
Persistent Activation of the P2X7 Receptor Underlies Chronic Inflammation and Carcinogenic Changes in the Intestine

 , and
, and
Abstract
1. Introduction
2. Rise of Inflammatory Bowel Disease and Colorectal Cancer
3. Purinergic Signaling in Inflammatory Bowel Diseases and Colorectal Cancer
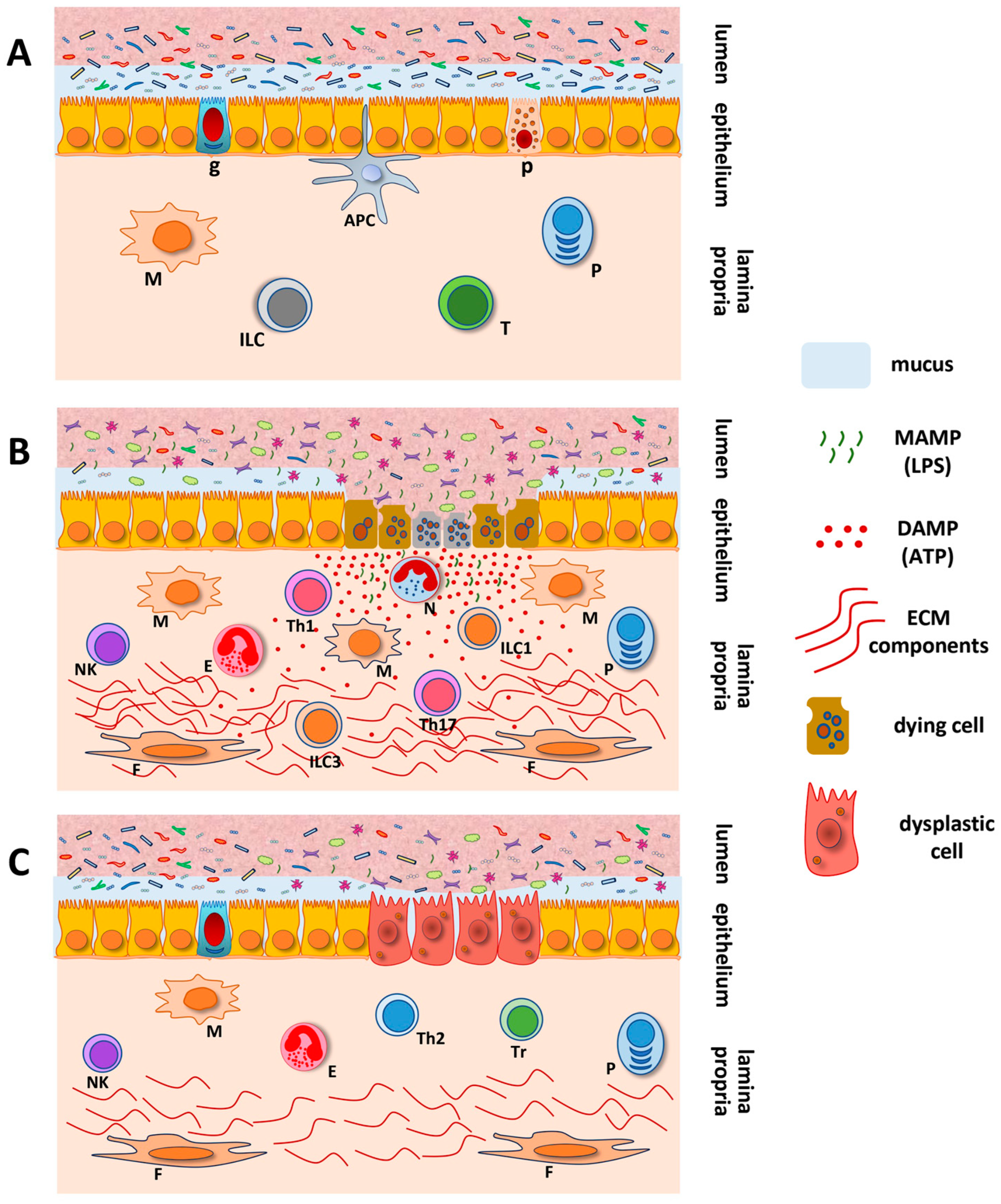
4. Abnormal Innate Immunity in Inflammatory Bowel Diseases and Colorectal Cancer
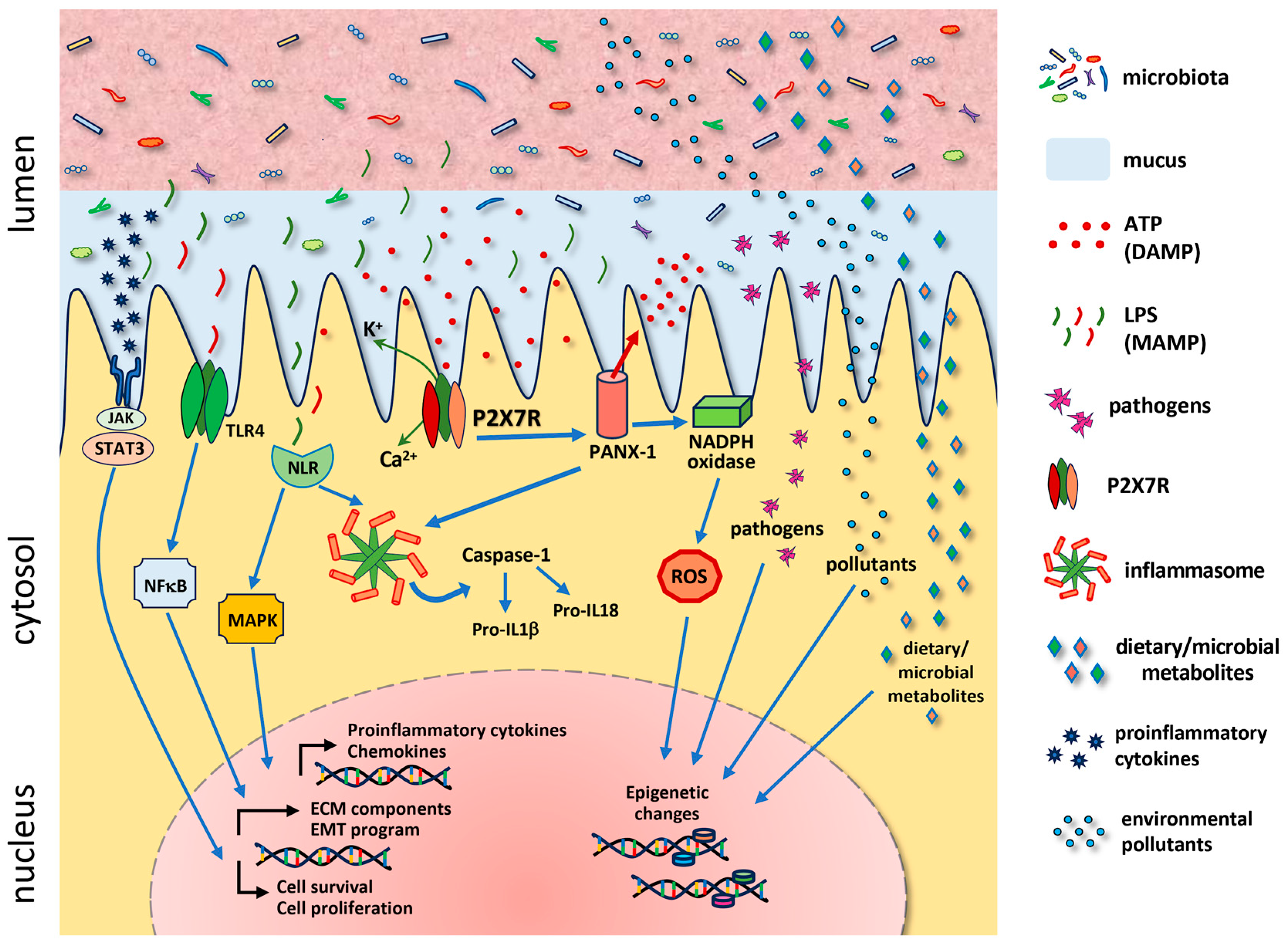
5. Inflammasome and DAMPS in Inflammatory Bowel Diseases and Colorectal Cancer
6. P2X7R and Cell Fate in the Gastrointestinal Tract
7. P2X7R and the Intestinal Microbiota
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boyapati, R.K.; Rossi, A.G.; Satsangi, J.; Ho, G.T. Gut mucosal DAMPs in IBD: From mechanisms to therapeutic implications. Mucosal Immunol. 2016, 9, 567–582. [Google Scholar] [CrossRef] [PubMed]
- Miyake, Y.; Yamasaki, S. Sensing necrotic cells. Adv. Exp. Med. Biol. 2012, 738, 144–152. [Google Scholar] [CrossRef]
- Fucikova, J.; Moserova, I.; Urbanova, L.; Bezu, L.; Kepp, O.; Cremer, I.; Salek, C.; Strnad, P.; Kroemer, G.; Galluzzi, L.; et al. Prognostic and Predictive Value of DAMPs and DAMP-Associated Processes in Cancer. Front. Immunol. 2015, 6, 402. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Yang, K.; Yu, G.; Hao, W.; Zhu, X.; Ge, A.; Chen, J.; Sun, L. Advances in research on immunocyte iron metabolism, ferroptosis, and their regulatory roles in autoimmune and autoinflammatory diseases. Cell Death Dis. 2024, 15, 481. [Google Scholar] [CrossRef] [PubMed]
- Leinardi, R.; Longo Sanchez-Calero, C.; Huaux, F. Think Beyond Particle Cytotoxicity: When Self-Cellular Components Released After Immunogenic Cell Death Explain Chronic Disease Development. Front. Toxicol. 2022, 4, 887228. [Google Scholar] [CrossRef] [PubMed]
- de Carvalho Braga, G.; Francisco, G.R.; Bagatini, M.D. Current treatment of Psoriasis triggered by Cytokine Storm and future immunomodulation strategies. J. Mol. Med. 2024, 102, 1187–1198. [Google Scholar] [CrossRef] [PubMed]
- Milicevic, K.D.; Bataveljic, D.B.; Bogdanovic Pristov, J.J.; Andjus, P.R.; Nikolic, L.M. Astroglial Cell-to-Cell Interaction with Autoreactive Immune Cells in Experimental Autoimmune Encephalomyelitis Involves P2X7 Receptor, beta(3)-Integrin, and Connexin-43. Cells 2023, 12, 1786. [Google Scholar] [CrossRef]
- Li, M.; Yang, C.; Wang, Y.; Song, W.; Jia, L.; Peng, X.; Zhao, R. The Expression of P2X7 Receptor on Th1, Th17, and Regulatory T Cells in Patients with Systemic Lupus Erythematosus or Rheumatoid Arthritis and Its Correlations with Active Disease. J. Immunol. 2020, 205, 1752–1762. [Google Scholar] [CrossRef]
- Liu, X.; Li, Y.; Huang, L.; Kuang, Y.; Wu, X.; Ma, X.; Zhao, B.; Lan, J. Unlocking the therapeutic potential of P2X7 receptor: A comprehensive review of its role in neurodegenerative disorders. Front. Pharmacol. 2024, 15, 1450704. [Google Scholar] [CrossRef]
- North, R.A. P2X receptors. Philos. Trans. R. Soc. B. Biol. Sci. 2016, 371, 20150427. [Google Scholar] [CrossRef]
- Burnstock, G.; Knight, G.E. The potential of P2X7 receptors as a therapeutic target, including inflammation and tumour progression. Purinergic Signal. 2018, 14, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Ma, W.; Zhang, Y.; Zhang, L. P2X7 Receptor (P2X7R) of Microglia Mediates Neuroinflammation by Regulating (NOD)-Like Receptor Protein 3 (NLRP3) Inflammasome-Dependent Inflammation After Spinal Cord Injury. Med. Sci. Monit. 2020, 26, e925491. [Google Scholar] [CrossRef] [PubMed]
- Di Virgilio, F.; Jiang, L.H.; Roger, S.; Falzoni, S.; Sarti, A.C.; Vultaggio-Poma, V.; Chiozzi, P.; Adinolfi, E. Structure, function and techniques of investigation of the P2X7 receptor (P2X7R) in mammalian cells. Methods Enzym. 2019, 629, 115–150. [Google Scholar] [CrossRef]
- Coutinho-Silva, R.; Correa, G.; Sater, A.A.; Ojcius, D.M. The P2X(7) receptor and intracellular pathogens: A continuing struggle. Purinergic Signal. 2009, 5, 197–204. [Google Scholar] [CrossRef]
- Cooper, G.S.; Stroehla, B.C. The epidemiology of autoimmune diseases. Autoimmun. Rev. 2003, 2, 119–125. [Google Scholar] [CrossRef]
- Zuckerman, M.K.; Harper, K.N.; Barrett, R.; Armelagos, G.J. The evolution of disease: Anthropological perspectives on epidemiologic transitions. Glob. Health Action 2014, 7, 23303. [Google Scholar] [CrossRef]
- Arnold, M.; Abnet, C.C.; Neale, R.E.; Vignat, J.; Giovannucci, E.L.; McGlynn, K.A.; Bray, F. Global Burden of 5 Major Types of Gastrointestinal Cancer. Gastroenterology 2020, 159, 335–349.e15. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Allender, S.; Foster, C.; Hutchinson, L.; Arambepola, C. Quantification of urbanization in relation to chronic diseases in developing countries: A systematic review. J. Urban Health 2008, 85, 938–951. [Google Scholar] [CrossRef]
- Costello, E.K.; Stagaman, K.; Dethlefsen, L.; Bohannan, B.J.; Relman, D.A. The application of ecological theory toward an understanding of the human microbiome. Science 2012, 336, 1255–1262. [Google Scholar] [CrossRef]
- Pasolli, E.; Asnicar, F.; Manara, S.; Zolfo, M.; Karcher, N.; Armanini, F.; Beghini, F.; Manghi, P.; Tett, A.; Ghensi, P.; et al. Extensive Unexplored Human Microbiome Diversity Revealed by Over 150,000 Genomes from Metagenomes Spanning Age, Geography, and Lifestyle. Cell 2019, 176, 649–662.e20. [Google Scholar] [CrossRef] [PubMed]
