
CRISPR-Cas12a for Highly Efficient and Marker-Free Targeted Integration in Human Pluripotent Stem Cells

 , , , , ,
, , , , ,  ,
,  , ,
, , 
Abstract
1. Introduction
2. Results
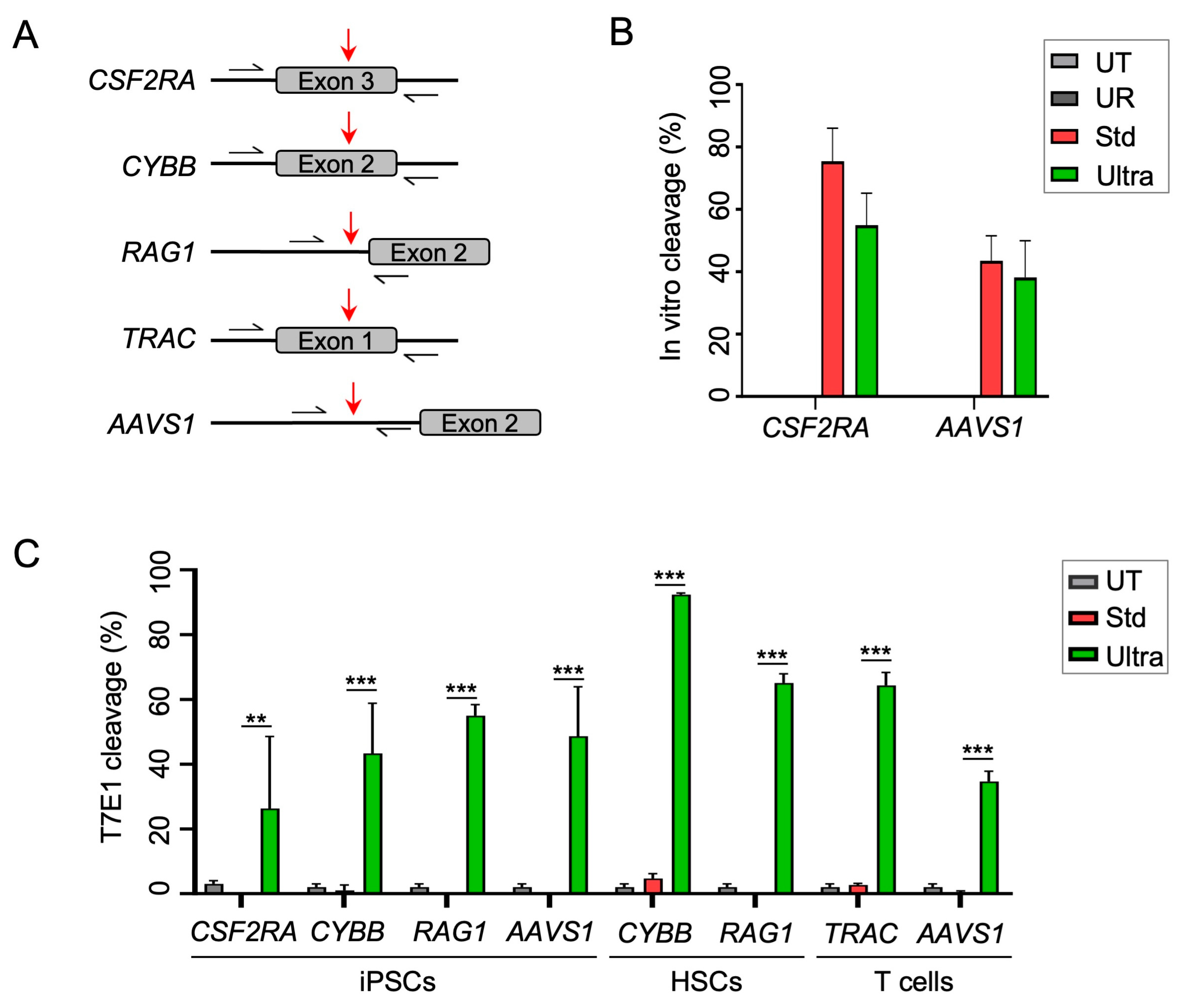
2.1. Cas12a Ultra Efficiently Induces Gene Disruption in Primary Human Cells and iPSCs
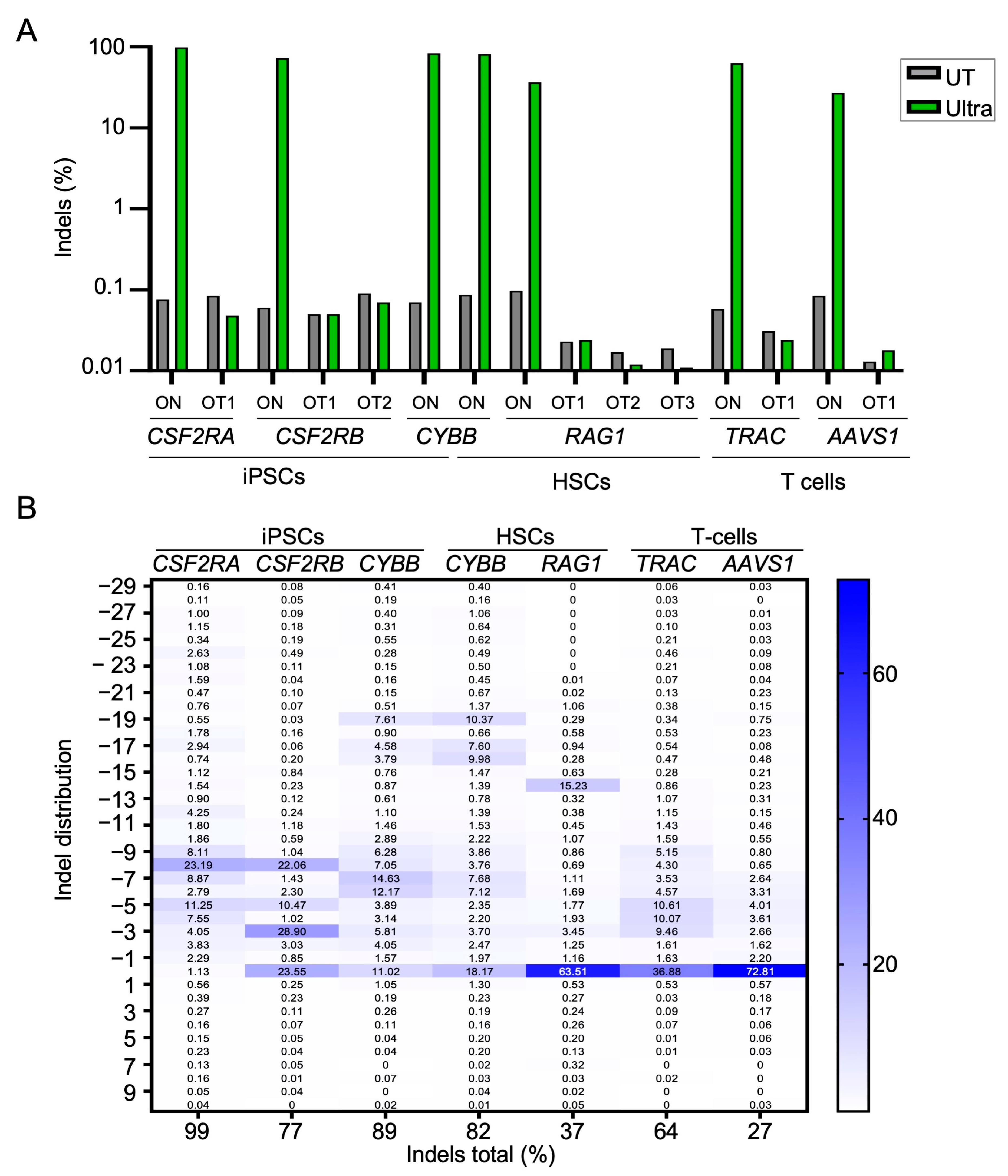
2.2. On- and Off-Target Activity of CRISPR-Cas12a Ultra
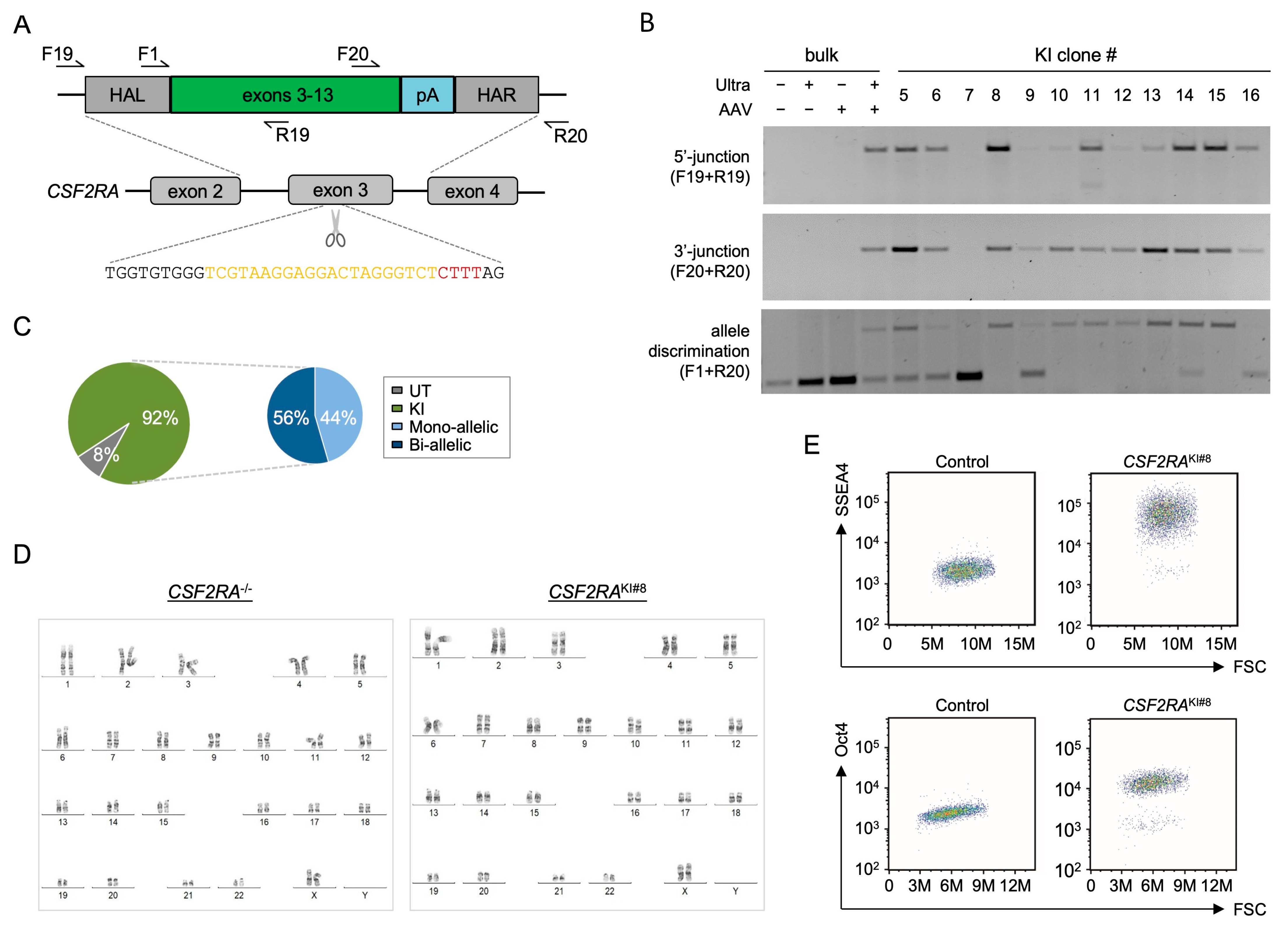
2.3. CRISPR-Cas12a Ultra Mediates Effective Targeted Genomic Integration in iPSCs
3. Discussion
4. Materials and Methods
4.1. HSPC Isolation and Culture
4.2. iPSC Generation and Culture
4.3. AAV Vector Production
4.4. Gene Editing of U2OS Cells
4.5. Gene Editing of Primary Human Cells
4.6. Genotyping
4.7. In Vitro Cleavage
4.8. Cell Analysis
4.9. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef] [PubMed]
- Cornu, T.I.; Mussolino, C.; Cathomen, T. Refining strategies to translate genome editing to the clinic. Nat. Med. 2017, 23, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yang, Y.; Hong, W.; Huang, M.; Wu, M.; Zhao, X. Applications of genome editing technology in the targeted therapy of human diseases: Mechanisms, advances and prospects. Signal Transduct. Target. Ther. 2020, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef]
- Anders, C.; Niewoehner, O.; Duerst, A.; Jinek, M. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 2014, 513, 569–573. [Google Scholar] [CrossRef]
- Kim, B.; Kim, J.; Lee, S. Unleashing the power of undifferentiated induced pluripotent stem cell bioprinting: Current progress and future prospects. Int. J. Stem Cells 2024, in press. [CrossRef]
- Zhang, L.; Zuris, J.A.; Viswanathan, R.; Edelstein, J.N.; Turk, R.; Thommandru, B.; Rube, H.T.; Glenn, S.E.; Collingwood, M.A.; Bode, N.M.; et al. AsCas12a ultra nuclease facilitates the rapid generation of therapeutic cell medicines. Nat. Commun. 2021, 12, 3908. [Google Scholar] [CrossRef]
- Paquet, D.; Kwart, D.; Chen, A.; Sproul, A.; Jacob, S.; Teo, S.; Olsen, K.M.; Gregg, A.; Noggle, S.; Tessier-Lavigne, M. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature 2016, 533, 125–129. [Google Scholar] [CrossRef]
