
The Role of Ligand Exchange in Salen Cobalt Complexes in the Alternating Copolymerization of Propylene Oxide and Carbon Dioxide

 , and
, and 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Polymer Synthesis
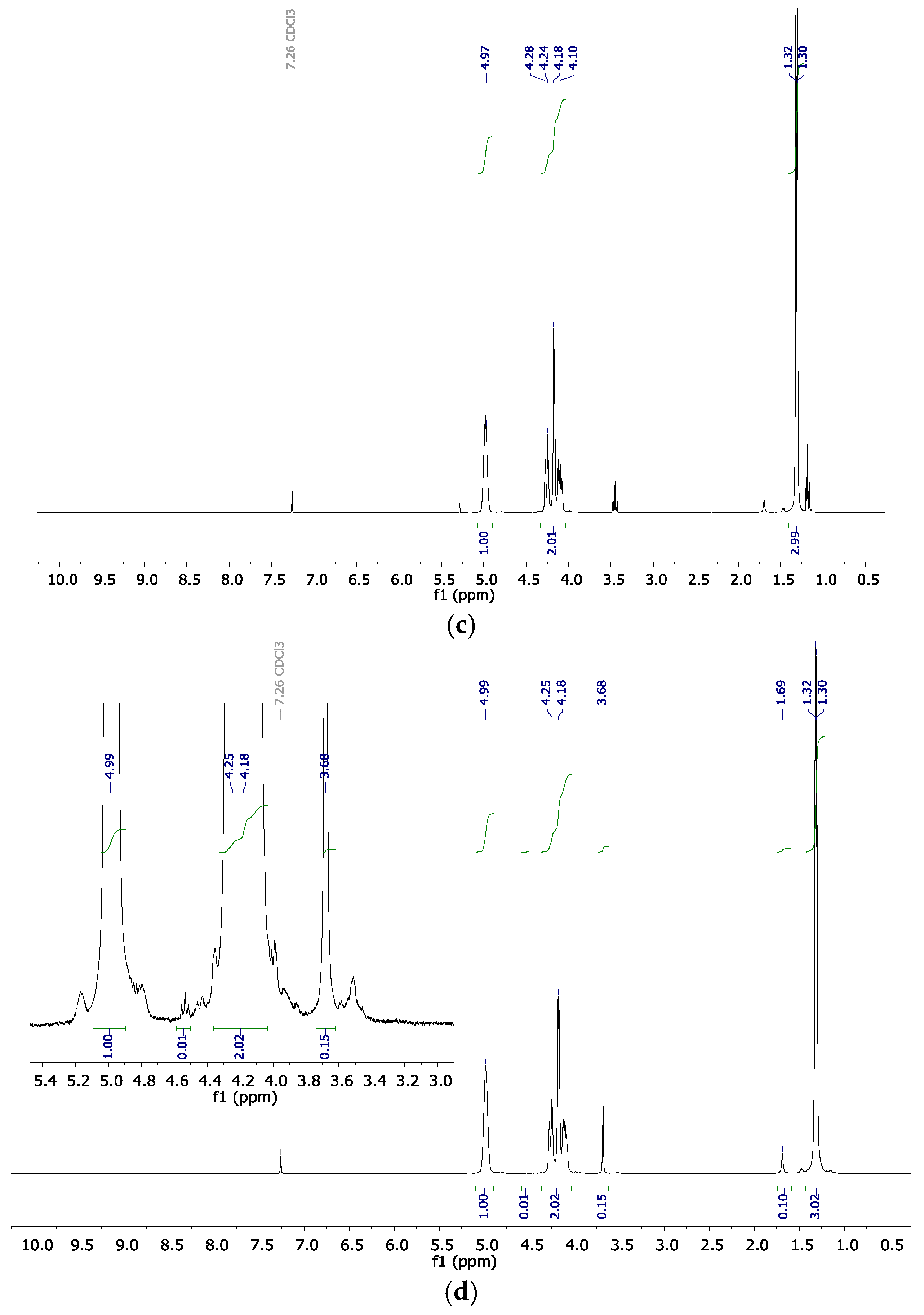
2.2. Copolymer Microstructure
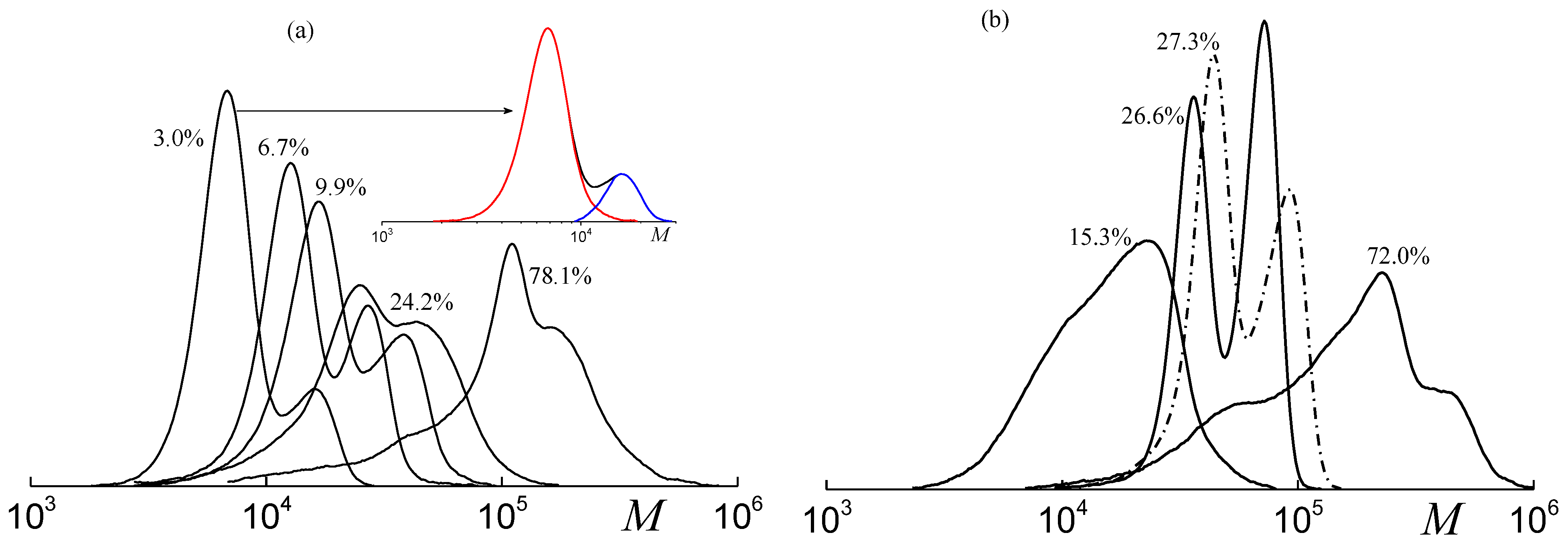
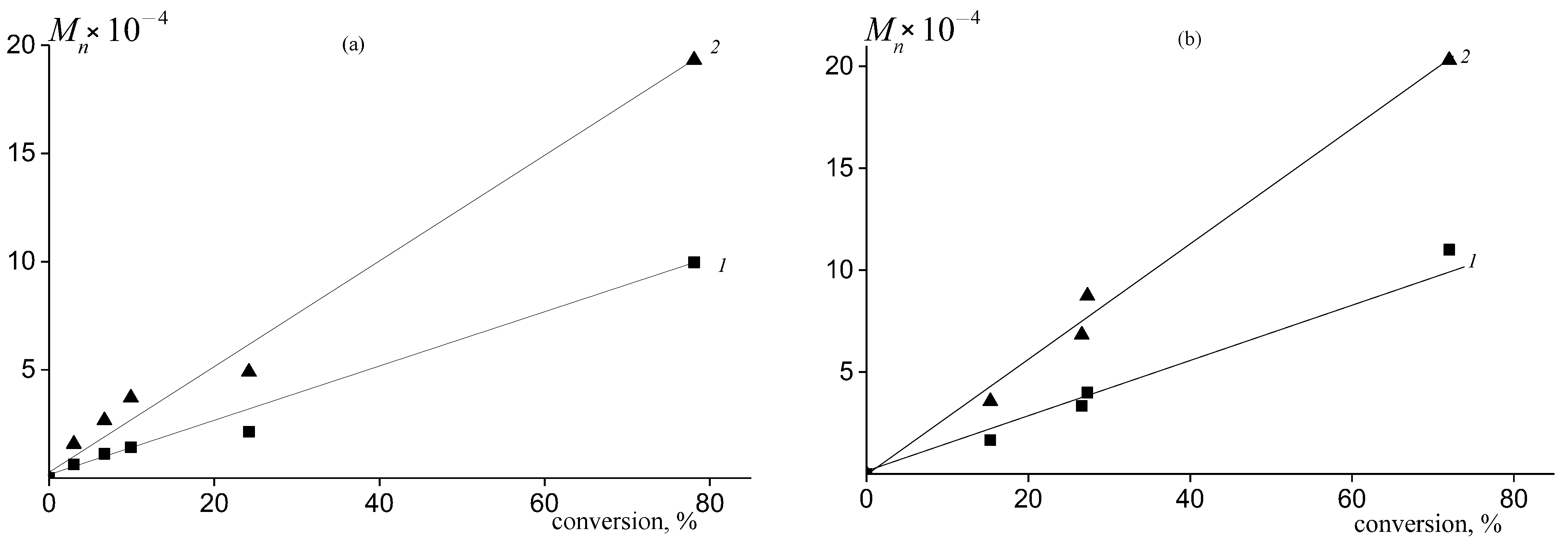
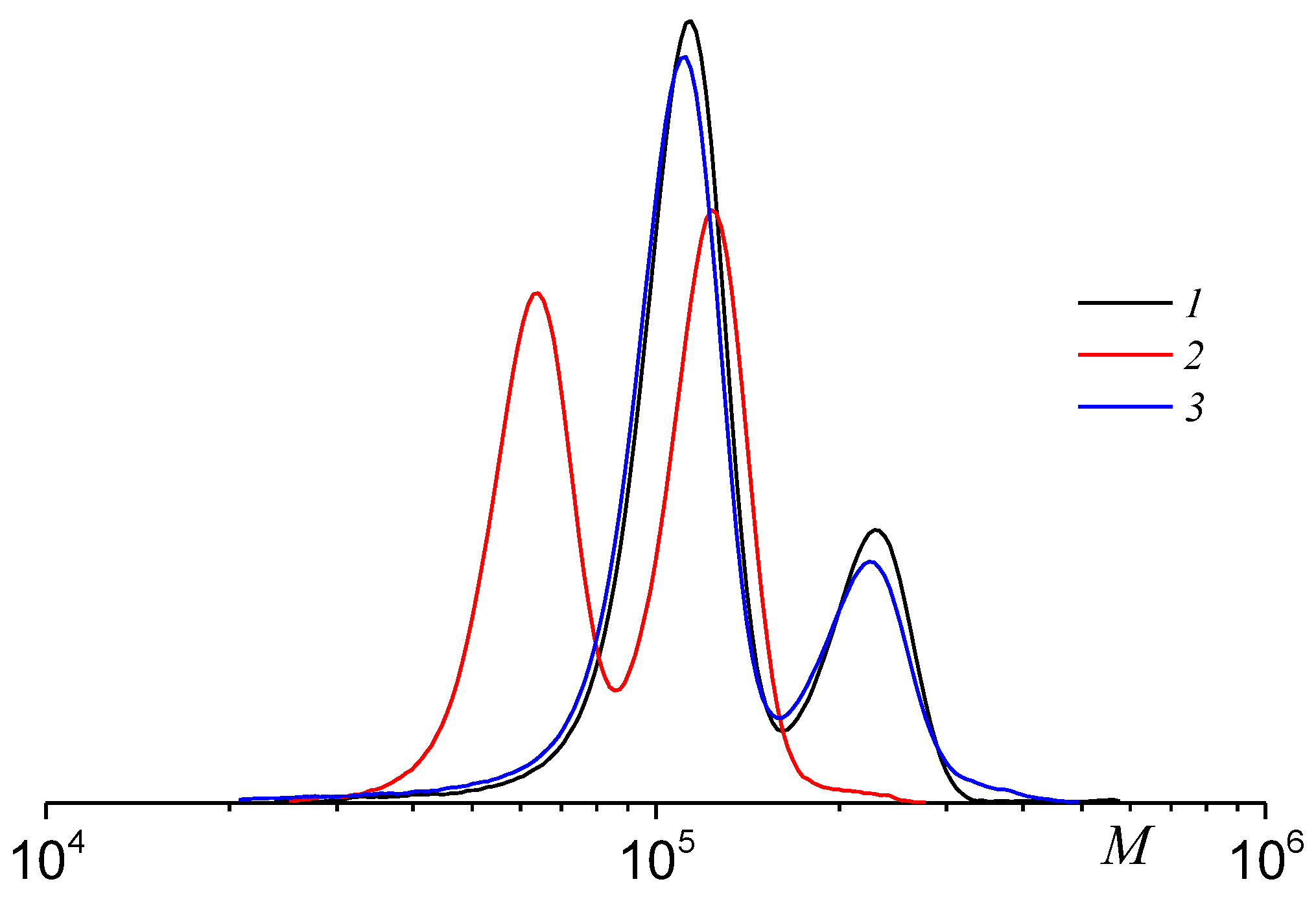
2.3. Molecular Weight Distribution of PPC
2.4. Thermal Stability of PPC
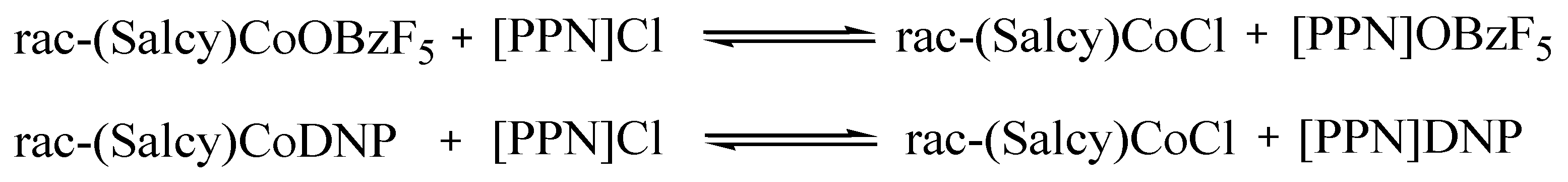
2.5. Polymerization Mechanism
3. Materials and Methods
3.1. Materials
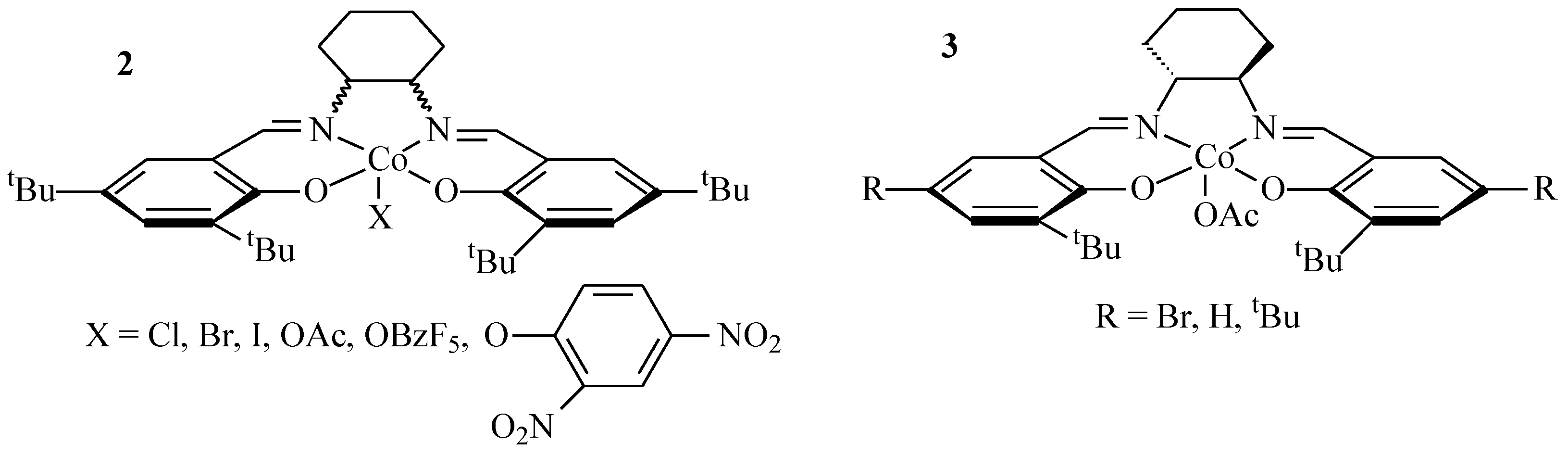
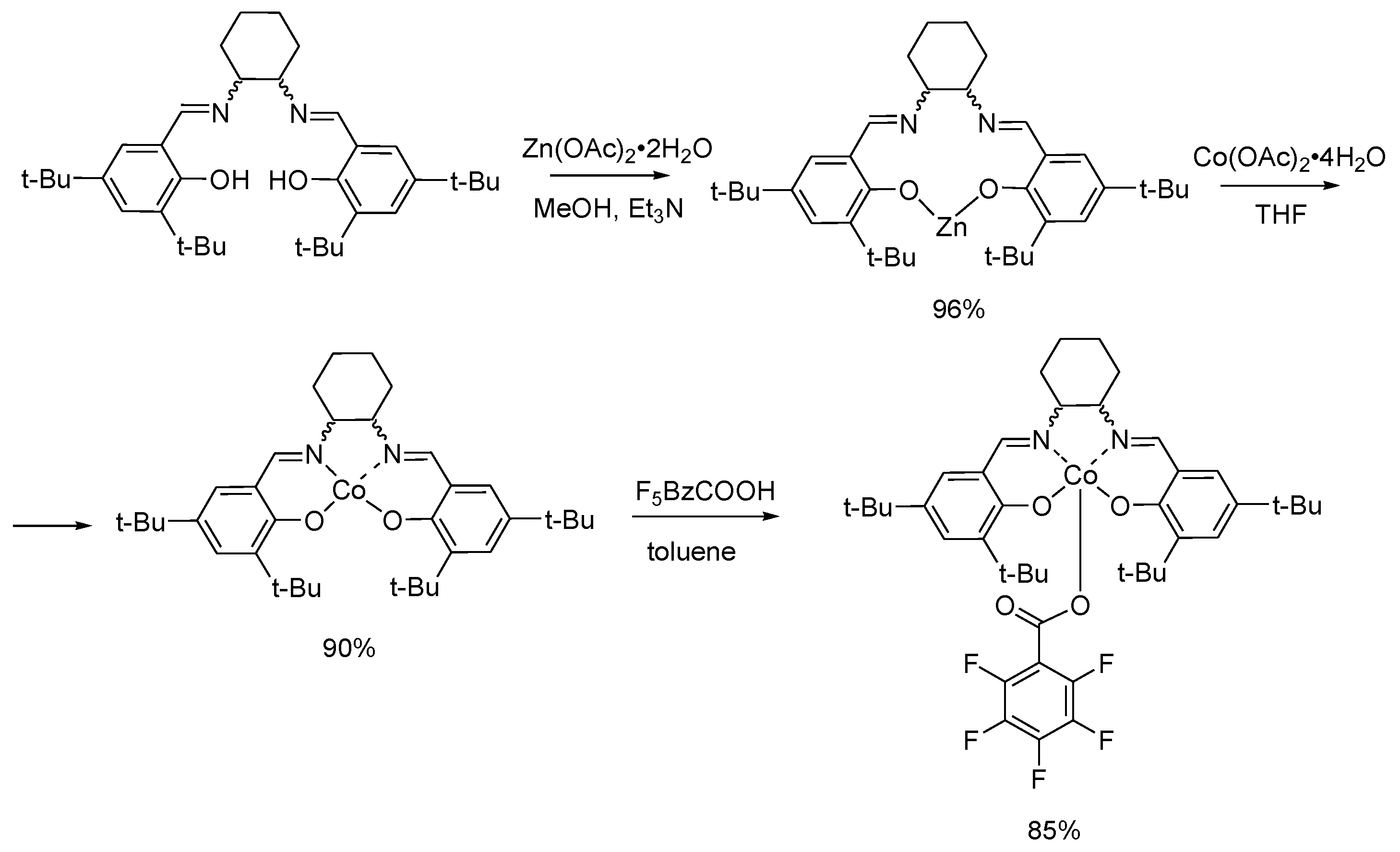
3.2. Synthesis of the Complexes
3.3. Polymerization Procedure
3.4. Instrumentation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cao, H.; Wang, X. Carbon dioxide copolymers: Emerging sustainable materials for versatile applications. SusMat 2021, 1, 88–104. [Google Scholar] [CrossRef]
- Artz, J.; Müller, T.E.; Thenert, K.; Kleinekorte, J.; Meys, R.; Sternberg, A.; Bardow, A.; Leitner, W. Sustainable Conversion of Carbon Dioxide: An Integrated Review of Catalysis and Life Cycle Assessment. Chem. Rev. 2018, 118, 434–504. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Romain, C.; Williams, C.K. Sustainable polymers from renewable resources. Nature 2016, 540, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Q.; Patel, M.K. Plastics derived from biological sources: Present and future: A technical and environmental review. Chem. Rev. 2012, 112, 2082–2099. [Google Scholar] [CrossRef]
- Muthuraj, R.; Mekonnen, T. Recent progress in carbon dioxide (CO2) as feedstock for sustainable materials development: Co-polymers and polymer blends. Polymer 2018, 145, 348–373. [Google Scholar] [CrossRef]