- Conrad, N.; Misra, S.; Verbakel, J.Y.; Verbeke, G.; Molenberghs, G.; Taylor, P.N.; Mason, J.; Sattar, N.; McMurray, J.J.V.; McInnes, I.B.; et al. Incidence, prevalence, and co-occurrence of autoimmune disorders over time and by age, sex, and socioeconomic status: A population-based cohort study of 22 million individuals in the UK. Lancet 2023, 401, 1878–1890. [Google Scholar] [CrossRef] [PubMed]
- Adolph, T.E.; Tilg, H. Western diets and chronic diseases. Nat. Med. 2024, 30, 2133–2147. [Google Scholar] [CrossRef] [PubMed]
- de Souza, H.S.P.; Fiocchi, C.; Iliopoulos, D. The IBD interactome: An integrated view of aetiology, pathogenesis and therapy. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 739–749. [Google Scholar] [CrossRef]
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.Y.; Chan, F.K.L.; et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet 2017, 390, 2769–2778. [Google Scholar] [CrossRef]
- Ananthakrishnan, A.N.; Bernstein, C.N.; Iliopoulos, D.; Macpherson, A.; Neurath, M.F.; Ali, R.A.R.; Vavricka, S.R.; Fiocchi, C. Environmental triggers in IBD: A review of progress and evidence. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 39–49. [Google Scholar] [CrossRef]
- Mishima, Y.; Sartor, R.B. Manipulating resident microbiota to enhance regulatory immune function to treat inflammatory bowel diseases. J. Gastroenterol. 2020, 55, 4–14. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Morgan, E.; Arnold, M.; Gini, A.; Lorenzoni, V.; Cabasag, C.J.; Laversanne, M.; Vignat, J.; Ferlay, J.; Murphy, N.; Bray, F. Global burden of colorectal cancer in 2020 and 2040: Incidence and mortality estimates from GLOBOCAN. Gut 2023, 72, 338–344. [Google Scholar] [CrossRef]
- Murphy, N.; Moreno, V.; Hughes, D.J.; Vodicka, L.; Vodicka, P.; Aglago, E.K.; Gunter, M.J.; Jenab, M. Lifestyle and dietary environmental factors in colorectal cancer susceptibility. Mol. Asp. Med. 2019, 69, 2–9. [Google Scholar] [CrossRef]
- Karahalios, A.; English, D.R.; Simpson, J.A. Weight change and risk of colorectal cancer: A systematic review and meta-analysis. Am. J. Epidemiol. 2015, 181, 832–845. [Google Scholar] [CrossRef] [PubMed]
- Keum, N.; Giovannucci, E. Global burden of colorectal cancer: Emerging trends, risk factors and prevention strategies. Nat Rev Gastroenterol. Hepatol. 2019, 16, 713–732. [Google Scholar] [CrossRef] [PubMed]
- Vuerich, M.; Mukherjee, S.; Robson, S.C.; Longhi, M.S. Control of Gut Inflammation by Modulation of Purinergic Signaling. Front. Immunol. 2020, 11, 1882. [Google Scholar] [CrossRef] [PubMed]
- Cheng, N.; Zhang, L.; Liu, L. Understanding the Role of Purinergic P2X7 Receptors in the Gastrointestinal System: A Systematic Review. Front. Pharmacol. 2021, 12, 786579. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G.; Jacobson, K.A.; Christofi, F.L. Purinergic drug targets for gastrointestinal disorders. Curr. Opin. Pharmacol. 2017, 37, 131–141. [Google Scholar] [CrossRef]
- Souza, C.O.; Santoro, G.F.; Figliuolo, V.R.; Nanini, H.F.; de Souza, H.S.; Castelo-Branco, M.T.; Abalo, A.A.; Paiva, M.M.; Coutinho, C.M.; Coutinho-Silva, R. Extracellular ATP induces cell death in human intestinal epithelial cells. Biochim. Biophys. Acta 2012, 1820, 1867–1878. [Google Scholar] [CrossRef]
- Zhang, R.; Kang, R.; Klionsky, D.J.; Tang, D. Ion Channels and Transporters in Autophagy. Autophagy 2022, 18, 4–23. [Google Scholar] [CrossRef]
- Kong, H.; Zhao, H.; Chen, T.; Song, Y.; Cui, Y. Targeted P2X7/NLRP3 signaling pathway against inflammation, apoptosis, and pyroptosis of retinal endothelial cells in diabetic retinopathy. Cell Death Dis. 2022, 13, 336. [Google Scholar] [CrossRef]
- Figliuolo, V.R.; Savio, L.E.B.; Safya, H.; Nanini, H.; Bernardazzi, C.; Abalo, A.; de Souza, H.S.P.; Kanellopoulos, J.; Bobe, P.; Coutinho, C.; et al. P2X7 receptor promotes intestinal inflammation in chemically induced colitis and triggers death of mucosal regulatory T cells. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1183–1194. [Google Scholar] [CrossRef]
- Marques, C.C.; Castelo-Branco, M.T.; Pacheco, R.G.; Buongusto, F.; do Rosario, A., Jr.; Schanaider, A.; Coutinho-Silva, R.; de Souza, H.S. Prophylactic systemic P2X7 receptor blockade prevents experimental colitis. Biochim. Biophys. Acta 2014, 1842, 65–78. [Google Scholar] [CrossRef]
- Neves, A.R.; Castelo-Branco, M.T.; Figliuolo, V.R.; Bernardazzi, C.; Buongusto, F.; Yoshimoto, A.; Nanini, H.F.; Coutinho, C.M.; Carneiro, A.J.; Coutinho-Silva, R.; et al. Overexpression of ATP-activated P2X7 receptors in the intestinal mucosa is implicated in the pathogenesis of Crohn’s disease. Inflamm. Bowel Dis. 2014, 20, 444–457. [Google Scholar] [CrossRef] [PubMed]
- Yaeger, R.; Shah, M.A.; Miller, V.A.; Kelsen, J.R.; Wang, K.; Heins, Z.J.; Ross, J.S.; He, Y.; Sanford, E.; Yantiss, R.K.; et al. Genomic Alterations Observed in Colitis-Associated Cancers Are Distinct From Those Found in Sporadic Colorectal Cancers and Vary by Type of Inflammatory Bowel Disease. Gastroenterology 2016, 151, 278–287.e6. [Google Scholar] [CrossRef] [PubMed]
- Bernardazzi, C.; Castelo-Branco, M.T.L.; Pego, B.; Ribeiro, B.E.; Rosas, S.L.B.; Santana, P.T.; Machado, J.C.; Leal, C.; Thompson, F.; Coutinho-Silva, R.; et al. The P2X7 Receptor Promotes Colorectal Inflammation and Tumorigenesis by Modulating Gut Microbiota and the Inflammasome. Int. J. Mol. Sci. 2022, 23, 4616. [Google Scholar] [CrossRef] [PubMed]
- Qian, F.; Xiao, J.; Hu, B.; Sun, N.; Yin, W.; Zhu, J. High expression of P2X7R is an independent postoperative indicator of poor prognosis in colorectal cancer. Hum. Pathol. 2017, 64, 61–68. [Google Scholar] [CrossRef]