- Alzubi, J.; Pallant, C.; Mussolino, C.; Howe, S.J.; Thrasher, A.J.; Cathomen, T. Targeted genome editing restores T cell differentiation in a humanized X-SCID pluripotent stem cell disease model. Sci. Rep. 2017, 7, 12475. [Google Scholar] [CrossRef] [PubMed]
- Dreyer, A.-K.; Hoffmann, D.; Lachmann, N.; Ackermann, M.; Steinemann, D.; Timm, B.; Siler, U.; Reichenbach, J.; Grez, M.; Moritz, T.; et al. TALEN-mediated functional correction of X-linked chronic granulomatous disease in patient-derived induced pluripotent stem cells. Biomaterials 2015, 69, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Merkert, S.; Haase, A.; Dahlmann, J.; Göhring, G.; Waqas, F.H.; Pessler, F.; Martin, U.; Olmer, R. Generation of two human NRF2 knockout iPSC clones using CRISPR/Cas9 editing. Stem Cell Res. 2023, 69, 103090. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.M.; Ikeda, K.; Cromer, M.K.; Uchida, N.; Nishimura, T.; Romano, R.; Tong, A.J.; Lemgart, V.T.; Camarena, J.; Pavel-Dinu, M.; et al. Highly Efficient and Marker-free Genome Editing of Human Pluripotent Stem Cells by CRISPR-Cas9 RNP and AAV6 Donor-Mediated Homologous Recombination. Cell Stem Cell 2019, 24, 821–828.e825. [Google Scholar] [CrossRef] [PubMed]
- Chupradit, K.; Thongsin, N.; Tayapiwatana, C.; Wattanapanitch, M. A precise gene delivery approach for human induced pluripotent stem cells using Cas9 RNP complex and recombinant AAV6 donor vectors. PLoS ONE 2022, 17, e0270963. [Google Scholar] [CrossRef]
- Müller, M.; Lee, C.M.; Gasiunas, G.; Davis, T.H.; Cradick, T.J.; Siksnys, V.; Bao, G.; Cathomen, T.; Mussolino, C. Streptococcus thermophilus CRISPR-Cas9 Systems Enable Specific Editing of the Human Genome. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 636–644. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.M.; Cradick, T.J.; Bao, G. The Neisseria meningitidis CRISPR-Cas9 System Enables Specific Genome Editing in Mammalian Cells. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 645–654. [Google Scholar] [CrossRef]
- Yang, Z.; Fu, Y.; Zhao, J.; Zhang, F.; Li, S.; Zhao, M.; Wen, W.; Zhang, L.; Cheng, T.; Zhang, J.; et al. Superior Fidelity and Distinct Editing Outcomes of SaCas9 Compared to SpCas9 in Genome Editing. Genom. Proteom. Bioinform. 2022, in press. [Google Scholar] [CrossRef]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; Regev, A.; et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef]
