Cao, J.; Xu, H.; Du, L.; Xu, B. Silencing ATF3 Might Delay TBHP-Induced Intervertebral Disc Degeneration by Repressing NPC Ferroptosis, Apoptosis, and ECM Degradation. Oxid. Med. Cell Longev. 2022, 2022, 4235126. [Google Scholar] [CrossRef] [PubMed]
- Inaba, Y.; Hashiuchi, E.; Watanabe, H.; Kimura, K.; Oshima, Y.; Tsuchiya, K.; Murai, S.; Takahashi, C.; Matsumoto, M.; Kitajima, S.; et al. The transcription factor ATF3 switches cell death from apoptosis to necroptosis in hepatic steatosis in male mice. Nat. Commun. 2023, 14, 167. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Hu, B.; Yang, C.; Zhao, L.; Wu, J.; Qi, N. ATF3 Promotes Arsenic-Induced Apoptosis and Oppositely Regulates DR5 and Bcl-xL Expression in Human Bronchial Epithelial Cells. Int. J. Mol. Sci. 2021, 22, 4223. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.Y.; Zong, C.S.; Xia, W.; Yamaguchi, H.; Ding, Q.; Xie, X.; Lang, J.Y.; Lai, C.C.; Chang, C.J.; Huang, W.C.; et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat. Cell Biol. 2008, 10, 138–148. [Google Scholar] [CrossRef]
- Maiese, A.; Spina, F.; Visi, G.; Del Duca, F.; De Matteis, A.; La Russa, R.; Di Paolo, M.; Frati, P.; Fineschi, V. The Expression of FOXO3a as a Forensic Diagnostic Tool in Cases of Traumatic Brain Injury: An Immunohistochemical Study. Int. J. Mol. Sci. 2023, 24, 2584. [Google Scholar] [CrossRef]
- Maiese, A.; Manetti, A.C.; Santoro, P.; Del Duca, F.; De Matteis, A.; Turillazzi, E.; Frati, P.; Fineschi, V. FOXO3 Depletion as a Marker of Compression-Induced Apoptosis in the Ligature Mark: An Immunohistochemical Study. Int. J. Mol. Sci. 2023, 24, 1396. [Google Scholar] [CrossRef]
- Li, D.; Qu, Y.; Mao, M.; Zhang, X.; Li, J.; Ferriero, D.; Mu, D. Involvement of the PTEN-AKT-FOXO3a pathway in neuronal apoptosis in developing rat brain after hypoxia-ischemia. J. Cereb. Blood Flow. Metab. 2009, 29, 1903–1913. [Google Scholar] [CrossRef] [PubMed]
- Van Der Heide, L.P.; Hoekman, M.F.; Smidt, M.P. The ins and outs of FoxO shuttling: Mechanisms of FoxO translocation and transcriptional regulation. Biochem. J. 2004, 380, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Lee, S.; Joo, J.C.; Hong, E.; Cui, Z.Y.; Jo, E.; Park, S.J.; Jang, H.J. Chelidonium majus Induces Apoptosis of Human Ovarian Cancer Cells via ATF3-Mediated Regulation of Foxo3a by Tip60. J. Microbiol. Biotechnol. 2022, 32, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Lu, G.; Ha, K.; Lin, H.; Du, Y.; Zuo, Q.; Fu, Y.; Zou, C.; Zhang, P. Acetylation of TIP60 at K104 is essential for metabolic stress-induced apoptosis in cells of hepatocellular cancer. Exp. Cell Res. 2018, 362, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Luo, J.; Zhang, W.; Gu, W. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol. Cell 2006, 24, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Qiu, M.; Liu, J.; Su, Y.; Guo, R.; Zhao, B.; Liu, J. Diosmetin Induces Apoptosis by Downregulating AKT Phosphorylation via P53 Activation in Human Renal Carcinoma ACHN Cells. Protein Pept. Lett. 2020, 27, 1022–1028. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Sun, J.; Chen, T.; Tao, Z.; Zhang, X.; Tian, F.; Zhou, X.; Lu, D. Tat-interactive Protein-60KDA (TIP60) Regulates the Tumorigenesis of Lung Cancer In Vitro. J. Cancer 2017, 8, 2277–2281. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ji, G.; Han, S.; Shao, Z.; Lu, Z.; Huo, L.; Zhang, J.; Yang, R.; Feng, Q.; Shen, H.; et al. Tip60 Suppresses Cholangiocarcinoma Proliferation and Metastasis via PI3k-AKT. Cell Physiol. Biochem. 2018, 50, 612–628. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Shimada, K.; Skouta, R.; Kaplan, A.; Yang, W.S.; Hayano, M.; Dixon, S.J.; Brown, L.M.; Valenzuela, C.A.; Wolpaw, A.J.; Stockwell, B.R. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 2016, 12, 497–503. [Google Scholar] [CrossRef]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Wang, C.; Yu, L.; Yu, R. Induction of ferroptosis by ATF3 elevation alleviates cisplatin resistance in gastric cancer by restraining Nrf2/Keap1/xCT signaling. Cell Mol. Biol. Lett. 2021, 26, 26. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Wang, X.Z.; He, C.; Wang, L.; Liang, S.P.; Wang, C.C.; Li, C.; Luo, T.F.; Feng, C.S.; Wang, Z.C.; et al. ATF3 contributes to brucine-triggered glioma cell ferroptosis via promotion of hydrogen peroxide and iron. Acta Pharmacol. Sin. 2021, 42, 1690–1702. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yan, J.; Zhao, Q.; Zhang, Y.; Zhang, Y. ATF3 promotes ferroptosis in sorafenib-induced cardiotoxicity by suppressing Slc7a11 expression. Front. Pharmacol. 2022, 13, 904314. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Du, Y.; Gao, Y.; Xu, Z.; Zhao, D.; Yang, M. ATF3 Regulates Osteogenic Function by Mediating Osteoblast Ferroptosis in Type 2 Diabetic Osteoporosis. Dis. Markers 2022, 2022, 9872243. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, D.; Xie, H.; Jia, M.; Sun, X.; Peng, F.; Guo, F.; Tang, D. AUF1 protects against ferroptosis to alleviate sepsis-induced acute lung injury by regulating NRF2 and ATF3. Cell Mol. Life Sci. 2022, 79, 228. [Google Scholar] [CrossRef]
- Wang, Y.; Quan, F.; Cao, Q.; Lin, Y.; Yue, C.; Bi, R.; Cui, X.; Yang, H.; Yang, Y.; Birnbaumer, L.; et al. Quercetin alleviates acute kidney injury by inhibiting ferroptosis. J. Adv. Res. 2021, 28, 231–243. [Google Scholar] [CrossRef]
- Zhu, B.; Ni, Y.; Gong, Y.; Kang, X.; Guo, H.; Liu, X.; Li, J.; Wang, L. Formononetin ameliorates ferroptosis-associated fibrosis in renal tubular epithelial cells and in mice with chronic kidney disease by suppressing the Smad3/ATF3/SLC7A11 signaling. Life Sci. 2023, 315, 121331. [Google Scholar] [CrossRef]
- Dong, H.; Qiang, Z.; Chai, D.; Peng, J.; Xia, Y.; Hu, R.; Jiang, H. Nrf2 inhibits ferroptosis and protects against acute lung injury due to intestinal ischemia reperfusion via regulating SLC7A11 and HO-1. Aging 2020, 12, 12943–12959. [Google Scholar] [CrossRef]
- Ge, M.H.; Tian, H.; Mao, L.; Li, D.Y.; Lin, J.Q.; Hu, H.S.; Huang, S.C.; Zhang, C.J.; Mei, X.F. Zinc attenuates ferroptosis and promotes functional recovery in contusion spinal cord injury by activating Nrf2/GPX4 defense pathway. CNS Neurosci. Ther. 2021, 27, 1023–1040. [Google Scholar] [CrossRef]
- Shao, C.J.; Zhou, H.L.; Gao, X.Z.; Xu, C.F. Downregulation of miR-221-3p promotes the ferroptosis in gastric cancer cells via upregulation of ATF3 to mediate the transcription inhibition of GPX4 and HRD1. Transl. Oncol. 2023, 32, 101649. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, M.; Bi, R.; Su, Y.; Quan, F.; Lin, Y.; Yue, C.; Cui, X.; Zhao, Q.; Liu, S.; et al. ACSL4 deficiency confers protection against ferroptosis-mediated acute kidney injury. Redox Biol. 2022, 51, 102262. [Google Scholar] [CrossRef] [PubMed]
- Miao, Z.; Tian, W.; Ye, Y.; Gu, W.; Bao, Z.; Xu, L.; Sun, G.; Li, C.; Tu, Y.; Chao, H.; et al. Hsp90 induces Acsl4-dependent glioma ferroptosis via dephosphorylating Ser637 at Drp1. Cell Death Dis. 2022, 13, 548. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tomso, D.J.; Chorley, B.N.; Cho, H.Y.; Cheung, V.G.; Kleeberger, S.R.; Bell, D.A. Identification of polymorphic antioxidant response elements in the human genome. Hum. Mol. Genet. 2007, 16, 1188–1200. [Google Scholar] [CrossRef] [PubMed]
