
Developing Advanced Chimeric Cell Therapy for Duchenne Muscular Dystrophy

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
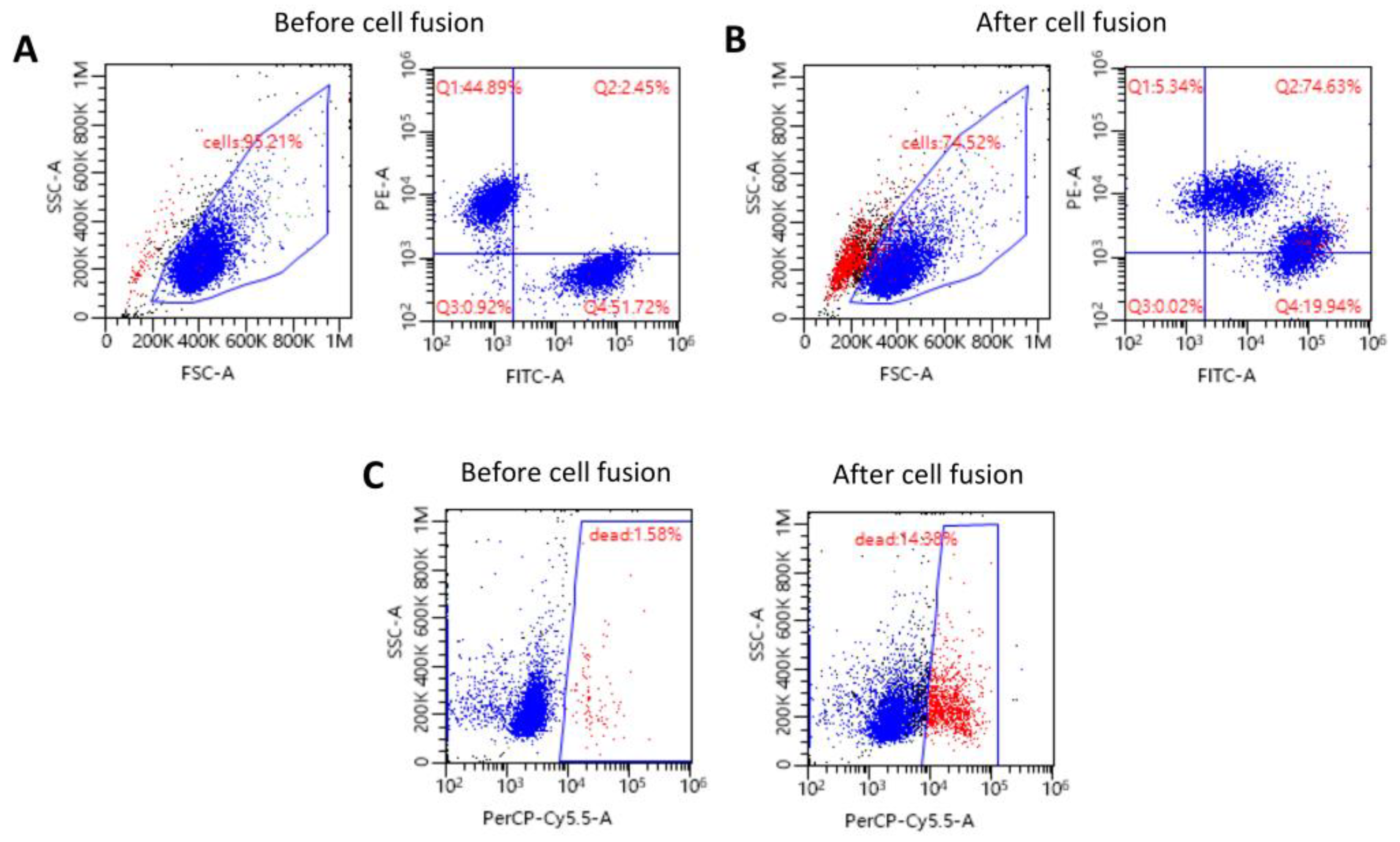
2.1. Confirmation of the Creation of High-Viability Human DEC Cells via Ex Vivo PEG-Mediated Fusion
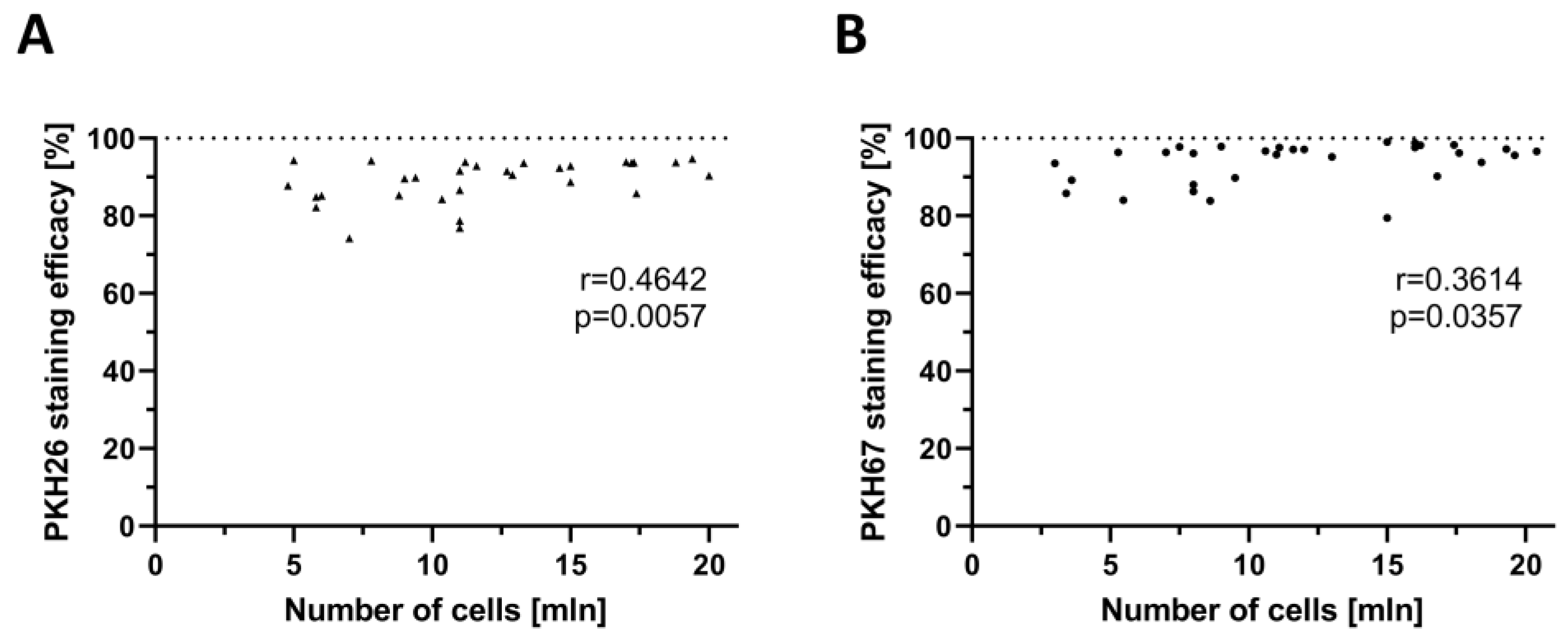
2.2. Confirmation of the Correlation between PKH Staining Efficacy and the Number of Myoblasts Used for Staining
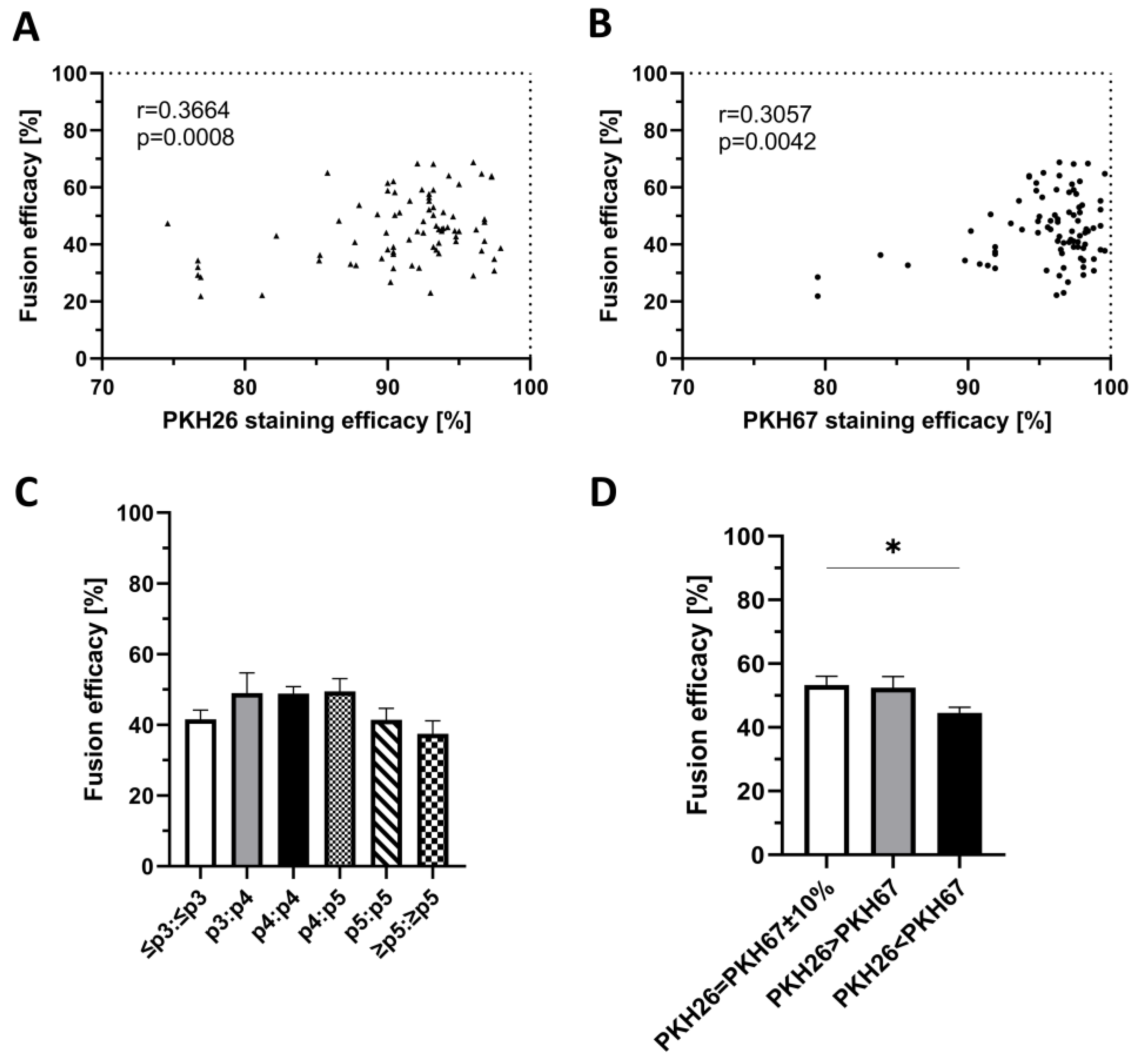
2.3. Confirmation of the Correlation between PKH Staining Efficacy and Fusion Efficacy
2.4. Confirmation of the Lack of Correlation between the Number of Myoblast Passages and Fusion Efficacy
2.5. Confirmation of the Correlation between the Ratio of Single-Stained Myoblasts before Cell Fusion and Fusion Efficacy
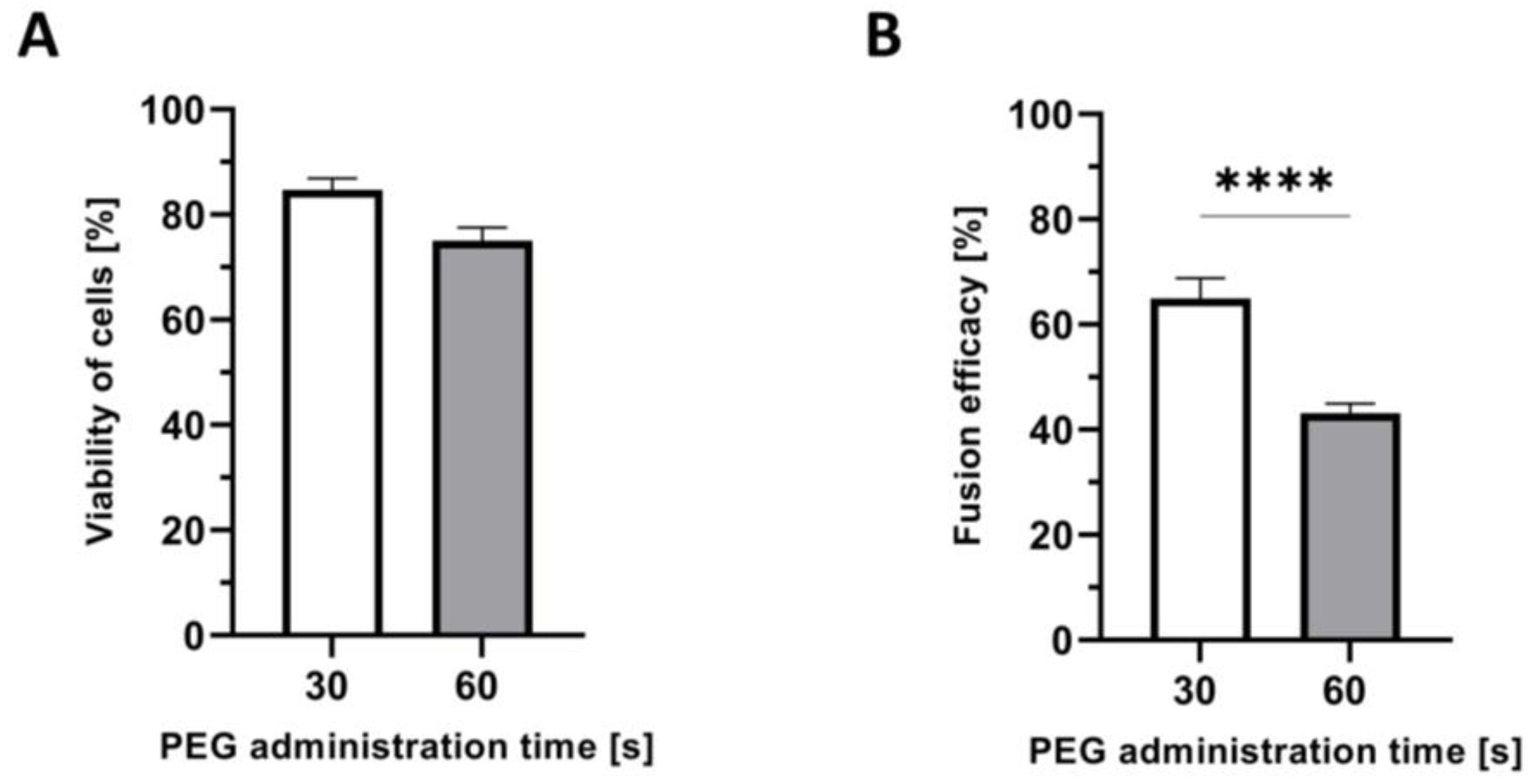
2.6. Confirmation of Shorter PEG Administration Time on Higher Myoblast Viability after Fusion
2.7. Confirmation of the Correlation between the Time of PEG Administration and Fusion Efficacy
3. Discussion
4. Materials and Methods
4.1. Myoblast Isolation
4.2. Myoblast Propagation and Passage
4.3. Myoblast Staining with PKH26 and PKH67 Dyes
4.4. Creation of Human DEC Cells via PEG-Mediated Cell Fusion Procedure and Cell Sorting
4.5. Cytometric Analysis of Fusion Efficacy and Cell Viability
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Primers 2021, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Bushby, K.; Finkel, R.; Birnkrant, D.J.; Case, L.E.; Clemens, P.R.; Cripe, L.; Kaul, A.; Kinnett, K.; McDonald, C.; Pandya, S.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010, 9, 77–93. [Google Scholar] [CrossRef] [PubMed]
- Emery, A.E. Population frequencies of inherited neuromuscular diseases—A world survey. Neuromuscul. Disord. 1991, 1, 19–29. [Google Scholar] [CrossRef]
- Crisafulli, S.; Sultana, J.; Fontana, A.; Salvo, F.; Messina, S.; Trifirò, G. Global epidemiology of Duchenne muscular dystrophy: An updated systematic review and meta-analysis. Orphanet J. Rare Dis. 2020, 15, 141. [Google Scholar] [CrossRef]
- Hoffman, E.P.; Brown, R.H.; Kunkel, L.M. Dystrophin: The protein product of the duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef]
- Kharraz, Y.; Guerra, J.; Pessina, P.; Serrano, A.L.; Muñoz-Cánoves, P. Understanding the process of fibrosis in Duchenne muscular dystrophy. Biomed. Res. Int. 2014, 2014, 965631. [Google Scholar] [CrossRef]
- Van Ruiten, H.J.; Marini Bettolo, C.; Cheetham, T.; Eagle, M.; Lochmuller, H.; Straub, V.; Bushby, K.; Guglieri, M. Why are some patients with Duchenne muscular dystrophy dying young: An analysis of causes of death in North East England. Eur. J. Paediatr. Neurol. 2016, 20, 904–909. [Google Scholar] [CrossRef] [PubMed]