- Broeders, M.; Herrero-Hernandez, P.; Ernst, M.P.T.; van der Ploeg, A.T.; Pijnappel, W.W.M.P. Sharpening the Molecular Scissors: Advances in Gene-Editing Technology. iScience 2020, 23, 100789. [Google Scholar] [CrossRef]
- Kim, D.; Kim, J.; Hur, J.K.; Been, K.W.; Yoon, S.H.; Kim, J.S. Genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells. Nat. Biotechnol. 2016, 34, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Swarts, D.C.; Jinek, M. Cas9 versus Cas12a/Cpf1: Structure–function comparisons and implications for genome editing. WIREs RNA 2018, 9, e1481. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Tsai, S.Q.; Prew, M.S.; Nguyen, N.T.; Welch, M.M.; Lopez, J.M.; McCaw, Z.R.; Aryee, M.J.; Joung, J.K. Genome-wide specificities of CRISPR-Cas Cpf1 nucleases in human cells. Nat. Biotechnol. 2016, 34, 869–874. [Google Scholar] [CrossRef]
- Gao, L.; Cox, D.B.T.; Yan, W.X.; Manteiga, J.C.; Schneider, M.W.; Yamano, T.; Nishimasu, H.; Nureki, O.; Crosetto, N.; Zhang, F. Engineered Cpf1 variants with altered PAM specificities. Nat. Biotechnol. 2017, 35, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Bin Moon, S.; Lee, J.M.; Kang, J.G.; Lee, N.-E.; Ha, D.-I.; Kim, D.Y.; Kim, S.H.; Yoo, K.; Kim, D.; Ko, J.-H.; et al. Highly efficient genome editing by CRISPR-Cpf1 using CRISPR RNA with a uridinylate-rich 3′-overhang. Nat. Commun. 2018, 9, 3651. [Google Scholar] [CrossRef] [PubMed]
- Safari, F.; Zare, K.; Negahdaripour, M.; Barekati-Mowahed, M.; Ghasemi, Y. CRISPR Cpf1 proteins: Structure, function and implications for genome editing. Cell Biosci. 2019, 9, 36. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Schiel, J.A.; Maksimova, E.; Strezoska, Ž.; Zhao, G.; Anderson, E.M.; Wu, Y.; Warren, J.; Bartels, A.; van Brabant Smith, A.; et al. ErCas12a CRISPR-MAD7 for Model Generation in Human Cells, Mice, and Rats. CRISPR J. 2020, 3, 97–108. [Google Scholar] [CrossRef]
- Pietralla, J.; Capdeville, N.; Schindele, P.; Puchta, H. Optimizing ErCas12a for efficient gene editing in Arabidopsis thaliana. Plant Biotechnol. J. 2023, in press. [Google Scholar] [CrossRef]
- Carey, B.; Trapnell, B.C. The molecular basis of pulmonary alveolar proteinosis. Clin. Immunol. 2010, 135, 223–235. [Google Scholar] [CrossRef]