- Pajares, M.; Rojo, A.I.; Arias, E.; Diaz-Carretero, A.; Cuervo, A.M.; Cuadrado, A. Transcription factor NFE2L2/NRF2 modulates chaperone-mediated autophagy through the regulation of LAMP2A. Autophagy 2018, 14, 1310–1322. [Google Scholar] [CrossRef] [PubMed]
- Feng, R.; Xiong, Y.; Lei, Y.; Huang, Q.; Liu, H.; Zhao, X.; Chen, Z.; Chen, H.; Liu, X.; Wang, L.; et al. Lysine-specific demethylase 1 aggravated oxidative stress and ferroptosis induced by renal ischemia and reperfusion injury through activation of TLR4/NOX4 pathway in mice. J. Cell Mol. Med. 2022, 26, 4254–4267. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Jia, M.; Xiao, L.; Wang, Z.; Yao, R.; Zhang, Y.; Gao, L. TRIM-containing 44 aggravates cardiac hypertrophy via TLR4/NOX4-induced ferroptosis. J. Mol. Med. 2023, 101, 685–697. [Google Scholar] [CrossRef]
- Liu, H.; Mo, H.; Yang, C.; Mei, X.; Song, X.; Lu, W.; Xiao, H.; Yan, J.; Wang, X.; Yan, J.; et al. A novel function of ATF3 in suppression of ferroptosis in mouse heart suffered ischemia/reperfusion. Free Radic. Biol. Med. 2022, 189, 122–135. [Google Scholar] [CrossRef]
- Park, T.J.; Park, J.H.; Lee, G.S.; Lee, J.Y.; Shin, J.H.; Kim, M.W.; Kim, Y.S.; Kim, J.Y.; Oh, K.J.; Han, B.S.; et al. Quantitative proteomic analyses reveal that GPX4 downregulation during myocardial infarction contributes to ferroptosis in cardiomyocytes. Cell Death Dis. 2019, 10, 835. [Google Scholar] [CrossRef]
- Sood, V.; Sharma, K.B.; Gupta, V.; Saha, D.; Dhapola, P.; Sharma, M.; Sen, U.; Kitajima, S.; Chowdhury, S.; Kalia, M.; et al. ATF3 negatively regulates cellular antiviral signaling and autophagy in the absence of type I interferons. Sci. Rep. 2017, 7, 8789. [Google Scholar] [CrossRef]
- Barnabas, S.; Hai, T.; Andrisani, O.M. The hepatitis B virus X protein enhances the DNA binding potential and transcription efficacy of bZip transcription factors. J. Biol. Chem. 1997, 272, 20684–20690. [Google Scholar] [CrossRef] [PubMed]
- Shiromoto, F.; Aly, H.H.; Kudo, H.; Watashi, K.; Murayama, A.; Watanabe, N.; Zheng, X.; Kato, T.; Chayama, K.; Muramatsu, M.; et al. IL-1beta/ATF3-mediated induction of Ski2 expression enhances hepatitis B virus x mRNA degradation. Biochem. Biophys. Res. Commun. 2018, 503, 1854–1860. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhang, Y.; Ye, S.; Wu, Q.; Lin, Y.; Sheng, K.; Chen, W.; Lin, X.; Lin, X. Chromatin remodelling factor BAF155 protects hepatitis B virus X protein (HBx) from ubiquitin-independent proteasomal degradation. Emerg. Microbes Infect. 2019, 8, 1393–1405. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Seong, B.L. Pro-apoptotic function of HBV X protein is mediated by interaction with c-FLIP and enhancement of death-inducing signal. EMBO J. 2003, 22, 2104–2116. [Google Scholar] [CrossRef] [PubMed]
- Orange, J.S.; Biron, C.A. An absolute and restricted requirement for IL-12 in natural killer cell IFN-gamma production and antiviral defense. Studies of natural killer and T cell responses in contrasting viral infections. J. Immunol. 1996, 156, 1138–1142. [Google Scholar] [CrossRef]
- Rosenberger, C.M.; Clark, A.E.; Treuting, P.M.; Johnson, C.D.; Aderem, A. ATF3 regulates MCMV infection in mice by modulating IFN-gamma expression in natural killer cells. Proc. Natl. Acad. Sci. USA 2008, 105, 2544–2549. [Google Scholar] [CrossRef] [PubMed]
- Pien, G.C.; Satoskar, A.R.; Takeda, K.; Akira, S.; Biron, C.A. Cutting edge: Selective IL-18 requirements for induction of compartmental IFN-gamma responses during viral infection. J. Immunol. 2000, 165, 4787–4791. [Google Scholar] [CrossRef]