- Koeks, Z.; Bladen, C.L.; Salgado, D.; van Zwet, E.; Pogoryelova, O.; McMacken, G.; Monges, S.; Foncuberta, M.E.; Kekou, K.; Kosma, K.; et al. Clinical Outcomes in Duchenne Muscular Dystrophy: A Study of 5345 Patients from the TREAT-NMD DMD Global Database. J. Neuromuscul. Dis. 2017, 4, 293–306. [Google Scholar] [CrossRef] [PubMed]
- Matthews, E.; Brassington, R.; Kuntzer, T.; Jichi, F.; Manzur, A.Y. Corticosteroids for the treatment of Duchenne muscular dystrophy. Cochrane Database Syst. Rev. 2016, 2016, CD003725. [Google Scholar] [CrossRef]
- Patterson, G.; Conner, H.; Groneman, M.; Blavo, C.; Parmar, M.S. Duchenne muscular dystrophy: Current treatment and emerging exon skipping and gene therapy approach. Eur. J. Pharmacol. 2023, 947, 175675. [Google Scholar] [CrossRef]
- Sun, C.; Shen, L.; Zhang, Z.; Xie, X. Therapeutic Strategies for Duchenne Muscular Dystrophy: An Update. Genes 2020, 11, 837. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Z.; Wu, P.; Shi, Z.M.; Xu, Y.L.; Liu, Z.J. The AAV-mediated and RNA-guided CRISPR/Cas9 system for gene therapy of DMD and BMD. Brain Dev. 2017, 39, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Happi Mbakam, C.; Lamothe, G.; Tremblay, G.; Tremblay, J.P. CRISPR-Cas9 Gene Therapy for Duchenne Muscular Dystrophy. Neurotherapeutics 2022, 19, 931–941. [Google Scholar] [CrossRef] [PubMed]
- Duan, D. Systemic AAV Micro-dystrophin Gene Therapy for Duchenne Muscular Dystrophy. Mol. Ther. 2018, 26, 2337–2356. [Google Scholar] [CrossRef] [PubMed]
- Skuk, D.; Tremblay, J.P. Cell therapy in muscular dystrophies: Many promises in mice and dogs, few facts in patients. Expert Opin. Biol. Ther. 2015, 15, 1307–1319. [Google Scholar] [CrossRef]
- Sun, C.; Serra, C.; Lee, G.; Wagner, K.R. Stem cell-based therapies for Duchenne muscular dystrophy. Exp. Neurol. 2020, 323, 113086. [Google Scholar] [CrossRef]
- Bajek, A.; Porowinska, D.; Kloskowski, T.; Brzoska, E.; Ciemerych, M.A.; Drewa, T. Cell therapy in Duchenne muscular dystrophy treatment: Clinical trials overview. Crit. Rev. Eukaryot. Gene Expr. 2015, 25, 1–11. [Google Scholar] [CrossRef]
- Sienkiewicz, D.; Kulak, W.; Okurowska-Zawada, B.; Paszko-Patej, G.; Kawnik, K. Duchenne muscular dystrophy: Current cell therapies. Ther. Adv. Neurol. Disord. 2015, 8, 166–177. [Google Scholar] [CrossRef]
- Torrente, Y.; Belicchi, M.; Marchesi, C.; D’Antona, G.; Cogiamanian, F.; Pisati, F.; Gavina, M.; Giordano, R.; Tonlorenzi, R.; Fagiolari, G.; et al. Autologous Transplantation of Muscle-Derived CD133+ Stem Cells in Duchenne Muscle Patients. Cell Transplant. 2007, 16, 563–577. [Google Scholar] [CrossRef]