- Zhang, Y.; Ding, J.; Wang, L. The role of P2X7 receptor in prognosis and metastasis of colorectal cancer. Adv. Med. Sci. 2019, 64, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Calik, I.; Calik, M.; Turken, G.; Ozercan, I.H. A promising independent prognostic biomarker in colorectal cancer: P2X7 receptor. Int. J. Clin. Exp. Pathol. 2020, 13, 107–121. [Google Scholar]
- Adinolfi, E.; Capece, M.; Franceschini, A.; Falzoni, S.; Giuliani, A.L.; Rotondo, A.; Sarti, A.C.; Bonora, M.; Syberg, S.; Corigliano, D.; et al. Accelerated tumor progression in mice lacking the ATP receptor P2X7. Cancer Res. 2015, 75, 635–644. [Google Scholar] [CrossRef]
- Hofman, P.; Cherfils-Vicini, J.; Bazin, M.; Ilie, M.; Juhel, T.; Hebuterne, X.; Gilson, E.; Schmid-Alliana, A.; Boyer, O.; Adriouch, S.; et al. Genetic and pharmacological inactivation of the purinergic P2RX7 receptor dampens inflammation but increases tumor incidence in a mouse model of colitis-associated cancer. Cancer Res. 2015, 75, 835–845. [Google Scholar] [CrossRef]
- Sainz, R.M.; Rodriguez-Quintero, J.H.; Maldifassi, M.C.; Stiles, B.M.; Wennerberg, E. Tumour immune escape via P2X7 receptor signalling. Front. Immunol. 2023, 14, 1287310. [Google Scholar] [CrossRef]
- Yang, C.; Shi, S.; Su, Y.; Tong, J.S.; Li, L. P2X7R promotes angiogenesis and tumour-associated macrophage recruitment by regulating the NF-kappaB signalling pathway in colorectal cancer cells. J. Cell. Mol. Med. 2020, 24, 10830–10841. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, F.; Wang, L.; Lou, Y. A438079 affects colorectal cancer cell proliferation, migration, apoptosis, and pyroptosis by inhibiting the P2X7 receptor. Biochem. Biophys. Res. Commun. 2021, 558, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.J.; Luo, C.; Huang, C.; Pu, F.Q.; Zhu, J.F.; Zhu, Z.M. PI3K/Akt/GSK-3beta signal pathway is involved in P2X7 receptor-induced proliferation and EMT of colorectal cancer cells. Eur. J. Pharmacol. 2021, 899, 174041. [Google Scholar] [CrossRef] [PubMed]
- Monteleone, G.; Pallone, F.; Stolfi, C. The dual role of inflammation in colon carcinogenesis. Int. J. Mol. Sci. 2012, 13, 11071–11084. [Google Scholar] [CrossRef]
- Huang, K.C.; Chiang, S.F.; Lin, P.C.; Hong, W.Z.; Yang, P.C.; Chang, H.P.; Peng, S.L.; Chen, T.W.; Ke, T.W.; Liang, J.A.; et al. TNFalpha modulates PANX1 activation to promote ATP release and enhance P2RX7-mediated antitumor immune responses after chemotherapy in colorectal cancer. Cell Death Dis. 2024, 15, 24. [Google Scholar] [CrossRef]
- Qiu, Y.; Li, W.H.; Zhang, H.Q.; Liu, Y.; Tian, X.X.; Fang, W.G. P2X7 mediates ATP-driven invasiveness in prostate cancer cells. PLoS ONE 2014, 9, e114371. [Google Scholar] [CrossRef]
- Pegoraro, A.; De Marchi, E.; Ferracin, M.; Orioli, E.; Zanoni, M.; Bassi, C.; Tesei, A.; Capece, M.; Dika, E.; Negrini, M.; et al. P2X7 promotes metastatic spreading and triggers release of miRNA-containing exosomes and microvesicles from melanoma cells. Cell Death Dis. 2021, 12, 1088. [Google Scholar] [CrossRef]
- Wang, Z.; Zhu, S.; Tan, S.; Zeng, Y.; Zeng, H. The P2 purinoceptors in prostate cancer. Purinergic Signal. 2023, 19, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Tafani, M.; Schito, L.; Pellegrini, L.; Villanova, L.; Marfe, G.; Anwar, T.; Rosa, R.; Indelicato, M.; Fini, M.; Pucci, B.; et al. Hypoxia-increased RAGE and P2X7R expression regulates tumor cell invasion through phosphorylation of Erk1/2 and Akt and nuclear translocation of NF-kappaB. Carcinogenesis 2011, 32, 1167–1175. [Google Scholar] [CrossRef]
- Demaria, O.; Cornen, S.; Daeron, M.; Morel, Y.; Medzhitov, R.; Vivier, E. Harnessing innate immunity in cancer therapy. Nature 2019, 574, 45–56. [Google Scholar] [CrossRef]
- Maiorino, L.; Dassler-Plenker, J.; Sun, L.; Egeblad, M. Innate Immunity and Cancer Pathophysiology. Annu. Rev. Pathol. 2022, 17, 425–457. [Google Scholar] [CrossRef]
- Morris, R.M.; Mortimer, T.O.; O’Neill, K.L. Cytokines: Can Cancer Get the Message? Cancers 2022, 14, 2178. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Bardelcikova, A.; Soltys, J.; Mojzis, J. Oxidative Stress, Inflammation and Colorectal Cancer: An Overview. Antioxidants 2023, 12, 901. [Google Scholar] [CrossRef] [PubMed]
- Itzkowitz, S.H.; Yio, X. Inflammation and cancer IV. Colorectal cancer in inflammatory bowel disease: The role of inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G7–G17. [Google Scholar] [CrossRef]
- Basak, D.; Uddin, M.N.; Hancock, J. The Role of Oxidative Stress and Its Counteractive Utility in Colorectal Cancer (CRC). Cancers 2020, 12, 3336. [Google Scholar] [CrossRef]
- Boakye, D.; Jansen, L.; Schottker, B.; Jansen, E.; Schneider, M.; Halama, N.; Gao, X.; Chang-Claude, J.; Hoffmeister, M.; Brenner, H. Blood markers of oxidative stress are strongly associated with poorer prognosis in colorectal cancer patients. Int. J. Cancer 2020, 147, 2373–2386. [Google Scholar] [CrossRef] [PubMed]
- Azer, S.A. Overview of molecular pathways in inflammatory bowel disease associated with colorectal cancer development. Eur. J. Gastroenterol. Hepatol. 2013, 25, 271–281. [Google Scholar] [CrossRef]
- Adinolfi, E.; Callegari, M.G.; Ferrari, D.; Bolognesi, C.; Minelli, M.; Wieckowski, M.R.; Pinton, P.; Rizzuto, R.; Di Virgilio, F. Basal activation of the P2X7 ATP receptor elevates mitochondrial calcium and potential, increases cellular ATP levels, and promotes serum-independent growth. Mol. Biol. Cell 2005, 16, 3260–3272. [Google Scholar] [CrossRef]
- Amoroso, F.; Falzoni, S.; Adinolfi, E.; Ferrari, D.; Di Virgilio, F. The P2X7 receptor is a key modulator of aerobic glycolysis. Cell Death Dis. 2012, 3, e370. [Google Scholar] [CrossRef]