- Orange, J.S.; Salazar-Mather, T.P.; Opal, S.M.; Biron, C.A. Mechanisms for virus-induced liver disease: Tumor necrosis factor-mediated pathology independent of natural killer and T cells during murine cytomegalovirus infection. J. Virol. 1997, 71, 9248–9258. [Google Scholar] [CrossRef]
- Orange, J.S.; Wang, B.; Terhorst, C.; Biron, C.A. Requirement for natural killer cell-produced interferon gamma in defense against murine cytomegalovirus infection and enhancement of this defense pathway by interleukin 12 administration. J. Exp. Med. 1995, 182, 1045–1056. [Google Scholar] [CrossRef]
- Tay, C.H.; Welsh, R.M. Distinct organ-dependent mechanisms for the control of murine cytomegalovirus infection by natural killer cells. J. Virol. 1997, 71, 267–275. [Google Scholar] [CrossRef]
- Kang, W.; Mukerjee, R.; Fraser, N.W. Establishment and maintenance of HSV latent infection is mediated through correct splicing of the LAT primary transcript. Virology 2003, 312, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Shu, M.; Du, T.; Zhou, G.; Roizman, B. Role of activating transcription factor 3 in the synthesis of latency-associated transcript and maintenance of herpes simplex virus 1 in latent state in ganglia. Proc. Natl. Acad. Sci. USA 2015, 112, E5420–E5426. [Google Scholar] [CrossRef] [PubMed]
- Kiryu-Seo, S.; Kato, R.; Ogawa, T.; Nakagomi, S.; Nagata, K.; Kiyama, H. Neuronal injury-inducible gene is synergistically regulated by ATF3, c-Jun, and STAT3 through the interaction with Sp1 in damaged neurons. J. Biol. Chem. 2008, 283, 6988–6996. [Google Scholar] [CrossRef] [PubMed]
- Schiffman, M.; Castle, P.E.; Jeronimo, J.; Rodriguez, A.C.; Wacholder, S. Human papillomavirus and cervical cancer. Lancet 2007, 370, 890–907. [Google Scholar] [CrossRef] [PubMed]
- zur Hausen, H. Papillomaviruses causing cancer: Evasion from host-cell control in early events in carcinogenesis. J. Natl. Cancer Inst. 2000, 92, 690–698. [Google Scholar] [CrossRef]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Zapien, D.; Ruiz, F.X.; Poirson, J.; Mitschler, A.; Ramirez, J.; Forster, A.; Cousido-Siah, A.; Masson, M.; Vande Pol, S.; Podjarny, A.; et al. Structure of the E6/E6AP/p53 complex required for HPV-mediated degradation of p53. Nature 2016, 529, 541–545. [Google Scholar] [CrossRef]
- Wang, H.; Mo, P.; Ren, S.; Yan, C. Activating transcription factor 3 activates p53 by preventing E6-associated protein from binding to E6. J. Biol. Chem. 2010, 285, 13201–13210. [Google Scholar] [CrossRef]
- Kooti, A.; Abuei, H.; Farhadi, A.; Behzad-Behbahani, A.; Zarrabi, M. Activating transcription factor 3 mediates apoptotic functions through a p53-independent pathway in human papillomavirus 18 infected HeLa cells. Virus Genes. 2022, 58, 88–97. [Google Scholar] [CrossRef]
- Badu, P.; Pager, C.T. Activation of ATF3 via the Integrated Stress Response Pathway Regulates Innate Immune and Autophagy Processes to Restrict Zika Virus. bioRxiv 2023. [Google Scholar] [CrossRef]
- Hwang, H.Y.; Kim, J.Y.; Lim, J.Y.; Chung, S.K.; Nam, J.H.; Park, S.I. Coxsackievirus B3 modulates cell death by downregulating activating transcription factor 3 in HeLa cells. Virus Res. 2007, 130, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Mishra, R.; Lahon, A.; Banerjea, A.C. Dengue Virus Degrades USP33-ATF3 Axis via Extracellular Vesicles to Activate Human Microglial Cells. J. Immunol. 2020, 205, 1787–1798. [Google Scholar] [CrossRef] [PubMed]