- Lachmann, N.; Happle, C.; Ackermann, M.; Luttge, D.; Wetzke, M.; Merkert, S.; Hetzel, M.; Kensah, G.; Jara-Avaca, M.; Mucci, A.; et al. Gene correction of human induced pluripotent stem cells repairs the cellular phenotype in pulmonary alveolar proteinosis. Am. J. Respir. Crit. Care Med. 2014, 189, 167–182. [Google Scholar] [CrossRef]
- Yang, L.; Grishin, D.; Wang, G.; Aach, J.; Zhang, C.-Z.; Chari, R.; Homsy, J.; Cai, X.; Zhao, Y.; Fan, J.-B.; et al. Targeted and genome-wide sequencing reveal single nucleotide variations impacting specificity of Cas9 in human stem cells. Nat. Commun. 2014, 5, 5507. [Google Scholar] [CrossRef]
- Assou, S.; Bouckenheimer, J.; De Vos, J. Concise Review: Assessing the Genome Integrity of Human Induced Pluripotent Stem Cells: What Quality Control Metrics? Stem Cells 2018, 36, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Hansen, N.F.; Zhao, L.; Du, Y.; Zou, C.; Donovan, F.X.; Chou, B.K.; Zhou, G.; Li, S.; Dowey, S.N.; et al. Low incidence of DNA sequence variation in human induced pluripotent stem cells generated by nonintegrating plasmid expression. Cell Stem Cell 2012, 10, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Zhang, Y. Genetic and epigenetic variations in iPSCs: Potential causes and implications for application. Cell Stem Cell 2013, 13, 149–159. [Google Scholar] [CrossRef]
- Chou, B.K.; Gu, H.; Gao, Y.; Dowey, S.N.; Wang, Y.; Shi, J.; Li, Y.; Ye, Z.; Cheng, T.; Cheng, L. A facile method to establish human induced pluripotent stem cells from adult blood cells under feeder-free and xeno-free culture conditions: A clinically compliant approach. Stem Cells Transl. Med. 2015, 4, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Alzubi, J.; Lock, D.; Rhiel, M.; Schmitz, S.; Wild, S.; Mussolino, C.; Hildenbeutel, M.; Brandes, C.; Rositzka, J.; Lennartz, S.; et al. Automated generation of gene-edited CAR T cells at clinical scale. Mol. Ther.—Methods Clin. Dev. 2021, 20, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, C.L.; Pavel-Dinu, M.; Choi, U.; Brault, J.; Liu, T.; Koontz, S.; Li, L.; Theobald, N.; Lee, J.; Bello, E.A.; et al. Correction of X-CGD patient HSPCs by targeted CYBB cDNA insertion using CRISPR/Cas9 with 53BP1 inhibition for enhanced homology-directed repair. Gene Ther. 2021, 28, 373–390. [Google Scholar] [CrossRef]