- Meregalli, M.; Farini, A.; Belicchi, M.; Parolini, D.; Cassinelli, L.; Razini, P.; Sitzia, C.; Torrente, Y. Perspectives of stem cell therapy in Duchenne muscular dystrophy. FEBS J. 2013, 280, 4251–4262. [Google Scholar] [CrossRef]
- Sitzia, C.; Farini, A.; Jardim, L.; Razini, P.; Belicchi, M.; Cassinelli, L.; Villa, C.; Erratico, S.; Parolini, D.; Bella, P.; et al. Adaptive Immune Response Impairs the Efficacy of Autologous Transplantation of Engineered Stem Cells in Dystrophic Dogs. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 1949–1964. [Google Scholar] [CrossRef] [PubMed]
- Skuk, D.; Tremblay, J.P. Myoblast transplantation: The current status of a potential therapeutic tool for myopathies. J. Muscle Res. Cell Motil. 2003, 24, 287–302. [Google Scholar] [CrossRef]
- Collins, C.A.; Olsen, I.; Zammit, P.S.; Heslop, L.; Petrie, A.; Partridge, T.A.; Morgan, J.E. Stem cell function, self-renewal, and behavioral heterogeneity of cells from the adult muscle satellite cell niche. Cell 2005, 122, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Shen, M.H. Polyethylene glycol-mediated cell fusion. Methods Mol. Biol. 2006, 325, 59–66. [Google Scholar] [CrossRef]
- Kou, J.; Shen, J.; Wang, Z.; Yu, W. Advances in hybridoma preparation using electrofusion technology. Biotechnol. J. 2023, 18, e2200428. [Google Scholar] [CrossRef]
- Leroy, H.; Han, M.; Woottum, M.; Bracq, L.; Bouchet, J.; Xie, M.; Benichou, S. Virus-Mediated Cell-Cell Fusion. Int. J. Mol. Sci. 2020, 21, 9644. [Google Scholar] [CrossRef]
- Cwykiel, J.; Siemionow, M.Z. Cellular Therapy Models: Ex Vivo Chimera Model by Cell Fusion. In Plastic and Reconstructive Surgery; Siemionow, M., Ed.; Springer: London, UK, 2014; pp. 593–603. [Google Scholar] [CrossRef]
- Cwykiel, J.; Madajka-Niemeyer, M.; Siemionow, M. Development of Donor Recipient Chimeric Cells of bone marrow origin as a novel approach for tolerance induction in transplantation. Stem Cell Investig. 2021, 8, 8. [Google Scholar] [CrossRef]
- Siemionow, M.; Cwykiel, J.; Heydemann, A.; Garcia-Martinez, J.; Siemionow, K.; Szilagyi, E. Creation of Dystrophin Expressing Chimeric Cells of Myoblast Origin as a Novel Stem Cell Based Therapy for Duchenne Muscular Dystrophy. Stem Cell Rev. Rep. 2018, 14, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Siemionow, M.; Langa, P.; Brodowska, S.; Kozlowska, K.; Zalants, K.; Budzynska, K.; Heydemann, A. Long-Term Protective Effect of Human Dystrophin Expressing Chimeric (DEC) Cell Therapy on Amelioration of Function of Cardiac, Respiratory and Skeletal Muscles in Duchenne Muscular Dystrophy. Stem Cell Rev. Rep. 2022, 18, 2872–2892. [Google Scholar] [CrossRef]