- Henderson, A.; Holloway, A.; Reeves, R.; Tremethick, D.J. Recruitment of SWI/SNF to the human immunodeficiency virus type 1 promoter. Mol. Cell Biol. 2004, 24, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Calton, C.M.; Wade, L.K.; So, M. Upregulation of ATF3 inhibits expression of the pro-inflammatory cytokine IL-6 during Neisseria gonorrhoeae infection. Cell Microbiol. 2013, 15, 1837–1850. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Luo, Y.F.; Tian, M.; Xiao, Y.L.; Cai, H.R.; Li, H. Activating transcription factor 3 involved in Pseudomonas aeruginosa PAO1-induced macrophage senescence. Mol. Immunol. 2021, 133, 122–127. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, X.; Qian, L.; Guo, L.; Liao, J.; Wu, X. Activating transcription factor 3 protects mice against pseudomonas aeruginosa-induced acute lung injury by interacting with lipopolysaccharide binding protein. Mol. Immunol. 2017, 90, 27–32. [Google Scholar] [CrossRef]
- Zhang, B.; Li, H.; Zhang, J.; Hang, Y.; Xu, Y. Activating transcription factor 3 protects alveolar epithelial type II cells from Mycobacterium tuberculosis infection-induced inflammation. Tuberculosis 2022, 135, 102227. [Google Scholar] [CrossRef]
- Wang, J.; Cheng, W.; Wang, Z.; Xin, L.; Zhang, W. ATF3 inhibits the inflammation induced by Mycoplasma pneumonia in vitro and in vivo. Pediatr. Pulmonol. 2017, 52, 1163–1170. [Google Scholar] [CrossRef]
- Kwon, O.; Soung, N.K.; Thimmegowda, N.R.; Jeong, S.J.; Jang, J.H.; Moon, D.O.; Chung, J.K.; Lee, K.S.; Kwon, Y.T.; Erikson, R.L.; et al. Patulin induces colorectal cancer cells apoptosis through EGR-1 dependent ATF3 up-regulation. Cell Signal 2012, 24, 943–950. [Google Scholar] [CrossRef]
- Yuan, L.; Mu, P.; Huang, B.; Li, H.; Mu, H.; Deng, Y. EGR1 is essential for deoxynivalenol-induced G2/M cell cycle arrest in HepG2 cells via the ATF3DeltaZip2a/2b-EGR1-p21 pathway. Toxicol. Lett. 2018, 299, 95–103. [Google Scholar] [CrossRef]
- Abhishek, K.; Das, S.; Kumar, A.; Kumar, A.; Kumar, V.; Saini, S.; Mandal, A.; Verma, S.; Kumar, M.; Das, P. Leishmania donovani induced Unfolded Protein Response delays host cell apoptosis in PERK dependent manner. PLoS Negl. Trop. Dis. 2018, 12, e0006646. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Roy, S.; Dutta, A.; Jana, K.; Ukil, A. Leishmania donovani Targets Host Transcription Factor NRF2 To Activate Antioxidant Enzyme HO-1 and Transcriptional Repressor ATF3 for Establishing Infection. Infect. Immun. 2021, 89, e0076420. [Google Scholar] [CrossRef] [PubMed]
- Schindler, C.; Levy, D.E.; Decker, T. JAK-STAT signaling: From interferons to cytokines. J. Biol. Chem. 2007, 282, 20059–20063. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Bhattacharyya, S.; Nain, M.; Kaur, M.; Sood, V.; Gupta, V.; Khasa, R.; Abdin, M.Z.; Vrati, S.; Kalia, M. Japanese encephalitis virus replication is negatively regulated by autophagy and occurs on LC3-I- and EDEM1-containing membranes. Autophagy 2014, 10, 1637–1651. [Google Scholar] [CrossRef] [PubMed]
- McFadden, M.J.; Gokhale, N.S.; Horner, S.M. Protect this house: Cytosolic sensing of viruses. Curr. Opin. Virol. 2017, 22, 36–43. [Google Scholar] [CrossRef]
- Gratton, R.; Agrelli, A.; Tricarico, P.M.; Brandao, L.; Crovella, S. Autophagy in Zika Virus Infection: A Possible Therapeutic Target to Counteract Viral Replication. Int. J. Mol. Sci. 2019, 20, 1048. [Google Scholar] [CrossRef]
- Echavarria-Consuegra, L.; Smit, J.M.; Reggiori, F. Role of autophagy during the replication and pathogenesis of common mosquito-borne flavi- and alphaviruses. Open Biol. 2019, 9, 190009. [Google Scholar] [CrossRef]
- Lin, H.; Li, H.F.; Chen, H.H.; Lai, P.F.; Juan, S.H.; Chen, J.J.; Cheng, C.F. Activating transcription factor 3 protects against pressure-overload heart failure via the autophagy molecule Beclin-1 pathway. Mol. Pharmacol. 2014, 85, 682–691. [Google Scholar] [CrossRef]