- Grignard, B.; Gennen, S.; Jérôme, C.; Kleij, A.W.; Detrembleur, C. Advances in the use of CO2 as a renewable feedstock for the synthesis of polymers. Chem. Soc. Rev. 2019, 48, 4466–4514. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, H.; Guo, J.Z.; Ren, W.M.; Lu, X.B. Completely recyclable monomers and polycarbonate: Approach to sustainable polymers. Angew. Chem. Int. Ed. 2017, 56, 4862–4866. [Google Scholar] [CrossRef]
- Qin, Y.; Wang, X.H. Carbon dioxide-based copolymers: Environmental benefits of PPC, an industrially viable catalyst. Biotechnol. J. 2010, 5, 1164–1180. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Wilson, S.J. What’s new with CO2? Recent advances in its copolymerization with oxiranes. Green Chem. 2012, 14, 2665–2671. [Google Scholar] [CrossRef]
- Lu, X.B.; Darensbourg, D.J. Cobalt catalysts for the coupling of CO2 and epoxides to provide polycarbonates and cyclic carbonates. Chem. Soc. Rev. 2012, 41, 1462–1484. [Google Scholar] [CrossRef]
- Paul, S.; Zhu, Y.; Romain, S.; Brooks, R.; Saini, P.K.; Williams, C.K. Ring-opening copolymerization (ROCOP): Synthesis and properties of polyesters and polycarbonates. Chem. Commun. 2015, 51, 6459–6479. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Worch, J.C.; Dove, A.P.; Coulembier, O. Update and challenges in carbon dioxide-based polycarbonate synthesis. ChemSusChem 2020, 13, 469–487. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.B.; Ren, B.H. Partners in Epoxide Copolymerization Catalysis: Approach to High Activity and Selectivity. Chin. J. Polym. Sci. 2022, 40, 1331–1348. [Google Scholar] [CrossRef]