- Siemionow, M.; Brodowska, S.; Langa, P.; Zalants, K.; Kozlowska, K.; Grau-Kazmierczak, W.; Heydemann, A. Long-Term Biodistribution and Safety of Human Dystrophin Expressing Chimeric Cell Therapy After Systemic-Intraosseous Administration to Duchenne Muscular Dystrophy Model. Arch. Immunol. Ther. Exp. 2022, 70, 20. [Google Scholar] [CrossRef]
- Siemionow, M.; Biegański, G.; Niezgoda, A.; Wachowiak, J.; Czarnota, J.; Siemionow, K.; Ziemiecka, A.; Sikorska, M.H.; Bożyk, K.; Heydemann, A. Safety and Efficacy of DT-DEC01 Therapy in Duchenne Muscular Dystrophy Patients: A 12-Month Follow-Up Study After Systemic Intraosseous Administration. Stem Cell Rev. Rep. 2023, 19, 2724–2740. [Google Scholar] [CrossRef] [PubMed]
- Silva, D.N.; Chrobok, M.; Ahlén, G.; Blomberg, P.; Sällberg, M.; Pasetto, A. ATMP development and pre-GMP environment in academia: A safety net for early cell and gene therapy development and manufacturing. Immunooncol Technol. 2022, 16, 100099. [Google Scholar] [CrossRef] [PubMed]
- Pearce, K.F.; Hildebrandt, M.; Greinix, H.; Scheding, S.; Koehl, U.; Worel, N.; Apperley, J.; Edinger, M.; Hauser, A.; Mischak-Weissinger, E.; et al. Regulation of advanced therapy medicinal products in Europe and the role of academia. Cytotherapy 2014, 16, 289–297. [Google Scholar] [CrossRef]
- Detela, G.; Lodge, A. EU Regulatory Pathways for ATMPs: Standard, Accelerated and Adaptive Pathways to Marketing Authorisation. Mol. Ther. Methods Clin. Dev. 2019, 13, 205–232. [Google Scholar] [CrossRef]
- Schuessler-Lenz, M.; Herberts, C.; Reischl, I.; Ruiz, S.; Celis, P.; Beuneu, C.; Kjeken, R.; Timón, M. Marketing Regulatory Oversight of Advanced Therapy Medicinal Products in Europe. Adv. Exp. Med. Biol. 2023, 1430, 1–21. [Google Scholar] [CrossRef]
- Stirm, M.; Klymiuk, N.; Nagashima, H.; Kupatt, C.; Wolf, E. Pig models for translational Duchenne muscular dystrophy research. Trends Mol. Med. 2024; ahead of print. [Google Scholar] [CrossRef]
- Guglieri, M.; Bushby, K.; McDermott, M.P.; Hart, K.A.; Tawil, R.; Martens, W.B.; Herr, B.E.; McColl, E.; Speed, C.; Wilkinson, J.; et al. Effect of Different Corticosteroid Dosing Regimens on Clinical Outcomes in Boys with Duchenne Muscular Dystrophy: A Randomized Clinical Trial. JAMA 2022, 327, 1456–1468. [Google Scholar] [CrossRef] [PubMed]
- Strehle, E.M.; Straub, V. Recent advances in the management of Duchenne muscular dystrophy. Arch. Dis. Child. 2015, 100, 1173–1177. [Google Scholar] [CrossRef]