- Carthy, C.M.; Granville, D.J.; Watson, K.A.; Anderson, D.R.; Wilson, J.E.; Yang, D.; Hunt, D.W.; McManus, B.M. Caspase activation and specific cleavage of substrates after coxsackievirus B3-induced cytopathic effect in HeLa cells. J. Virol. 1998, 72, 7669–7675. [Google Scholar] [CrossRef]
- Rothman, A.L. Immunity to dengue virus: A tale of original antigenic sin and tropical cytokine storms. Nat. Rev. Immunol. 2011, 11, 532–543. [Google Scholar] [CrossRef]
- Li, J.; Yan, X.; Li, B.; Huang, L.; Wang, X.; He, B.; Xie, H.; Wu, Q.; Chen, L. Identification and validation of ferroptosis-related genes in patients infected with dengue virus: Implication in the pathogenesis of DENV. Virus Genes. 2023, 59, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Tripathy, M.K.; Abbas, W.; Herbein, G. Epigenetic regulation of HIV-1 transcription. Epigenomics 2011, 3, 487–502. [Google Scholar] [CrossRef] [PubMed]
- Verdin, E.; Paras, P., Jr.; Van Lint, C. Chromatin disruption in the promoter of human immunodeficiency virus type 1 during transcriptional activation. EMBO J. 1993, 12, 3249–3259. [Google Scholar] [CrossRef] [PubMed]
- el Kharroubi, A.; Martin, M.A. cis-acting sequences located downstream of the human immunodeficiency virus type 1 promoter affect its chromatin structure and transcriptional activity. Mol. Cell Biol. 1996, 16, 2958–2966. [Google Scholar] [CrossRef] [PubMed]
- Rabbi, M.F.; Saifuddin, M.; Gu, D.S.; Kagnoff, M.F.; Roebuck, K.A. U5 region of the human immunodeficiency virus type 1 long terminal repeat contains TRE-like cAMP-responsive elements that bind both AP-1 and CREB/ATF proteins. Virology 1997, 233, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Van Lint, C.; Amella, C.A.; Emiliani, S.; John, M.; Jie, T.; Verdin, E. Transcription factor binding sites downstream of the human immunodeficiency virus type 1 transcription start site are important for virus infectivity. J. Virol. 1997, 71, 6113–6127. [Google Scholar] [CrossRef]
- Henderson, A.; Bunce, M.; Siddon, N.; Reeves, R.; Tremethick, D.J. High-mobility-group protein I can modulate binding of transcription factors to the U5 region of the human immunodeficiency virus type 1 proviral promoter. J. Virol. 2000, 74, 10523–10534. [Google Scholar] [CrossRef]
- Wallace, V.C.; Blackbeard, J.; Pheby, T.; Segerdahl, A.R.; Davies, M.; Hasnie, F.; Hall, S.; McMahon, S.B.; Rice, A.S. Pharmacological, behavioural and mechanistic analysis of HIV-1 gp120 induced painful neuropathy. Pain 2007, 133, 47–63. [Google Scholar] [CrossRef]
- Bekeredjian-Ding, I.; Stein, C.; Uebele, J. The Innate Immune Response Against Staphylococcus aureus. Curr. Top. Microbiol. Immunol. 2017, 409, 385–418. [Google Scholar] [CrossRef]
- De Luca, A.; Pariano, M.; Cellini, B.; Costantini, C.; Villella, V.R.; Jose, S.S.; Palmieri, M.; Borghi, M.; Galosi, C.; Paolicelli, G.; et al. The IL-17F/IL-17RC Axis Promotes Respiratory Allergy in the Proximal Airways. Cell Rep. 2017, 20, 1667–1680. [Google Scholar] [CrossRef]
- Choi, S.M.; McAleer, J.P.; Zheng, M.; Pociask, D.A.; Kaplan, M.H.; Qin, S.; Reinhart, T.A.; Kolls, J.K. Innate Stat3-mediated induction of the antimicrobial protein Reg3gamma is required for host defense against MRSA pneumonia. J. Exp. Med. 2013, 210, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Robinson, K.M.; Choi, S.M.; McHugh, K.J.; Mandalapu, S.; Enelow, R.I.; Kolls, J.K.; Alcorn, J.F. Influenza A exacerbates Staphylococcus aureus pneumonia by attenuating IL-1beta production in mice. J. Immunol. 2013, 191, 5153–5159. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, N.D.; Ritchie, R.; Bayes, H.K.; Mitchell, T.J.; Evans, T.J. IL-17 can be protective or deleterious in murine pneumococcal pneumonia. PLoS Pathog. 2018, 14, e1007099. [Google Scholar] [CrossRef] [PubMed]