- Hill, L.M.; Gavala, M.L.; Lenertz, L.Y.; Bertics, P.J. Extracellular ATP may contribute to tissue repair by rapidly stimulating purinergic receptor X7-dependent vascular endothelial growth factor release from primary human monocytes. J. Immunol. 2010, 185, 3028–3034. [Google Scholar] [CrossRef]
- Bingle, L.; Brown, N.J.; Lewis, C.E. The role of tumour-associated macrophages in tumour progression: Implications for new anticancer therapies. J. Pathol. 2002, 196, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Bergamin, L.S.; Braganhol, E.; Figueiro, F.; Casali, E.A.; Zanin, R.F.; Sevigny, J.; Battastini, A.M. Involvement of purinergic system in the release of cytokines by macrophages exposed to glioma-conditioned medium. J. Cell Biochem. 2015, 116, 721–729. [Google Scholar] [CrossRef]
- Qin, J.; Zhang, X.; Tan, B.; Zhang, S.; Yin, C.; Xue, Q.; Zhang, Z.; Ren, H.; Chen, J.; Liu, M.; et al. Blocking P2X7-Mediated Macrophage Polarization Overcomes Treatment Resistance in Lung Cancer. Cancer Immunol. Res. 2020, 8, 1426–1439. [Google Scholar] [CrossRef]
- Ren, W.; Rubini, P.; Tang, Y.; Engel, T.; Illes, P. Inherent P2X7 Receptors Regulate Macrophage Functions during Inflammatory Diseases. Int. J. Mol. Sci. 2021, 23, 232. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, X. Research progress of P2X7 receptor in inflammatory bowel disease. Scand. J. Gastroenterol. 2019, 54, 521–527. [Google Scholar] [CrossRef]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef]
- Oberg, H.H.; Wesch, D.; Kalyan, S.; Kabelitz, D. Regulatory Interactions between Neutrophils, Tumor Cells and T Cells. Front. Immunol. 2019, 10, 1690. [Google Scholar] [CrossRef]
- Asadirad, A.; Baghaei, K.; Hashemi, S.M.; Dehnavi, S.; Ghanbarian, H.; Mortaz, E.; Anissian, A.; Asadzadeh Aghdaei, H.; Amani, D. Dendritic cell immunotherapy with miR-155 enriched tumor-derived exosome suppressed cancer growth and induced antitumor immune responses in murine model of colorectal cancer induced by CT26 cell line. Int. Immunopharmacol. 2022, 104, 108493. [Google Scholar] [CrossRef]
- Zitvogel, L.; Kepp, O.; Galluzzi, L.; Kroemer, G. Inflammasomes in carcinogenesis and anticancer immune responses. Nat. Immunol. 2012, 13, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat. Med. 2009, 15, 1170–1178. [Google Scholar] [CrossRef] [PubMed]
- Aymeric, L.; Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Martins, I.; Kroemer, G.; Smyth, M.J.; Zitvogel, L. Tumor cell death and ATP release prime dendritic cells and efficient anticancer immunity. Cancer Res. 2010, 70, 855–858. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Mao, Z.; Wang, W.; Ma, J.; Tian, J.; Wang, S.; Yin, K. Netrin-1 Promotes the Immunosuppressive Activity of MDSCs in Colorectal Cancer. Cancer Immunol. Res. 2023, 11, 600–613. [Google Scholar] [CrossRef]
- Idzko, M.; Ferrari, D.; Eltzschig, H.K. Nucleotide signalling during inflammation. Nature 2014, 509, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Principi, E.; Raffaghello, L. The role of the P2X7 receptor in myeloid-derived suppressor cells and immunosuppression. Curr. Opin. Pharmacol. 2019, 47, 82–89. [Google Scholar] [CrossRef]
- Grassi, F.; De Ponte Conti, B. The P2X7 Receptor in Tumor Immunity. Front. Cell Dev. Biol. 2021, 9, 694831. [Google Scholar] [CrossRef]
- Grassi, F.; Salina, G. The P2X7 Receptor in Autoimmunity. Int. J. Mol. Sci. 2023, 24, 14116. [Google Scholar] [CrossRef]
- Romagnani, A.; Rottoli, E.; Mazza, E.M.C.; Rezzonico-Jost, T.; De Ponte Conti, B.; Proietti, M.; Perotti, M.; Civanelli, E.; Perruzza, L.; Catapano, A.L.; et al. P2X7 Receptor Activity Limits Accumulation of T Cells within Tumors. Cancer Res. 2020, 80, 3906–3919. [Google Scholar] [CrossRef]
- Schenk, U.; Frascoli, M.; Proietti, M.; Geffers, R.; Traggiai, E.; Buer, J.; Ricordi, C.; Westendorf, A.M.; Grassi, F. ATP inhibits the generation and function of regulatory T cells through the activation of purinergic P2X receptors. Sci. Signal. 2011, 4, ra12. [Google Scholar] [CrossRef]
- Yip, L.; Woehrle, T.; Corriden, R.; Hirsh, M.; Chen, Y.; Inoue, Y.; Ferrari, V.; Insel, P.A.; Junger, W.G. Autocrine regulation of T-cell activation by ATP release and P2X7 receptors. FASEB J. 2009, 23, 1685–1693. [Google Scholar] [CrossRef] [PubMed]
- Elinav, E.; Henao-Mejia, J.; Flavell, R.A. Integrative inflammasome activity in the regulation of intestinal mucosal immune responses. Mucosal Immunol. 2013, 6, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Mu, C.; Yang, Y.; Zhu, W. Crosstalk between The Immune Receptors and Gut Microbiota. Curr. Protein Pept. Sci. 2015, 16, 622–631. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, C.; Huebener, P.; Schwabe, R.F. Damage-associated molecular patterns in cancer: A double-edged sword. Oncogene 2016, 35, 5931–5941. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; MacFadyen, J.G.; Thuren, T.; Everett, B.M.; Libby, P.; Glynn, R.J.; Group, C.T. Effect of interleukin-1beta inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: Exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 2017, 390, 1833–1842. [Google Scholar] [CrossRef]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 2007, 13, 1050–1059. [Google Scholar] [CrossRef]
- Chiba, S.; Baghdadi, M.; Akiba, H.; Yoshiyama, H.; Kinoshita, I.; Dosaka-Akita, H.; Fujioka, Y.; Ohba, Y.; Gorman, J.V.; Colgan, J.D.; et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat. Immunol. 2012, 13, 832–842. [Google Scholar] [CrossRef]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef]