- Ten Ham, R.M.T.; Hoekman, J.; Hövels, A.M.; Broekmans, A.W.; Leufkens, H.G.M.; Klungel, O.H. Challenges in Advanced Therapy Medicinal Product Development: A Survey among Companies in Europe. Mol. Ther. Methods Clin. Dev. 2018, 11, 121–130. [Google Scholar] [CrossRef]
- Nelson, C.E.; Hakim, C.H.; Ousterout, D.G.; Thakore, P.I.; Moreb, E.A.; Castellanos Rivera, R.M.; Madhavan, S.; Pan, X.; Ran, F.A.; Yan, W.X.; et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 2016, 351, 403–407. [Google Scholar] [CrossRef]
- Mendell, J.R.; Al-Zaidy, S.A.; Rodino-Klapac, L.R.; Goodspeed, K.; Gray, S.J.; Kay, C.N.; Boye, S.L.; Boye, S.E.; George, L.A.; Salabarria, S.; et al. Current Clinical Applications of In Vivo Gene Therapy with AAVs. Mol. Ther. 2021, 29, 464–488. [Google Scholar] [CrossRef]
- Yokota, T.; Duddy, W.; Partridge, T. Optimizing exon skipping therapies for DMD. Acta Myol. 2007, 26, 179–184. Available online: https://pubmed.ncbi.nlm.nih.gov/18646569 (accessed on 6 October 2024). [PubMed]
- Aartsma-Rus, A.; Straub, V.; Hemmings, R.; Haas, M.; Schlosser-Weber, G.; Stoyanova-Beninska, V.; Mercuri, E.; Muntoni, F.; Sepodes, B.; Vroom, E.; et al. Development of Exon Skipping Therapies for Duchenne Muscular Dystrophy: A Critical Review and a Perspective on the Outstanding Issues. Nucleic Acid. Ther. 2017, 27, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.C.; Wood, M.J.A.; Davies, K.E. Therapeutic approaches for Duchenne muscular dystrophy. Nat. Rev. Drug Discov. 2023, 22, 917–934. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Chun, S.; Asfahani, R.; Lochmüller, H.; Muntoni, F.; Morgan, J. Human skeletal muscle-derived CD133+ cells form functional satellite cells after intramuscular transplantation in immunodeficient host mice. Mol. Ther. 2014, 22, 1008–1017. [Google Scholar] [CrossRef]
- Salmaninejad, A.; Valilou, S.F.; Bayat, H.; Ebadi, N.; Daraei, A.; Yousefi, M.; Nesaei, A.; Mojarrad, M. Duchenne muscular dystrophy: An updated review of common available therapies. Int. J. Neurosci. 2018, 128, 854–864. [Google Scholar] [CrossRef]
- Yamanaka, S. Pluripotent Stem Cell-Based Cell Therapy-Promise and Challenges. Cell Stem Cell 2020, 27, 523–531. [Google Scholar] [CrossRef]
- Skuk, D.; Tremblay, J.P. Confirmation of donor-derived dystrophin in a duchenne muscular dystrophy patient allotransplanted with normal myoblasts. Muscle Nerve 2016, 54, 979–981. [Google Scholar] [CrossRef]
- Gussoni, E.; Blau, H.M.; Kunkel, L.M. The fate of individual myoblasts after transplantation into muscles of DMD patients. Nat. Med. 1997, 3, 970–977. [Google Scholar] [CrossRef]