- Wonnenberg, B.; Jungnickel, C.; Honecker, A.; Wolf, L.; Voss, M.; Bischoff, M.; Tschernig, T.; Herr, C.; Bals, R.; Beisswenger, C. IL-17A attracts inflammatory cells in murine lung infection with P. aeruginosa. Innate Immun. 2016, 22, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.J.; Gross, J.; Bogaert, D.; Finn, A.; Bagrade, L.; Zhang, Q.; Kolls, J.K.; Srivastava, A.; Lundgren, A.; Forte, S.; et al. Interleukin-17A mediates acquired immunity to pneumococcal colonization. PLoS Pathog. 2008, 4, e1000159. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Ng, V.H.; Maeda, S.; Rest, R.F.; Karin, M. Anthrolysin O and other gram-positive cytolysins are toll-like receptor 4 agonists. J. Exp. Med. 2004, 200, 1647–1655. [Google Scholar] [CrossRef] [PubMed]
- Annane, D.; Bellissant, E.; Cavaillon, J.M. Septic shock. Lancet 2005, 365, 63–78. [Google Scholar] [CrossRef]
- Sawa, T. The molecular mechanism of acute lung injury caused by Pseudomonas aeruginosa: From bacterial pathogenesis to host response. J. Intensive Care 2014, 2, 10. [Google Scholar] [CrossRef]
- Deshpande, R.; Zou, C. Pseudomonas Aeruginosa Induced Cell Death in Acute Lung Injury and Acute Respiratory Distress Syndrome. Int. J. Mol. Sci. 2020, 21, 5356. [Google Scholar] [CrossRef]
- Peyron, P.; Vaubourgeix, J.; Poquet, Y.; Levillain, F.; Botanch, C.; Bardou, F.; Daffe, M.; Emile, J.F.; Marchou, B.; Cardona, P.J.; et al. Foamy macrophages from tuberculous patients’ granulomas constitute a nutrient-rich reservoir for M. tuberculosis persistence. PLoS Pathog. 2008, 4, e1000204. [Google Scholar] [CrossRef]
- Russell, D.G.; Cardona, P.J.; Kim, M.J.; Allain, S.; Altare, F. Foamy macrophages and the progression of the human tuberculosis granuloma. Nat. Immunol. 2009, 10, 943–948. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.P.; Cao, C.; Wu, Y.F.; Li, M.; Lai, T.W.; Zhu, C.; Wang, Y.; Ying, S.M.; Chen, Z.H.; Shen, H.H.; et al. Activating transcription factor 3 represses cigarette smoke-induced IL6 and IL8 expression via suppressing NF-kappaB activation. Toxicol. Lett. 2017, 270, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yang, K.; Zhou, L.; Minhaowu; Wu, Y.; Zhu, M.; Lai, X.; Chen, T.; Feng, L.; Li, M.; et al. MicroRNA-155 promotes autophagy to eliminate intracellular mycobacteria by targeting Rheb. PLoS Pathog. 2013, 9, e1003697. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Hu, J.; Zhang, W.; Dong, Y.; Xiong, S.; Dong, C. RELL1 inhibits autophagy pathway and regulates Mycobacterium tuberculosis survival in macrophages. Tuberculosis 2020, 120, 101900. [Google Scholar] [CrossRef] [PubMed]
- Glaser, N.; Stopper, H. Patulin: Mechanism of genotoxicity. Food Chem. Toxicol. 2012, 50, 1796–1801. [Google Scholar] [CrossRef] [PubMed]
- Heussner, A.H.; Dietrich, D.R.; O’Brien, E. In vitro investigation of individual and combined cytotoxic effects of ochratoxin A and other selected mycotoxins on renal cells. Toxicol Vitr. 2006, 20, 332–341. [Google Scholar] [CrossRef]
- Wu, T.S.; Liao, Y.C.; Yu, F.Y.; Chang, C.H.; Liu, B.H. Mechanism of patulin-induced apoptosis in human leukemia cells (HL-60). Toxicol. Lett. 2008, 183, 105–111. [Google Scholar] [CrossRef]
- Wu, Q.; Dohnal, V.; Kuca, K.; Yuan, Z. Trichothecenes: Structure-toxic activity relationships. Curr. Drug Metab. 2013, 14, 641–660. [Google Scholar] [CrossRef]