- Bettum, I.J.; Vasiliauskaite, K.; Nygaard, V.; Clancy, T.; Pettersen, S.J.; Tenstad, E.; Maelandsmo, G.M.; Prasmickaite, L. Metastasis-associated protein S100A4 induces a network of inflammatory cytokines that activate stromal cells to acquire pro-tumorigenic properties. Cancer Lett. 2014, 344, 28–39. [Google Scholar] [CrossRef]
- Ghavami, S.; Rashedi, I.; Dattilo, B.M.; Eshraghi, M.; Chazin, W.J.; Hashemi, M.; Wesselborg, S.; Kerkhoff, C.; Los, M. S100A8/A9 at low concentration promotes tumor cell growth via RAGE ligation and MAP kinase-dependent pathway. J. Leukoc. Biol. 2008, 83, 1484–1492. [Google Scholar] [CrossRef]
- Jovanovic, I.P.; Pejnovic, N.N.; Radosavljevic, G.D.; Pantic, J.M.; Milovanovic, M.Z.; Arsenijevic, N.N.; Lukic, M.L. Interleukin-33/ST2 axis promotes breast cancer growth and metastases by facilitating intratumoral accumulation of immunosuppressive and innate lymphoid cells. Int. J. Cancer 2014, 134, 1669–1682. [Google Scholar] [CrossRef] [PubMed]
- Rider, P.; Carmi, Y.; Voronov, E.; Apte, R.N. Interleukin-1alpha. Semin. Immunol. 2013, 25, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Eun, H.S.; Jo, E.K. Roles of Autophagy-Related Genes in the Pathogenesis of Inflammatory Bowel Disease. Cells 2019, 8, 77. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.L.; Li, C.M.; Li, W.; Liu, Q.S.; Hu, S.Y.; Zhao, M.Y.; Hu, D.S.; Hao, Y.W.; Zeng, J.H.; Zhang, Y. How autophagy, a potential therapeutic target, regulates intestinal inflammation. Front. Immunol. 2023, 14, 1087677. [Google Scholar] [CrossRef]
- Khan, S.U.; Rayees, S.; Sharma, P.; Malik, F. Targeting redox regulation and autophagy systems in cancer stem cells. Clin. Exp. Med. 2023, 23, 1405–1423. [Google Scholar] [CrossRef]
- Vanaja, S.K.; Rathinam, V.A.; Fitzgerald, K.A. Mechanisms of inflammasome activation: Recent advances and novel insights. Trends Cell Biol. 2015, 25, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Prochnicki, T.; Latz, E. Inflammasomes on the Crossroads of Innate Immune Recognition and Metabolic Control. Cell Metab. 2017, 26, 71–93. [Google Scholar] [CrossRef]
- Awad, F.; Assrawi, E.; Louvrier, C.; Jumeau, C.; Georgin-Lavialle, S.; Grateau, G.; Amselem, S.; Giurgea, I.; Karabina, S.A. Inflammasome biology, molecular pathology and therapeutic implications. Pharmacol. Ther. 2018, 187, 133–149. [Google Scholar] [CrossRef]
- Protti, M.P.; De Monte, L. Dual Role of Inflammasome Adaptor ASC in Cancer. Front. Cell Dev. Biol. 2020, 8, 40. [Google Scholar] [CrossRef]
- Miller, C.M.; Zakrzewski, A.M.; Robinson, D.P.; Fuller, S.J.; Walker, R.A.; Ikin, R.J.; Bao, S.J.; Grigg, M.E.; Wiley, J.S.; Smith, N.C. Lack of a Functioning P2X7 Receptor Leads to Increased Susceptibility to Toxoplasmic Ileitis. PLoS ONE 2015, 10, e0129048. [Google Scholar] [CrossRef] [PubMed]
- Boucher, D.; Monteleone, M.; Coll, R.C.; Chen, K.W.; Ross, C.M.; Teo, J.L.; Gomez, G.A.; Holley, C.L.; Bierschenk, D.; Stacey, K.J.; et al. Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J. Exp. Med. 2018, 215, 827–840. [Google Scholar] [CrossRef]
- Platnich, J.M.; Muruve, D.A. NOD-like receptors and inflammasomes: A review of their canonical and non-canonical signaling pathways. Arch. Biochem. Biophys. 2019, 670, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Keshavarz Shahbaz, S.; Koushki, K.; Ayati, S.H.; Bland, A.R.; Bezsonov, E.E.; Sahebkar, A. Inflammasomes and Colorectal Cancer. Cells 2021, 10, 2172. [Google Scholar] [CrossRef]
- Zaki, M.H.; Boyd, K.L.; Vogel, P.; Kastan, M.B.; Lamkanfi, M.; Kanneganti, T.D. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 2010, 32, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Dupaul-Chicoine, J.; Yeretssian, G.; Doiron, K.; Bergstrom, K.S.; McIntire, C.R.; LeBlanc, P.M.; Meunier, C.; Turbide, C.; Gros, P.; Beauchemin, N.; et al. Control of intestinal homeostasis, colitis, and colitis-associated colorectal cancer by the inflammatory caspases. Immunity 2010, 32, 367–378. [Google Scholar] [CrossRef]
- Huber, S.; Gagliani, N.; Zenewicz, L.A.; Huber, F.J.; Bosurgi, L.; Hu, B.; Hedl, M.; Zhang, W.; O’Connor, W., Jr.; Murphy, A.J.; et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature 2012, 491, 259–263. [Google Scholar] [CrossRef]
- Williams, T.M.; Leeth, R.A.; Rothschild, D.E.; Coutermarsh-Ott, S.L.; McDaniel, D.K.; Simmons, A.E.; Heid, B.; Cecere, T.E.; Allen, I.C. The NLRP1 inflammasome attenuates colitis and colitis-associated tumorigenesis. J. Immunol. 2015, 194, 3369–3380. [Google Scholar] [CrossRef]
- Hu, B.; Elinav, E.; Huber, S.; Booth, C.J.; Strowig, T.; Jin, C.; Eisenbarth, S.C.; Flavell, R.A. Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proc. Natl. Acad. Sci. USA 2010, 107, 21635–21640. [Google Scholar] [CrossRef]
- Wilson, J.E.; Petrucelli, A.S.; Chen, L.; Koblansky, A.A.; Truax, A.D.; Oyama, Y.; Rogers, A.B.; Brickey, W.J.; Wang, Y.; Schneider, M.; et al. Inflammasome-independent role of AIM2 in suppressing colon tumorigenesis via DNA-PK and Akt. Nat. Med. 2015, 21, 906–913. [Google Scholar] [CrossRef]
- Ohashi, K.; Wang, Z.; Yang, Y.M.; Billet, S.; Tu, W.; Pimienta, M.; Cassel, S.L.; Pandol, S.J.; Lu, S.C.; Sutterwala, F.S.; et al. NOD-like receptor C4 Inflammasome Regulates the Growth of Colon Cancer Liver Metastasis in NAFLD. Hepatology 2019, 70, 1582–1599. [Google Scholar] [CrossRef] [PubMed]
- Zakeri, Z.F.; Ahuja, H.S. Cell death/apoptosis: Normal, chemically induced, and teratogenic effect. Mutat. Res. 1997, 396, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.P.; Choi, D.W. Ions, cell volume, and apoptosis. Proc. Natl. Acad. Sci. USA 2000, 97, 9360–9362. [Google Scholar] [CrossRef] [PubMed]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef]