- Skuk, D.; Goulet, M.; Roy, B.; Piette, V.; Côté, C.H.; Chapdelaine, P.; Hogrel, J.Y.; Paradis, M.; Bouchard, J.P.; Sylvain, M.; et al. First test of a “high-density injection” protocol for myogenic cell transplantation throughout large volumes of muscles in a Duchenne muscular dystrophy patient: Eighteen months follow-up. Neuromuscul. Disord. 2007, 17, 38–46. [Google Scholar] [CrossRef]
- Gussoni, E.; Bennett, R.R.; Muskiewicz, K.R.; Meyerrose, T.; Nolta, J.A.; Gilgoff, I.; Stein, J.; Chan, Y.M.; Lidov, H.G.; Bönnemann, C.G.; et al. Long-term persistence of donor nuclei in a Duchenne muscular dystrophy patient receiving bone marrow transplantation. J. Clin. Investig. 2002, 110, 807–814. [Google Scholar] [CrossRef]
- Rajput, B.S.; Chakrabarti, S.K.; Dongare, V.S.; Ramirez, C.M.; Deb, K.D. Human Umbilical Cord Mesenchymal Stem Cells in the Treatment of Duchenne Muscular Dystrophy: Safety and Feasibility Study in India. J. Stem Cells 2015, 10, 141–156. Available online: http://www.ncbi.nlm.nih.gov/pubmed/27125141 (accessed on 6 October 2024). [PubMed]
- Ogle, B.M.; Cascalho, M.; Platt, J.L. Biological implications of cell fusion. Nat. Rev. Mol. Cell Biol. 2005, 6, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Dittmar, T.; Zänker, K.S. Introduction. In Cell Fusion in Health and Disease. II: Cell Fusion in Disease; Advances in Experimental Medicine and Biology; Springer: Dordrecht, The Netherlands, 2011; Volume 714, pp. 1–3. [Google Scholar] [CrossRef]
- Iosilevskii, Y.; Podbilewicz, B. Programmed cell fusion in development and homeostasis. Curr. Top. Dev. Biol. 2021, 144, 215–244. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.Y.; Tai, J.A.; Sakaguchi, Y.; Nishikawa, T.; Hirayama, Y.; Yamashita, K. Enhancement of polyethylene glycol-cell fusion efficiency by novel application of transient pressure using a jet injector. FEBS Open Bio 2023, 13, 478–489. [Google Scholar] [CrossRef]
- Rems, L.; Ušaj, M.; Kandušer, M.; Reberšek, M.; Miklavčič, D.; Pucihar, G. Cell electrofusion using nanosecond electric pulses. Sci. Rep. 2013, 3, 3382. [Google Scholar] [CrossRef]
- Jaroszeski, M.J.; Gilbert, R.; Fallon, P.G.; Heller, R. Mechanically facilitated cell-cell electrofusion. Biophys J. 1994, 67, 1574–1581. [Google Scholar] [CrossRef]
- Kandušer, M.; Ušaj, M. Cell electrofusion: Past and future perspectives for antibody production and cancer cell vaccines. Expert Opin. Drug Deliv. 2014, 11, 1885–1898. [Google Scholar] [CrossRef]
- Zimmermann, U.; Vienken, J. Electric field-induced cell-to-cell fusion. J. Membr. Biol. 1982, 67, 165–182. [Google Scholar] [CrossRef]