- Yang, G.W.; Wang, Y.; Qi, H.; Zhang, Y.Y.; Zhu, X.F.; Lu, C.; Yang, L.; Wu, G.P. Highly Selective Preparation and Depolymerization of Chemically Recyclable Poly (cyclopentene carbonate) Enabled by Organoboron Catalysts. Angew. Chem. Int. Ed. 2022, 61, e202210243. [Google Scholar] [CrossRef]
- Chen, C.; Gnanou, Y.; Feng, X. Ultra-Productive Upcycling CO2 into Polycarbonate Polyols via Borinane-Based Bifunctional Organocatalysts. Macromolecules 2023, 56, 892–898. [Google Scholar] [CrossRef]
- Zhao, M.; Zhu, S.; Zhang, G.; Wang, Y.; Liao, Y.; Xu, J.; Zhou, X.; Xie, X. One-Step Synthesis of Linear and Hyperbranched CO2-Based Block Copolymers via Organocatalytic Switchable Polymerization. Macromolecules 2023, 56, 2379–2387. [Google Scholar] [CrossRef]
- Naumann, S. Borane catalysis for epoxide (co)polymerization. Polym. Chem. 2023, 14, 1834–1862. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, Z.; Guo, W.; Zhang, C.; Zhang, X. Phosphine-Borane Frustrated Lewis Pairs for Metal-Free CO2/Epoxide Copolymerization. Macromolecules 2023, 56, 4901–4909. [Google Scholar] [CrossRef]
- Zhou, X.; Zhai, Y.; Ren, K.; Cheng, Z.; Shen, X.; Zhang, T.; Bai, Y.; Jia, Y.; Hong, J. Life cycle assessment of polycarbonate production: Proposed optimization toward sustainability. Resour. Conserv. Recycl. 2023, 189, 106765. [Google Scholar] [CrossRef]
- Inoue, S.; Koinuma, H.; Tsuruta, T. Copolymerization of carbon dioxide and epoxide with organometallic compounds. Macromol. Chem. 1969, 130, 210–220. [Google Scholar] [CrossRef]
- Trott, G.; Saini, P.K.; Williams, C.K. Catalysts for CO2/epoxide ring-opening copolymerization. Philos. Trans. R. Soc. A 2016, 374, 20150085. [Google Scholar] [CrossRef] [PubMed]