- Meier, P.; Finch, A.; Evan, G. Apoptosis in development. Nature 2000, 407, 796–801. [Google Scholar] [CrossRef]
- Martinvalet, D.; Zhu, P.; Lieberman, J. Granzyme A induces caspase-independent mitochondrial damage, a required first step for apoptosis. Immunity 2005, 22, 355–370. [Google Scholar] [CrossRef]
- Strasser, A.; Pellegrini, M. T-lymphocyte death during shutdown of an immune response. Trends Immunol. 2004, 25, 610–615. [Google Scholar] [CrossRef]
- Nunes, T.; Bernardazzi, C.; de Souza, H.S. Cell death and inflammatory bowel diseases: Apoptosis, necrosis, and autophagy in the intestinal epithelium. Biomed. Res. Int. 2014, 2014, 218493. [Google Scholar] [CrossRef]
- Okada, H.; Mak, T.W. Pathways of apoptotic and non-apoptotic death in tumour cells. Nat. Rev. Cancer 2004, 4, 592–603. [Google Scholar] [CrossRef]
- Thompson, A.I.; Lees, C.W. Genetics of ulcerative colitis. Inflamm. Bowel Dis. 2011, 17, 831–848. [Google Scholar] [CrossRef]
- Garza-Hernandez, D.; Estrada, K.; Trevino, V. Multivariate genome-wide association study models to improve prediction of Crohn’s disease risk and identification of potential novel variants. Comput. Biol. Med. 2022, 145, 105398. [Google Scholar] [CrossRef]
- Souza, H.S.; Tortori, C.J.; Castelo-Branco, M.T.; Carvalho, A.T.; Margallo, V.S.; Delgado, C.F.; Dines, I.; Elia, C.C. Apoptosis in the intestinal mucosa of patients with inflammatory bowel disease: Evidence of altered expression of FasL and perforin cytotoxic pathways. Int. J. Colorectal Dis. 2005, 20, 277–286. [Google Scholar] [CrossRef]
- Di Sabatino, A.; Ciccocioppo, R.; Luinetti, O.; Ricevuti, L.; Morera, R.; Cifone, M.G.; Solcia, E.; Corazza, G.R. Increased enterocyte apoptosis in inflamed areas of Crohn’s disease. Dis. Colon Rectum 2003, 46, 1498–1507. [Google Scholar] [CrossRef]
- Gunther, C.; Neumann, H.; Neurath, M.F.; Becker, C. Apoptosis, necrosis and necroptosis: Cell death regulation in the intestinal epithelium. Gut 2013, 62, 1062–1071. [Google Scholar] [CrossRef]
- Dourmashkin, R.R.; Davies, H.; Wells, C.; Shah, D.; Price, A.; O’Morain, C.; Levi, J. Epithelial patchy necrosis in Crohn’s disease. Hum. Pathol. 1983, 14, 643–648. [Google Scholar] [CrossRef]
- Levine, B.; Deretic, V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat. Rev. Immunol. 2007, 7, 767–777. [Google Scholar] [CrossRef]
- Lapaquette, P.; Glasser, A.L.; Huett, A.; Xavier, R.J.; Darfeuille-Michaud, A. Crohn’s disease-associated adherent-invasive E. coli are selectively favoured by impaired autophagy to replicate intracellularly. Cell Microbiol. 2010, 12, 99–113. [Google Scholar] [CrossRef]
- Prescott, N.J.; Fisher, S.A.; Franke, A.; Hampe, J.; Onnie, C.M.; Soars, D.; Bagnall, R.; Mirza, M.M.; Sanderson, J.; Forbes, A.; et al. A nonsynonymous SNP in ATG16L1 predisposes to ileal Crohn’s disease and is independent of CARD15 and IBD5. Gastroenterology 2007, 132, 1665–1671. [Google Scholar] [CrossRef]
- Cadwell, K.; Liu, J.Y.; Brown, S.L.; Miyoshi, H.; Loh, J.; Lennerz, J.K.; Kishi, C.; Kc, W.; Carrero, J.A.; Hunt, S.; et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 2008, 456, 259–263. [Google Scholar] [CrossRef]
- Saitoh, T.; Fujita, N.; Jang, M.H.; Uematsu, S.; Yang, B.G.; Satoh, T.; Omori, H.; Noda, T.; Yamamoto, N.; Komatsu, M.; et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 2008, 456, 264–268. [Google Scholar] [CrossRef]
- Kubisch, J.; Turei, D.; Foldvari-Nagy, L.; Dunai, Z.A.; Zsakai, L.; Varga, M.; Vellai, T.; Csermely, P.; Korcsmaros, T. Complex regulation of autophagy in cancer-integrated approaches to discover the networks that hold a double-edged sword. Semin Cancer Biol. 2013, 23, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, J.; Levine, B.; Debnath, J. Autophagy and cancer metabolism. Methods Enzymol. 2014, 542, 25–57. [Google Scholar] [CrossRef] [PubMed]
- Taraborrelli, L.; Senbabaoglu, Y.; Wang, L.; Lim, J.; Blake, K.; Kljavin, N.; Gierke, S.; Scherl, A.; Ziai, J.; McNamara, E.; et al. Tumor-intrinsic expression of the autophagy gene Atg16l1 suppresses anti-tumor immunity in colorectal cancer. Nat. Commun. 2023, 14, 5945. [Google Scholar] [CrossRef]
- Di Virgilio, F.; Adinolfi, E. Extracellular purines, purinergic receptors and tumor growth. Oncogene 2017, 36, 293–303. [Google Scholar] [CrossRef]
- Coutinho-Silva, R.; Stahl, L.; Cheung, K.K.; de Campos, N.E.; de Oliveira Souza, C.; Ojcius, D.M.; Burnstock, G. P2X and P2Y purinergic receptors on human intestinal epithelial carcinoma cells: Effects of extracellular nucleotides on apoptosis and cell proliferation. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G1024–G1035. [Google Scholar] [CrossRef]
- Yoo, B.H.; Wu, X.; Derouet, M.; Haniff, M.; Eskelinen, E.L.; Rosen, K. Hypoxia-induced downregulation of autophagy mediator Beclin 1 reduces the susceptibility of malignant intestinal epithelial cells to hypoxia-dependent apoptosis. Autophagy 2009, 5, 1166–1179. [Google Scholar] [CrossRef]
- Jiang, Z.F.; Wu, W.; Hu, H.B.; Li, Z.Y.; Zhong, M.; Zhang, L. P2X7 receptor as the regulator of T-cell function in intestinal barrier disruption. World J. Gastroenterol. 2022, 28, 5265–5279. [Google Scholar] [CrossRef] [PubMed]
- Gulbransen, B.D.; Bashashati, M.; Hirota, S.A.; Gui, X.; Roberts, J.A.; MacDonald, J.A.; Muruve, D.A.; McKay, D.M.; Beck, P.L.; Mawe, G.M.; et al. Activation of neuronal P2X7 receptor-pannexin-1 mediates death of enteric neurons during colitis. Nat. Med. 2012, 18, 600–604. [Google Scholar] [CrossRef] [PubMed]