- Mohr, M.; Damas, N.; Gudmand-Hoyer, J.; Zeeberg, K.; Jedrzejczyk, D.; Vlassis, A.; Morera-Gomez, M.; Pereira-Schoning, S.; Pus, U.; Oliver-Almirall, A.; et al. The CRISPR-Cas12a Platform for Accurate Genome Editing, Gene Disruption, and Efficient Transgene Integration in Human Immune Cells. ACS Synth. Biol. 2023, 12, 375–389. [Google Scholar] [CrossRef]
- Teng, F.; Li, J.; Cui, T.; Xu, K.; Guo, L.; Gao, Q.; Feng, G.; Chen, C.; Han, D.; Zhou, Q.; et al. Enhanced mammalian genome editing by new Cas12a orthologs with optimized crRNA scaffolds. Genome Biol. 2019, 20, 15. [Google Scholar] [CrossRef]
- Murugan, K.; Seetharam, A.S.; Severin, A.J.; Sashital, D.G. CRISPR-Cas12a has widespread off-target and dsDNA-nicking effects. J. Biol. Chem. 2020, 295, 5538–5553. [Google Scholar] [CrossRef]
- Chen, J.S.; Ma, E.; Harrington, L.B.; Da Costa, M.; Tian, X.; Palefsky, J.M.; Doudna, J.A. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 2018, 360, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Turchiano, G.; Andrieux, G.; Klermund, J.; Blattner, G.; Pennucci, V.; El Gaz, M.; Monaco, G.; Poddar, S.; Mussolino, C.; Cornu, T.I.; et al. Quantitative evaluation of chromosomal rearrangements in gene-edited human stem cells by CAST-Seq. Cell Stem Cell 2021, 28, 1136–1147.e1135. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Zhang, L.; Li, Z.; Xu, C.; Du, X.; Wu, S. Cas12a mediates efficient and precise endogenous gene tagging via MITI: Microhomology-dependent targeted integrations. Cell. Mol. Life Sci. 2020, 77, 3875–3884. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Edwards, H.; Xu, P. CRISPR-Cas12a/Cpf1-assisted precise, efficient and multiplexed genome-editing in Yarrowia lipolytica. Metab. Eng. Commun. 2020, 10, e00112. [Google Scholar] [CrossRef] [PubMed]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.C.