- Kernbichl, S.; Rieger, B. Aliphatic Polycarbonates Derived from Epoxides and CO2. In Engineering Solutions for CO2 Conversion; Reina, T.R., Arellano-Garcia, H., Odriozola, J.A., Eds.; Wiley-VCH: Weinheim, Germany, 2021; p. 385. [Google Scholar] [CrossRef]
- Liang, X.; Tan, F.; Zhu, Y. Recent Developments in Ring-Opening Copolymerization of Epoxides with CO2 and Cyclic Anhydrides for Biomedical Applications. Front. Chem. 2021, 9, 647245. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Qin, A.; Tang, B.Z. Syntheses, properties, and applications of CO2-based functional polymers. Cell Rep. Phys. Sci. 2022, 3, 100719. [Google Scholar] [CrossRef]
- Lidston, C.A.L.; Severson, S.M.; Abel, B.A.; Coates, G.W. Multifunctional Catalysts for Ring-Opening Copolymerizations. ACS Catal. 2022, 12, 11037–11070. [Google Scholar] [CrossRef]
- Nakano, K.; Hashimoto, S.; Nakamura, M.; Kamada, T.; Nozaki, K. Stereocomplex of Poly (propylene carbonate): Synthesis of Stereogradient Poly(propylene carbonate) by Regio- and Enantioselective Copolymerization of Propylene Oxide with Carbon Dioxide. Angew. Chem. Int. Ed. 2011, 50, 4868–4871. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.B.; Wang, Y. Highly Active, Binary Catalyst Systems for the Alternating Copolymerization of CO2 and Epoxides under Mild Conditions. Angew. Chem. Int. Ed. 2004, 43, 3574–3577. [Google Scholar] [CrossRef] [PubMed]
- Paddock, R.L.; Nguyen, S.T. Chiral (salen) CoIII catalyst for the synthesis of cyclic carbonates. Chem. Commun. 2004, 14, 1622–1623. [Google Scholar] [CrossRef] [PubMed]