- da Silva, M.V.; Marosti, A.R.; Mendes, C.E.; Palombit, K.; Castelucci, P. Differential effects of experimental ulcerative colitis on P2X7 receptor expression in enteric neurons. Histochem. Cell Biol. 2015, 143, 171–184. [Google Scholar] [CrossRef]
- Heiss, K.; Janner, N.; Mahnss, B.; Schumacher, V.; Koch-Nolte, F.; Haag, F.; Mittrucker, H.W. High sensitivity of intestinal CD8+ T cells to nucleotides indicates P2X7 as a regulator for intestinal T cell responses. J. Immunol. 2008, 181, 3861–3869. [Google Scholar] [CrossRef]
- Hope, J.M.; Greenlee, J.D.; King, M.R. Mechanosensitive Ion Channels: TRPV4 and P2X7 in Disseminating Cancer Cells. Cancer J 2018, 24, 84–92. [Google Scholar] [CrossRef]
- Coutinho-Silva, R.; Perfettini, J.L.; Persechini, P.M.; Dautry-Varsat, A.; Ojcius, D.M. Modulation of P2Z/P2X(7) receptor activity in macrophages infected with Chlamydia psittaci. Am. J. Physiol. Cell. Physiol. 2001, 280, C81–C89. [Google Scholar] [CrossRef]
- Baricordi, O.R.; Melchiorri, L.; Adinolfi, E.; Falzoni, S.; Chiozzi, P.; Buell, G.; Di Virgilio, F. Increased proliferation rate of lymphoid cells transfected with the P2X(7) ATP receptor. J. Biol. Chem. 1999, 274, 33206–33208. [Google Scholar] [CrossRef]
- Antonioli, L.; Giron, M.C.; Colucci, R.; Pellegrini, C.; Sacco, D.; Caputi, V.; Orso, G.; Tuccori, M.; Scarpignato, C.; Blandizzi, C.; et al. Involvement of the P2X7 purinergic receptor in colonic motor dysfunction associated with bowel inflammation in rats. PLoS ONE 2014, 9, e116253. [Google Scholar] [CrossRef]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat Rev Mol Cell Biol 2010, 11, 700–714. [Google Scholar] [CrossRef]
- Bidula, S.; Dhuna, K.; Helliwell, R.; Stokes, L. Positive allosteric modulation of P2X7 promotes apoptotic cell death over lytic cell death responses in macrophages. Cell Death Dis. 2019, 10, 882. [Google Scholar] [CrossRef]
- Taurog, J.D.; Richardson, J.A.; Croft, J.T.; Simmons, W.A.; Zhou, M.; Fernandez-Sueiro, J.L.; Balish, E.; Hammer, R.E. The germfree state prevents development of gut and joint inflammatory disease in HLA-B27 transgenic rats. J. Exp. Med. 1994, 180, 2359–2364. [Google Scholar] [CrossRef]
- Miyoshi, J.; Bobe, A.M.; Miyoshi, S.; Huang, Y.; Hubert, N.; Delmont, T.O.; Eren, A.M.; Leone, V.; Chang, E.B. Peripartum Antibiotics Promote Gut Dysbiosis, Loss of Immune Tolerance, and Inflammatory Bowel Disease in Genetically Prone Offspring. Cell Rep. 2017, 20, 491–504. [Google Scholar] [CrossRef]
- de Martel, C.; Georges, D.; Bray, F.; Ferlay, J.; Clifford, G.M. Global burden of cancer attributable to infections in 2018: A worldwide incidence analysis. Lancet Glob. Heal. 2020, 8, e180–e190. [Google Scholar] [CrossRef]
- Moore, P.S.; Chang, Y. Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat. Rev. Cancer 2010, 10, 878–889. [Google Scholar] [CrossRef]
- Petrelli, F.; Ghidini, M.; Ghidini, A.; Perego, G.; Cabiddu, M.; Khakoo, S.; Oggionni, E.; Abeni, C.; Hahne, J.C.; Tomasello, G.; et al. Use of Antibiotics and Risk of Cancer: A Systematic Review and Meta-Analysis of Observational Studies. Cancers 2019, 11, 1174. [Google Scholar] [CrossRef] [PubMed]
- Schulz, M.D.; Atay, C.; Heringer, J.; Romrig, F.K.; Schwitalla, S.; Aydin, B.; Ziegler, P.K.; Varga, J.; Reindl, W.; Pommerenke, C.; et al. High-fat-diet-mediated dysbiosis promotes intestinal carcinogenesis independently of obesity. Nature 2014, 514, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Wallace, K.L.; Zheng, L.B.; Kanazawa, Y.; Shih, D.Q. Immunopathology of inflammatory bowel disease. World J. Gastroenterol. 2014, 20, 6–21. [Google Scholar] [CrossRef]
- Kennedy, J.M.; De Silva, A.; Walton, G.E.; Gibson, G.R. A review on the use of prebiotics in ulcerative colitis. Trends Microbiol. 2024, 32, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Tlaskalova-Hogenova, H.; Tuckova, L.; Stepankova, R.; Hudcovic, T.; Palova-Jelinkova, L.; Kozakova, H.; Rossmann, P.; Sanchez, D.; Cinova, J.; Hrncir, T.; et al. Involvement of innate immunity in the development of inflammatory and autoimmune diseases. Ann. NY Acad. Sci. 2005, 1051, 787–798. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Han, M.; Heinrich, B.; Fu, Q.; Zhang, Q.; Sandhu, M.; Agdashian, D.; Terabe, M.; Berzofsky, J.A.; Fako, V.; et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science 2018, 360, eaan5931. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Wang, K.; Mucida, D.; Stewart, C.A.; Schnabl, B.; Jauch, D.; Taniguchi, K.; Yu, G.Y.; Osterreicher, C.H.; Hung, K.E.; et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 2012, 491, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Eichele, D.D.; Kharbanda, K.K. Dextran sodium sulfate colitis murine model: An indispensable tool for advancing our understanding of inflammatory bowel diseases pathogenesis. World J. Gastroenterol. 2017, 23, 6016–6029. [Google Scholar] [CrossRef]
- Caamano, J.; Hunter, C.A. NF-kappaB family of transcription factors: Central regulators of innate and adaptive immune functions. Clin. Microbiol. Rev. 2002, 15, 414–429. [Google Scholar] [CrossRef]
- Hakansson, A.; Tormo-Badia, N.; Baridi, A.; Xu, J.; Molin, G.; Hagslatt, M.L.; Karlsson, C.; Jeppsson, B.; Cilio, C.M.; Ahrne, S. Immunological alteration and changes of gut microbiota after dextran sulfate sodium (DSS) administration in mice. Clin. Exp. Med. 2015, 15, 107–120. [Google Scholar] [CrossRef]
- Yacoub, E.; Saed Abdul-Wahab, O.M.; Al-Shyarba, M.H.; Ben Abdelmoumen Mardassi, B. The Relationship between Mycoplasmas and Cancer: Is It Fact or Fiction ? Narrative Review and Update on the Situation. J. Oncol. 2021, 2021, 9986550. [Google Scholar] [CrossRef] [PubMed]