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Iancu, O.; Allen, D.; Knop, O.; Zehavi, Y.; Breier, D.; Arbiv, A.; Lev, A.; Lee, Y.N.; Beider, K.; Nagler, A.; et al. Multiplex HDR for disease and correction modeling of SCID by CRISPR genome editing in human HSPCs. Mol. Ther. Nucleic Acids 2023, 31, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Lattanzi, A.; Camarena, J.; Lahiri, P.; Segal, H.; Srifa, W.; Vakulskas, C.A.; Frock, R.L.; Kenrick, J.; Lee, C.; Talbott, N.; et al. Development of β-globin gene correction in human hematopoietic stem cells as a potential durable treatment for sickle cell disease. Sci. Transl. Med. 2021, 13, eabf2444. [Google Scholar] [CrossRef]
- Mosti, L.; Langner, L.M.; Chmielewski, K.O.; Arbuthnot, P.; Alzubi, J.; Cathomen, T. Targeted multi-epitope switching enables straightforward positive/negative selection of CAR T cells. Gene Ther. 2021, 28, 602–612. [Google Scholar] [CrossRef]
- Schiroli, G.; Conti, A.; Ferrari, S.; Della Volpe, L.; Jacob, A.; Albano, L.; Beretta, S.; Calabria, A.; Vavassori, V.; Gasparini, P.; et al. Precise Gene Editing Preserves Hematopoietic Stem Cell Function following Transient p53-Mediated DNA Damage Response. Cell Stem Cell 2019, 24, 551–565.e558. [Google Scholar] [CrossRef]
- Romito, M.; Juillerat, A.; Kok, Y.L.; Hildenbeutel, M.; Rhiel, M.; Andrieux, G.; Geiger, J.; Rudolph, C.; Mussolino, C.; Duclert, A.; et al. Preclinical Evaluation of a Novel TALEN Targeting CCR5 Confirms Efficacy and Safety in Conferring Resistance to HIV-1 Infection. Biotechnol. J. 2021, 16, 2000023. [Google Scholar] [CrossRef]
- Craig-Mueller, N.; Hammad, R.; Elling, R.; Alzubi, J.; Timm, B.; Kolter, J.; Knelangen, N.; Bednarski, C.; Gläser, B.; Ammann, S.; et al. Modeling MyD88 Deficiency In Vitro Provides New Insights in Its Function. Front. Immunol. 2020, 11, 608802. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.E.; Liu, J.M.; Xiao, X.; Young, N.S.; Nienhuis, A.W.; Samulski, R.J. Regulated high level expression of a human gamma-globin gene introduced into erythroid cells by an adeno-associated virus vector. Proc. Natl. Acad. Sci. USA 1992, 89, 7257–7261. [Google Scholar] [CrossRef] [PubMed]
- Mussolino, C.; Alzubi, J.; Fine, E.J.; Morbitzer, R.; Cradick, T.J.; Lahaye, T.; Bao, G.; Cathomen, T. TALENs facilitate targeted genome editing in human cells with high specificity and low cytotoxicity. Nucleic Acids Res. 2014, 42, 6762–6773. [Google Scholar] [CrossRef] [PubMed]