- Naik, S.; Mohammed, A. Coexpression network analysis of human candida infection reveals key modules and hub genes responsible for host-pathogen interactions. Front. Genet. 2022, 13, 917636. [Google Scholar] [CrossRef]
- Zhu, G.D.; Xie, L.M.; Su, J.W.; Cao, X.J.; Yin, X.; Li, Y.P.; Gao, Y.M.; Guo, X.G. Identification of differentially expressed genes and signaling pathways with Candida infection by bioinformatics analysis. Eur. J. Med. Res. 2022, 27, 43. [Google Scholar] [CrossRef]
- Olivier, M.; Gregory, D.J.; Forget, G. Subversion mechanisms by which Leishmania parasites can escape the host immune response: A signaling point of view. Clin. Microbiol. Rev. 2005, 18, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Pham, N.K.; Mouriz, J.; Kima, P.E. Leishmania pifanoi amastigotes avoid macrophage production of superoxide by inducing heme degradation. Infect. Immun. 2005, 73, 8322–8333. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Kuang, X.; Zhang, Y.; Ye, Y.; Li, J.; Liang, L.; Xie, Z.; Weng, L.; Guo, J.; Li, H.; et al. ADORA1 Inhibition Promotes Tumor Immune Evasion by Regulating the ATF3-PD-L1 Axis. Cancer Cell 2020, 37, 324–339.e328. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; McBrearty, N.; Chen, J.; Tomar, V.S.; Zhang, H.; De Rosa, G.; Tan, A.; Weljie, A.M.; Beiting, D.P.; Miao, Z.; et al. ATF3 and CH25H regulate effector trogocytosis and anti-tumor activities of endogenous and immunotherapeutic cytotoxic T lymphocytes. Cell Metab. 2022, 34, 1342–1358.e7. [Google Scholar] [CrossRef] [PubMed]
- Di Marcantonio, D.; Martinez, E.; Kanefsky, J.S.; Huhn, J.M.; Gabbasov, R.; Gupta, A.; Krais, J.J.; Peri, S.; Tan, Y.; Skorski, T.; et al. ATF3 coordinates serine and nucleotide metabolism to drive cell cycle progression in acute myeloid leukemia. Mol. Cell 2021, 81, 2752–2764.e6. [Google Scholar] [CrossRef]
- Huo, S.; Wang, Q.; Shi, W.; Peng, L.; Jiang, Y.; Zhu, M.; Guo, J.; Peng, D.; Wang, M.; Men, L.; et al. ATF3/SPI1/SLC31A1 Signaling Promotes Cuproptosis Induced by Advanced Glycosylation End Products in Diabetic Myocardial Injury. Int. J. Mol. Sci. 2023, 24, 1667. [Google Scholar] [CrossRef]
- Ku, H.C.; Cheng, C.F. Master Regulator Activating Transcription Factor 3 (ATF3) in Metabolic Homeostasis and Cancer. Front. Endocrinol. 2020, 11, 556. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.; Li, Z.; Lan, S.; Hao, H.; Baz, A.A.; Yan, X.; Gao, P.; Chen, S.; Chu, Y. The Dual Roles of Activating Transcription Factor 3 (ATF3) in Inflammation, Apoptosis, Ferroptosis, and Pathogen Infection Responses. Int. J. Mol. Sci. 2024, 25, 824. https://doi.org/10.3390/ijms25020824
Liu S, Li Z, Lan S, Hao H, Baz AA, Yan X, Gao P, Chen S, Chu Y. The Dual Roles of Activating Transcription Factor 3 (ATF3) in Inflammation, Apoptosis, Ferroptosis, and Pathogen Infection Responses. International Journal of Molecular Sciences. 2024; 25(2):824. https://doi.org/10.3390/ijms25020824
Chicago/Turabian StyleLiu, Shuang, Zhangcheng Li, Shimei Lan, Huafang Hao, Ahmed Adel Baz, Xinmin Yan, Pengcheng Gao, Shengli Chen, and Yuefeng Chu. 2024. "The Dual Roles of Activating Transcription Factor 3 (ATF3) in Inflammation, Apoptosis, Ferroptosis, and Pathogen Infection Responses" International Journal of Molecular Sciences 25, no. 2: 824. https://doi.org/10.3390/ijms25020824
APA StyleLiu, S., Li, Z., Lan, S., Hao, H., Baz, A. A., Yan, X., Gao, P., Chen, S., & Chu, Y. (2024). The Dual Roles of Activating Transcription Factor 3 (ATF3) in Inflammation, Apoptosis, Ferroptosis, and Pathogen Infection Responses. International Journal of Molecular Sciences, 25(2), 824. https://doi.org/10.3390/ijms25020824