- Cohen, C.T.; Thomas, C.M.; Peretti, K.L.; Lobkovsky, E.B.; Coates, G.W. Copolymerization of cyclohexene oxide and carbon dioxide using (salen) Co(III) complexes: Synthesis and characterization of syndiotactic poly (cyclohexene carbonate). Dalton Trans. 2006, 1, 237–249. [Google Scholar] [CrossRef]
- Cohen, C.T.; Coates, G.W. Alternating copolymerization of propylene oxide and carbon dioxide with highly efficient and selective (salen) Co(III) catalysts: Effect of ligand and cocatalyst variation. J. Polym. Sci. Polym. Chem. 2006, 44, 5182–5191. [Google Scholar] [CrossRef]
- Cohen, C.T.; Chu, T.; Coates, G.W. Cobalt Catalysts for the Alternating Copolymerization of Propylene Oxide and Carbon Dioxide: Combining High Activity and Selectivity. J. Am. Chem. Soc. 2005, 127, 10869–10878. [Google Scholar] [CrossRef]
- Chukanova, O.M.; Belov, G.P. Reaction between carbon dioxide and propylene oxide catalyzed by cobalt and chromium porphyrin complexes: The effect of reaction conditions on the reaction rate. Kinet. Catal. 2017, 58, 397–401. [Google Scholar] [CrossRef]
- Chukanova, O.M.; Belov, G.P. Effect of the ligand nature in cobalt complexes on the selectivity of the reaction of carbon dioxide and propylene oxide. Kinet. Catal. 2016, 57, 821–825. [Google Scholar] [CrossRef]
- Chukanova, O.M.; Perepelitsina, E.O.; Belov, G.P. The influence of reagent concentration on the kinetics of carbon dioxide-propylene oxide copolymerization in the presence of a cobalt complex. Polym. Sci. Ser. B 2014, 56, 547–552. [Google Scholar] [CrossRef]
- Ajiro, H.; Peretti, K.; Lobkovsky, E.B.; Coates, G.W. On the mechanism of isospecific epoxide polymerization by salen cobalt(III) complexes: Evidence for solid-state catalysis. Dalton Trans. 2009, 8828–8830. [Google Scholar] [CrossRef]