- Kutton, S. The mechanism of virus-induced cell fusion. Micron 1978, 9, 133–154. [Google Scholar] [CrossRef]
- Okada, Y. Sendai virus-induced cell fusion. Methods Enzymol. 1993, 221, 18–41. [Google Scholar] [CrossRef]
- Duelli, D.M.; Padilla-Nash, H.M.; Berman, D.; Murphy, K.M.; Ried, T.; Lazebnik, Y. A virus causes cancer by inducing massive chromosomal instability through cell fusion. Curr. Biol. 2007, 17, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Kurooka, M.; Kaneda, Y. Inactivated Sendai virus particles eradicate tumors by inducing immune responses through blocking regulatory T cells. Cancer Res. 2007, 67, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Pontecorvo, G. Production of mammalian somatic cell hybrids by means of polyethylene glycol treatment. Somat. Cell Genet. 1975, 1, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Davidson, R.L.; Gerald, P.S. Improved techniques for the induction of mammalian cell hybridization by polyethylene glycol. Somat. Cell Genet. 1976, 2, 165–176. [Google Scholar] [CrossRef]
- Wojcieszyn, J.W.; Schlegel, R.A.; Lumley-Sapanski, K.; Jacobson, K.A. Studies on the mechanism of polyethylene glycol-mediated cell fusion using fluorescent membrane and cytoplasmic probes. J. Cell Biol. 1983, 96, 151–159. [Google Scholar] [CrossRef]
- Lentz, B.R.; Lee, J.K. Poly(ethylene glycol) (PEG)-mediated fusion between pure lipid bilayers: A mechanism in common with viral fusion and secretory vesicle release? Mol. Membr. Biol. 1999, 16, 279–296. [Google Scholar] [CrossRef]
- Mitra, S.; Tomar, P.C. Hybridoma technology; advancements, clinical significance, and future aspects. J. Genet. Eng. Biotechnol. 2021, 19, 159. [Google Scholar] [CrossRef]
- Ford, J.W.; Welling, T.H., 3rd; Stanley, J.C.; Messina, L.M. PKH26 and 125I-PKH95: Characterization and efficacy as labels for in vitro and in vivo endothelial cell localization and tracking. J. Surg. Res. 1996, 62, 23–28. [Google Scholar] [CrossRef]
- Li, J.; Wu, Z.; Zhao, L.; Liu, Y.; Su, Y.; Gong, X.; Liu, F.; Zhang, L. The heterogeneity of mesenchymal stem cells: An important issue to be addressed in cell therapy. Stem Cell Res. Ther. 2023, 14, 381. [Google Scholar] [CrossRef]
- Rozwadowska, N.; Sikorska, M.; Bozyk, K.; Jarosinska, K.; Cieciuch, A.; Brodowska, S.; Andrzejczak, M.; Siemionow, M. Optimization of human myoblasts culture under different media conditions for application in the in vitro studies. Am. J. Stem Cells 2022, 11, 1–11. [Google Scholar]