- Clement, K.; Rees, H.; Canver, M.C.; Gehrke, J.M.; Farouni, R.; Hsu, J.Y.; Cole, M.A.; Liu, D.R.; Joung, J.K.; Bauer, D.E.; et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 2019, 37, 224–226. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.; Park, J.; Kim, J.S. Cas-OFFinder: A fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 2014, 30, 1473–1475. [Google Scholar] [CrossRef]
- Kim, J.M.; Kim, D.; Kim, S.; Kim, J.-S. Genotyping with CRISPR-Cas-derived RNA-guided endonucleases. Nat. Commun. 2014, 5, 3157. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| crRNA Name | Target Sequence (5′-3′) |
|---|---|
| CSF2RA | TCTGGGATCAGGAGGAATGCT |
| CSF2RB | ACGGGGCCGCCGTGCTCAGCT |
| CYBB | TCTGGTATTACCGGGTTTATG |
| RAG1 | CACATTGTATTAGCCTCATTG |
| TRAC | CACATGCAAAGTCAGATTTGT |
| AAVS1 | TGTCACCAATCCTGTCCCTAG |
| Primer Use | Target | Sequence (5′-3′) | Serial No. |
|---|---|---|---|
| T7E1 assay | CSF2RA-F1 | tgtttccccaaatcacctcc | 3982 |
| CSF2RA-R1 | tcactacctggatggtcgttg | 3983 | |
| CYBB-F2 | agtggcctgctatcagctac | 4536 | |
| CYBB-R2 | atgtgtcactcctggatggattg | 4310 | |
| RAG1-F3 | gttgcaggtttagagttccgtg | 3363 | |
| RAG1-R3 | ggatctcacccggaacagct | 3364 | |
| TRAC-F4 | taaagcatgagaccgtgact | 3437 | |
| TRAC-R4 | tagacatcattgaccagagc | 3438 | |
| AAVS1-F5 | ccttcttgtaggcctgcatcatcacc | 1567 | |
| AAVS1-R5 | ggatcctctctggctccatcgtaag | 1568 | |
| 5′ and 3′ junction PCR | AAVS1-F6 | ccagctcccatagctcagtctg | 1207 |
| AAVS1-R6 | gacgtgaagaatgtgcgaga | 1405 | |
| AAVS1-F7 | ctacggcaagctgaccctgaa | 154 | |
| AAVS1-R7 | gggctcagtctgaagagcagag | 1208 | |
| CSF2RA-F19 | tcacgaggtcaggagatggag | 5393 | |
| CSF2RA-R19 | aaggttcatagttcgactgtcg | 5082 | |
| CSF2RA-F20 | acgatggcaacctcggttc | 5394 | |
| CSF2RA-R20 | aggaacgaaggaacgaaggaac | 5395 | |
| on-target analysis | CSF2RA-F8 | tgccaggaatgtcctgggag | 5042 |
| CSF2RA-R8 | gggttggctggtgcttattgg | 5043 | |
| CYBB-F9 | ctgactccagtcttgtgtggaatc | 5044 | |
| CYBB-R9 | gagggagtgaggctaatggtac | 5045 | |
| RAG1-F10 | caagtagtgaataattagtttctttgggtttgcagc | 5046 | |
| RAG1-R10 | attcatctttgcctccccaagggt | 5047 | |
| TRAC-F11 | acaagtctgtctgcctattcaccg | 5048 | |
| TRAC-R11 | tcattgaccagagctctgggc | 5049 | |
| AAVS1-F12 | ggcccctatgtccacttcag | 5050 | |
| AAVS1-R12 | ctggcaaggagagagatggc | 5051 | |
| CSF2RB-F21 | tgatgaatcacacggtgggc | 6301 | |