- Peretti, K.L.; Ajiro, H.; Cohen, C.T.; Lobkovsky, E.B.; Coates, G.W. A Highly Active, Isospecific Cobalt Catalyst for Propylene Oxide Polymerization. J. Am. Chem. Soc. 2005, 127, 11566–11567. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, M.; Larrow, J.F.; Kakiuchi, F.; Jacobsen, E.N. Asymmetric Catalysis with Water: Efficient Kinetic Resolution of Terminal Epoxides by Means of Catalytic Hydrolysis. Science 1997, 277, 936–938. [Google Scholar] [CrossRef]
- Nielsen, L.P.C.; Stevenson, C.P.; Blackmond, D.G.; Jacobsen, E.N. Mechanistic Investigation Leads to a Synthetic Improvement in the Hydrolytic Kinetic Resolution of Terminal Epoxides. J. Am. Chem. Soc. 2004, 126, 1360–1362. [Google Scholar] [CrossRef]
- Qin, Z.; Thomas, C.M.; Lee, S.; Coates, G.W. Cobalt-Based Complexes for the Copolymerization of Propylene Oxide and CO2: Active and Selective Catalysts for Polycarbonate Synthesis. Angew. Chem. Int. Ed. 2003, 42, 5484–5487. [Google Scholar] [CrossRef]
- Aida, T.; Inoue, S. Metalloporphyrins as Initiators for Living and Immortal Polymerizations. Acc. Chem. Res. 1996, 29, 39–48. [Google Scholar] [CrossRef]
- Lu, X.-B.; Shi, L.; Wang, Y.-M.; Zhang, R.; Zhang, Y.-J.; Peng, X.-J.; Zhang, Z.-C.; Li, B. Design of Highly Active Binary Catalyst Systems for CO2/Epoxide Copolymerization: Polymer Selectivity, Enantioselec-tivity, and Stereochemistry Control. J. Am. Chem. Soc. 2006, 128, 1664–1674. [Google Scholar] [CrossRef]
- Zhuang, X.; Oyaizu, K.; Niu, Y.; Koshika, K.; Chen, X.; Nishide, H. Synthesis and electrochemistry of Schiff base cobalt(III) complexes and their catalytic activity for copolymerization of epoxide and carbon dioxide. Macromol. Chem. Phys. 2010, 211, 669–676. [Google Scholar] [CrossRef]