- Moreira-Souza, A.C.A.; Nanini, H.F.; Rangel, T.P.; da Silva, S.R.B.; Damasceno, B.P.; Ribeiro, B.E.; Cascabulho, C.M.; Thompson, F.; Leal, C.; Santana, P.T.; et al. P2X7 Receptor Modulation of the Gut Microbiota and the Inflammasome Determines the Severity of Toxoplasma gondii-Induced Ileitis. Biomedicines 2023, 11, 555. [Google Scholar] [CrossRef] [PubMed]
- Santana, P.T.; Rosas, S.L.B.; Ribeiro, B.E.; Marinho, Y.; de Souza, H.S.P. Dysbiosis in Inflammatory Bowel Disease: Pathogenic Role and Potential Therapeutic Targets. Int. J. Mol. Sci. 2022, 23, 3464. [Google Scholar] [CrossRef] [PubMed]
- Loubinoux, J.; Bronowicki, J.P.; Pereira, I.A.; Mougenel, J.L.; Faou, A.E. Sulfate-reducing bacteria in human feces and their association with inflammatory bowel diseases. FEMS Microbiol. Ecol. 2002, 40, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Whitehead, R.N.; Griffiths, L.; Dawson, C.; Bai, H.; Waring, R.H.; Ramsden, D.B.; Hunter, J.O.; Cauchi, M.; Bessant, C.; et al. Diversity and distribution of sulphate-reducing bacteria in human faeces from healthy subjects and patients with inflammatory bowel disease. FEMS Immunol. Med. Microbiol. 2012, 65, 55–68. [Google Scholar] [CrossRef]
- Figliuolo, V.R.; Dos Santos, L.M.; Abalo, A.; Nanini, H.; Santos, A.; Brittes, N.M.; Bernardazzi, C.; de Souza, H.S.P.; Vieira, L.Q.; Coutinho-Silva, R.; et al. Sulfate-reducing bacteria stimulate gut immune responses and contribute to inflammation in experimental colitis. Life Sci. 2017, 189, 29–38. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Immune Cell | P2X7R Activation | Effect on IBD | Effect on CRC | References |
|---|---|---|---|---|
| Macrophages | Leads to the activation of the NLRP3 inflammasome and the ERK1/2, NFκB, and PI3K/AKT pathways | The M1 phenotype is pro-inflammatory, producing IL-1β, IL-18, TNF-α and induction of pyroptotic cell death, contributing to chronic inflammation and tissue damage | The M2 phenotype is immunosuppressive and facilitates tumor growth, angiogenesis, and metastasis by secreting anti-inflammatory cytokines (IL-10 and TGF-β) | [71,72,73,75,76] |
| T-cells | Plays a role in the homeostatic regulation of subpopulations by inducing P2X7R-dependent T-cell death; favors differentiation, migration, and induces the production of MMPs; and induces NFAT and MAPK signaling, promoting IL-2 expression and T-cell proliferation | Increased activated CD4+ T-cells orchestrates Th1/Th17 immune responses; survival and differentiation of CD8+ cytotoxic T-cells that promote tissue damage by releasing perforin and granzymes; inhibition of the suppressive potential of Treg | Reduction of Tregs and of CD8+ T-cell infiltration enhance anti-tumor responses; induction of cellular senescence in tumor-infiltrating T-cells | [87,88,89,90,91] |
| Neutrophils | Triggers the NLRP3 inflammasome and IL-1β release | Production of excessive proinflammatory cytokines, MMPs, and ROS, fueling inflammation and tissue damage | Reduced tumor-fighting capacity; differentiate into TANs in the tumor microenvironment; the N2 type promotes tumor growth and metastasis | [47,73,74] [78,79] |
| Dendritic Cells (DCs) | Enhances antigen presentation, amplifying the responses of CD4+ T cells and CD8+ T cells; triggers the NLRP3 inflammasome and IL-1β release in the context of the TME | Overactive DCs produce excessive pro-inflammatory cytokines, leading to chronic inflammation | Dysfunctional DCs fail to present antigens effectively, inducing tumor growth and immune evasion | [80,81,82,83] |
| Natural Killer (NK) Cells | Reduces the ability of NK cells to kill cancer cells | Reduced activity can lead to less effective immune surveillance and increased inflammation | Impaired NK cell function facilitates tumor expansion and metastasis | [47,73,74] |
| Myeloid-Derived Suppressor Cells | Leads to the production of immunosuppressive factors, including ROS, Arginase-1, and TGFβ | Suppression of the immune responses, exacerbating chronic inflammation that can worsen the tissue damage in the gut and IBD symptoms | Increases VEGF, stimulating angiogenesis and blood supply with nutrients and facilitating metastasis by inhibiting anti-tumor immunity | [84,85,86] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santana, P.T.; de Lima, I.S.; Silva e Souza, K.C.d.; Barbosa, P.H.S.; de Souza, H.S.P. Persistent Activation of the P2X7 Receptor Underlies Chronic Inflammation and Carcinogenic Changes in the Intestine. Int. J. Mol. Sci. 2024, 25, 10874. https://doi.org/10.3390/ijms252010874
Santana PT, de Lima IS, Silva e Souza KCd, Barbosa PHS, de Souza HSP. Persistent Activation of the P2X7 Receptor Underlies Chronic Inflammation and Carcinogenic Changes in the Intestine. International Journal of Molecular Sciences. 2024; 25(20):10874. https://doi.org/10.3390/ijms252010874
Chicago/Turabian StyleSantana, Patricia Teixeira, Isadora Schmukler de Lima, Karen Cristina da Silva e Souza, Pedro Henrique Sales Barbosa, and Heitor Siffert Pereira de Souza. 2024. "Persistent Activation of the P2X7 Receptor Underlies Chronic Inflammation and Carcinogenic Changes in the Intestine" International Journal of Molecular Sciences 25, no. 20: 10874. https://doi.org/10.3390/ijms252010874
APA StyleSantana, P. T., de Lima, I. S., Silva e Souza, K. C. d., Barbosa, P. H. S., & de Souza, H. S. P. (2024). Persistent Activation of the P2X7 Receptor Underlies Chronic Inflammation and Carcinogenic Changes in the Intestine. International Journal of Molecular Sciences, 25(20), 10874. https://doi.org/10.3390/ijms252010874