| CSF2RB-R21 | gagagcaaggccaagaggag | 6302 | |
| off-target analysis | CSF2RA-OT1-F13 | cacagaagtcacatgctcctatattaaattctacg | 5069 |
| CSF2RA-OT1-R13 | gagggaacataagcccatttgtaaacagaaaatatga | 5070 | |
| RAG1-OT1-F14 | tgcatacatgagcatccctaca | 5071 | |
| RAG1-OT1-R14 | ctgtgacactacaggaaaagga | 5072 | |
| RAG1-OT2-F15 | ggatttggctgctgcttctttctg | 5073 | |
| RAG1-OT2-R15 | ctctgccaactaacttatgtcatgctatcaac | 5074 | |
| RAG1-OT3-F16 | aagcaaggtccttgtgttgggg | 5075 | |
| RAG1-OT3-R16 | cttctccttccagaaggggtc | 5076 | |
| TRAC-OT1-F17 | ccatggcagtatagtggcacc | 5077 | |
| TRAC-OT1-R17 | ctgcacctggcgtgtttttatcc | 5078 | |
| AAVS1-OT1-F18 | tacagacacagcaggaggcc | 5079 | |
| AAVS1-OT1-R18 | ctctcttgattctaccaccacattctactc | 5080 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hammad, R.; Alzubi, J.; Rhiel, M.; Chmielewski, K.O.; Mosti, L.; Rositzka, J.; Heugel, M.; Lawrenz, J.; Pennucci, V.; Gläser, B.; et al. CRISPR-Cas12a for Highly Efficient and Marker-Free Targeted Integration in Human Pluripotent Stem Cells. Int. J. Mol. Sci. 2024, 25, 985. https://doi.org/10.3390/ijms25020985
Hammad R, Alzubi J, Rhiel M, Chmielewski KO, Mosti L, Rositzka J, Heugel M, Lawrenz J, Pennucci V, Gläser B, et al. CRISPR-Cas12a for Highly Efficient and Marker-Free Targeted Integration in Human Pluripotent Stem Cells. International Journal of Molecular Sciences. 2024; 25(2):985. https://doi.org/10.3390/ijms25020985
Chicago/Turabian StyleHammad, Ruba, Jamal Alzubi, Manuel Rhiel, Kay O. Chmielewski, Laura Mosti, Julia Rositzka, Marcel Heugel, Jan Lawrenz, Valentina Pennucci, Birgitta Gläser, and et al. 2024. "CRISPR-Cas12a for Highly Efficient and Marker-Free Targeted Integration in Human Pluripotent Stem Cells" International Journal of Molecular Sciences 25, no. 2: 985. https://doi.org/10.3390/ijms25020985
APA StyleHammad, R., Alzubi, J., Rhiel, M., Chmielewski, K. O., Mosti, L., Rositzka, J., Heugel, M., Lawrenz, J., Pennucci, V., Gläser, B., Fischer, J., Schambach, A., Moritz, T., Lachmann, N., Cornu, T. I., Mussolino, C., Schäfer, R., & Cathomen, T. (2024). CRISPR-Cas12a for Highly Efficient and Marker-Free Targeted Integration in Human Pluripotent Stem Cells. International Journal of Molecular Sciences, 25(2), 985. https://doi.org/10.3390/ijms25020985