- Sugimoto, H.; Kuroda, K. The Cobalt Porphyrin−Lewis Base System: A Highly Selective Catalyst for Alternating Copolymerization of CO2 and Epoxide under Mild Conditions. Macromolecules 2008, 41, 312–317. [Google Scholar] [CrossRef]
- Kukushkin, V.Y.; Moiseev, A.I. Convenient synthesis of μ-nitridobis(triphenylphosphonium) chloride ([PPN]Cl) with contribution of PCl5 as chlorinating (PCl5 + PPh3) or deoxygenating (PCl5 + OPPh3) reagent. Inorg. Chim. Acta 1990, 176, 79–81. [Google Scholar] [CrossRef]
- Larrow, J.F.; Jacobsen, E.N. (R,R)-N,N′-Bis(3,5-di-tert-Butylsalicylidene)-1,2-Cyclohexanediamino Manganese(III) Chloride, A Highly Enantioselective Epoxidation Catalyst. Org. Synth. Coll. 1998, 75, 1. [Google Scholar] [CrossRef]









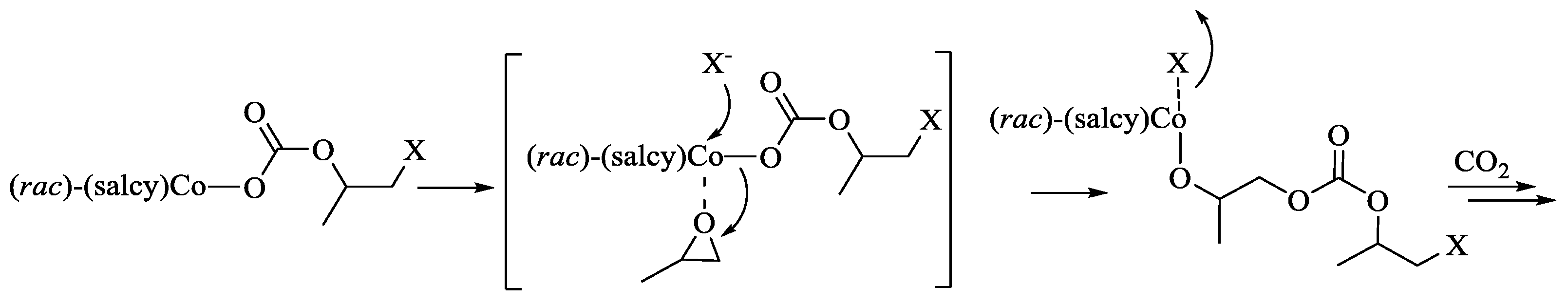


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | PO | Microstructure |
|---|---|---|
| (R,R)-(salcy)CoBr | (rac)-PO | Regioregular atactic |
| (S)-PO | Regioregular isotactic | |
| (R)-PO | Regiorandom heterotactic | |
| (rac)-(salcy)CoBr | (R)-PO | Regioregular isotactic |
| (rac)-PO | Regioregular syndio-enriched | |
| (R,R)-(salcy)CoOBzF5 | (rac)-PO | Regioregular iso-enriched |
| (S)-PO | Regioregular isotactic | |
| (rac)-(salcy)CoOBzF5 | (R)-PO | Regioregular isotactic |
| (rac)-PO | Regioregular atactic |
| Catalyst | [PO]/[Cat] | [Cat]/[[PPN]Cl] | Time, h | Selectivity, PPC% | TON | TOF, h−1 |
|---|---|---|---|---|---|---|
| (rac)-(salcy)CoOBzF5 | 6420 | 0.87 | 2 | >97 | 195 | 98 |
| 6630 | 0.86 | 4 | >97 | 440 | 110 | |
| 6350 | 0.86 | 6 | >97 | 630 | 105 | |
| 6500 | 0.86 | 24 | >50 | 1570 | 65 | |