- Liu, A.; Yang, G.; Liu, Y.; Liu, T. Research progress in membrane fusion-based hybrid exosomes for drug delivery systems. Front. Bioeng. Biotechnol. 2022, 10, 939441. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, A.; Watanabe, S.; Goel, I.; Ishihara, K.; Ekdahl, K.N.; Nilsson, B.; Teramura, Y. Promotion of cell membrane fusion by cell-cell attachment through cell surface modification with functional peptide-PEG-lipids. Biomaterials 2020, 253, 120113. [Google Scholar] [CrossRef]
- Nakajima, N.; Ikada, Y. Effects of Concentration, Molecular Weight, and Exposure Time of Poly(ethylene glycol) on Cell Fusion. Polym. J. 1995, 27, 211–219. [Google Scholar] [CrossRef]
- Messineo, E.; Pollins, A.; Thayer, W. Optimization and evaluation of an in vitro model of PEG-mediated fusion of nerve cell bodies. J. Clin. Neurosci. 2019, 63, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Jankowski, R.J.; Haluszczak, C.; Trucco, M.; Huard, J. Flow cytometric characterization of myogenic cell populations obtained via the preplate technique: Potential for rapid isolation of muscle-derived stem cells. Hum. Gene Ther. 2001, 12, 619–628. [Google Scholar] [CrossRef]
- Gharaibeh, B.; Lu, A.; Tebbets, J.; Zheng, B.; Feduska, J.; Crisan, M.; Péault, B.; Cummins, J.; Huard, J. Isolation of a slowly adhering cell fraction containing stem cells from murine skeletal muscle by the preplate technique. Nat. Protoc. 2008, 3, 1501–1509. [Google Scholar] [CrossRef]
- Chan, Y.H. Biostatistics 104: Correlational analysis. Singap. Med. J. 2003, 44, 614–619. [Google Scholar] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Budzynska, K.; Bozyk, K.T.; Jarosinska, K.; Ziemiecka, A.; Siemionow, K.; Siemionow, M. Developing Advanced Chimeric Cell Therapy for Duchenne Muscular Dystrophy. Int. J. Mol. Sci. 2024, 25, 10947. https://doi.org/10.3390/ijms252010947
Budzynska K, Bozyk KT, Jarosinska K, Ziemiecka A, Siemionow K, Siemionow M. Developing Advanced Chimeric Cell Therapy for Duchenne Muscular Dystrophy. International Journal of Molecular Sciences. 2024; 25(20):10947. https://doi.org/10.3390/ijms252010947
Chicago/Turabian StyleBudzynska, Katarzyna, Katarzyna T. Bozyk, Klaudia Jarosinska, Anna Ziemiecka, Krzysztof Siemionow, and Maria Siemionow. 2024. "Developing Advanced Chimeric Cell Therapy for Duchenne Muscular Dystrophy" International Journal of Molecular Sciences 25, no. 20: 10947. https://doi.org/10.3390/ijms252010947
APA StyleBudzynska, K., Bozyk, K. T., Jarosinska, K., Ziemiecka, A., Siemionow, K., & Siemionow, M. (2024). Developing Advanced Chimeric Cell Therapy for Duchenne Muscular Dystrophy. International Journal of Molecular Sciences, 25(20), 10947. https://doi.org/10.3390/ijms252010947