| 6460 | 0.86 | 72 | >50 | 5050 | 79 | |
| (rac)-(salcy)CoDNP | 6170 | 0.90 | 2 | 96.0 | 950 | 470 |
| 6480 | 0.88 | 4 | 97.2 | 1730 | 430 | |
| 7080 | 0.74 | 4.5 | 98.4 | 1930 | 430 | |
| 6500 | 0.81 | 24 | >75 | 4784 | 199 |
| Cat/Co-Cat | [PO]/[Cat] | Conversion (PO), % | Selectivity, PPC% | TON | TOF, h−1 | Mn1/Mn2, (a) kDa | ĐM1/ĐM2 (a) |
|---|---|---|---|---|---|---|---|
| (rac)-(salcy)CoDNP [PPN]DNP | 6670 | 38.2 | 99.0 | 2548 | 637 | 103.2/220.6 | 1.05/1.03 |
| (rac)-(salcy)CoCl [PPN]Cl | 5990 | 40.4 | 96.4 | 2422 | 605 | 60.0/121.1 | 1.04/1.03 |
| (rac)-(salcy)CoCl [PPN]DNP | 6000 | 43.0 | 97.5 | 2582 | 645 | 98.2/220.8 | 1.07/1.04 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rzhevskiy, S.A.; Shurupova, O.V.; Asachenko, A.F.; Plutalova, A.V.; Chernikova, E.V.; Beletskaya, I.P. The Role of Ligand Exchange in Salen Cobalt Complexes in the Alternating Copolymerization of Propylene Oxide and Carbon Dioxide. Int. J. Mol. Sci. 2024, 25, 10946. https://doi.org/10.3390/ijms252010946
Rzhevskiy SA, Shurupova OV, Asachenko AF, Plutalova AV, Chernikova EV, Beletskaya IP. The Role of Ligand Exchange in Salen Cobalt Complexes in the Alternating Copolymerization of Propylene Oxide and Carbon Dioxide. International Journal of Molecular Sciences. 2024; 25(20):10946. https://doi.org/10.3390/ijms252010946
Chicago/Turabian StyleRzhevskiy, Sergey A., Olga V. Shurupova, Andrey F. Asachenko, Anna V. Plutalova, Elena V. Chernikova, and Irina P. Beletskaya. 2024. "The Role of Ligand Exchange in Salen Cobalt Complexes in the Alternating Copolymerization of Propylene Oxide and Carbon Dioxide" International Journal of Molecular Sciences 25, no. 20: 10946. https://doi.org/10.3390/ijms252010946