

NF-κB in Alzheimer’s Disease: Friend or Foe? Opposite Functions in Neurons and Glial Cells

Abstract
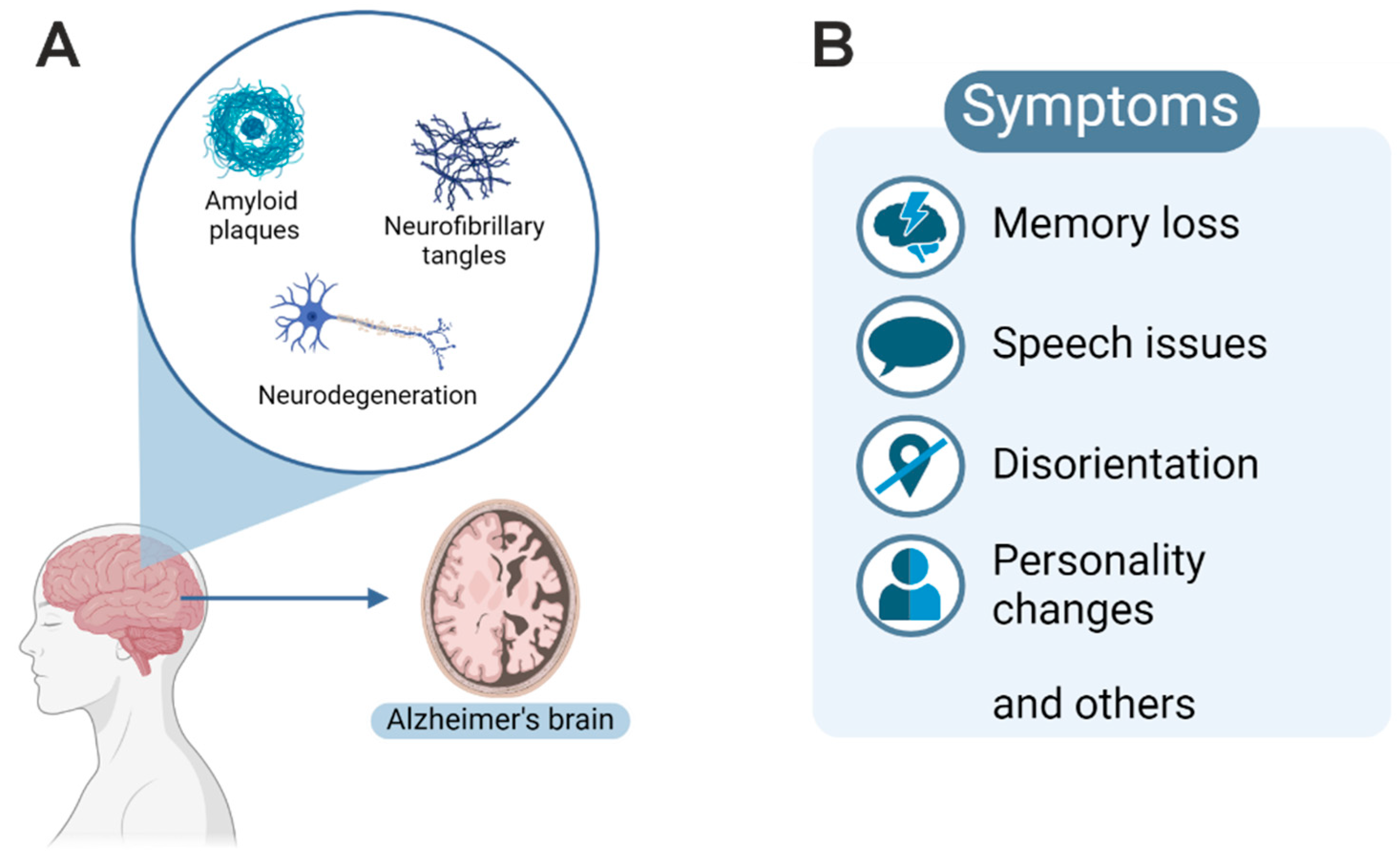
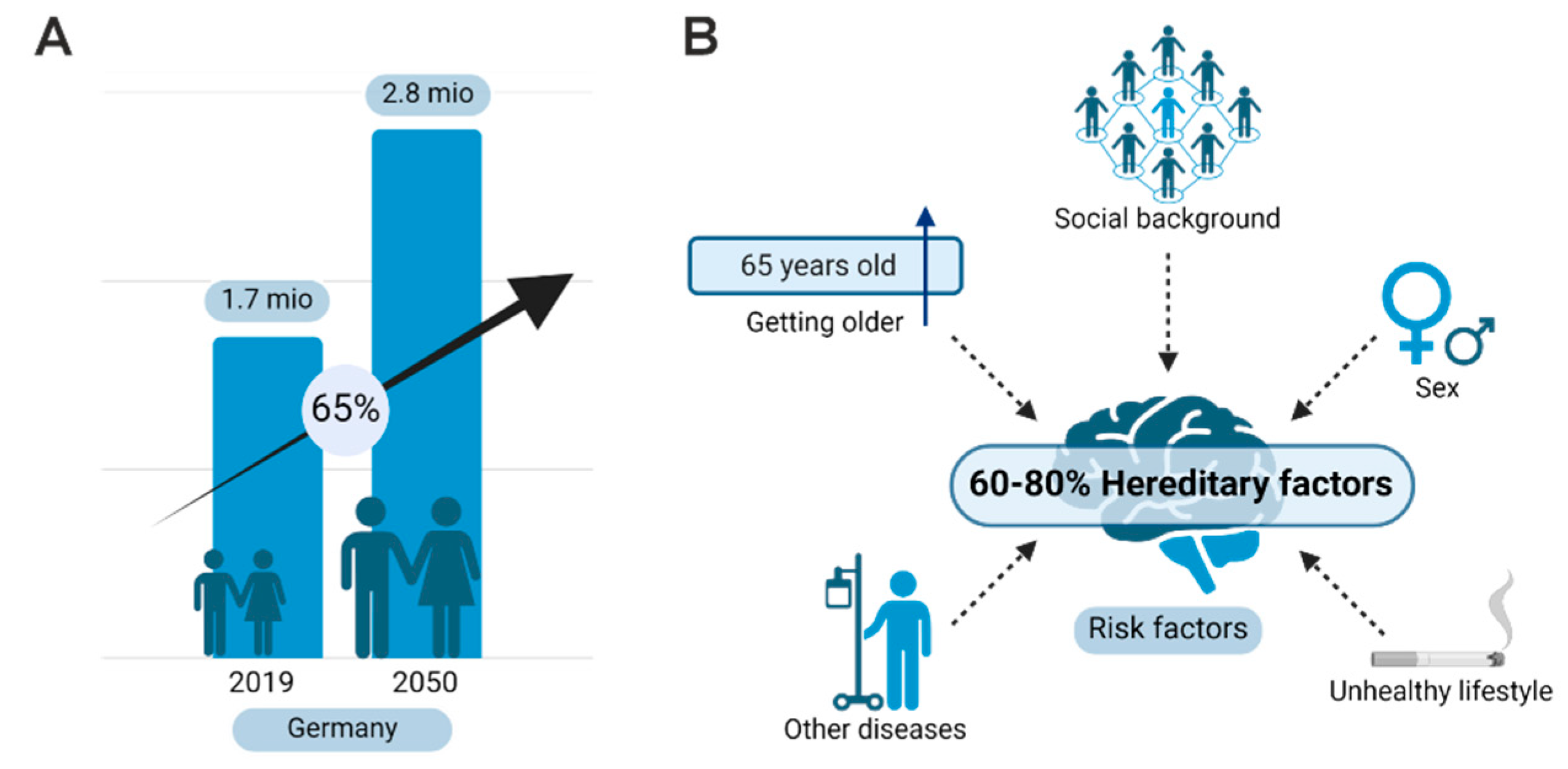
1. Introduction: Alzheimer’s Disease
2. Definition of Disease-Relevant Bits and Pieces
3. Neuropathological Diagnosis
4. Biomarkers and Genetics
4.1. Biomarkers
4.2. Genetics
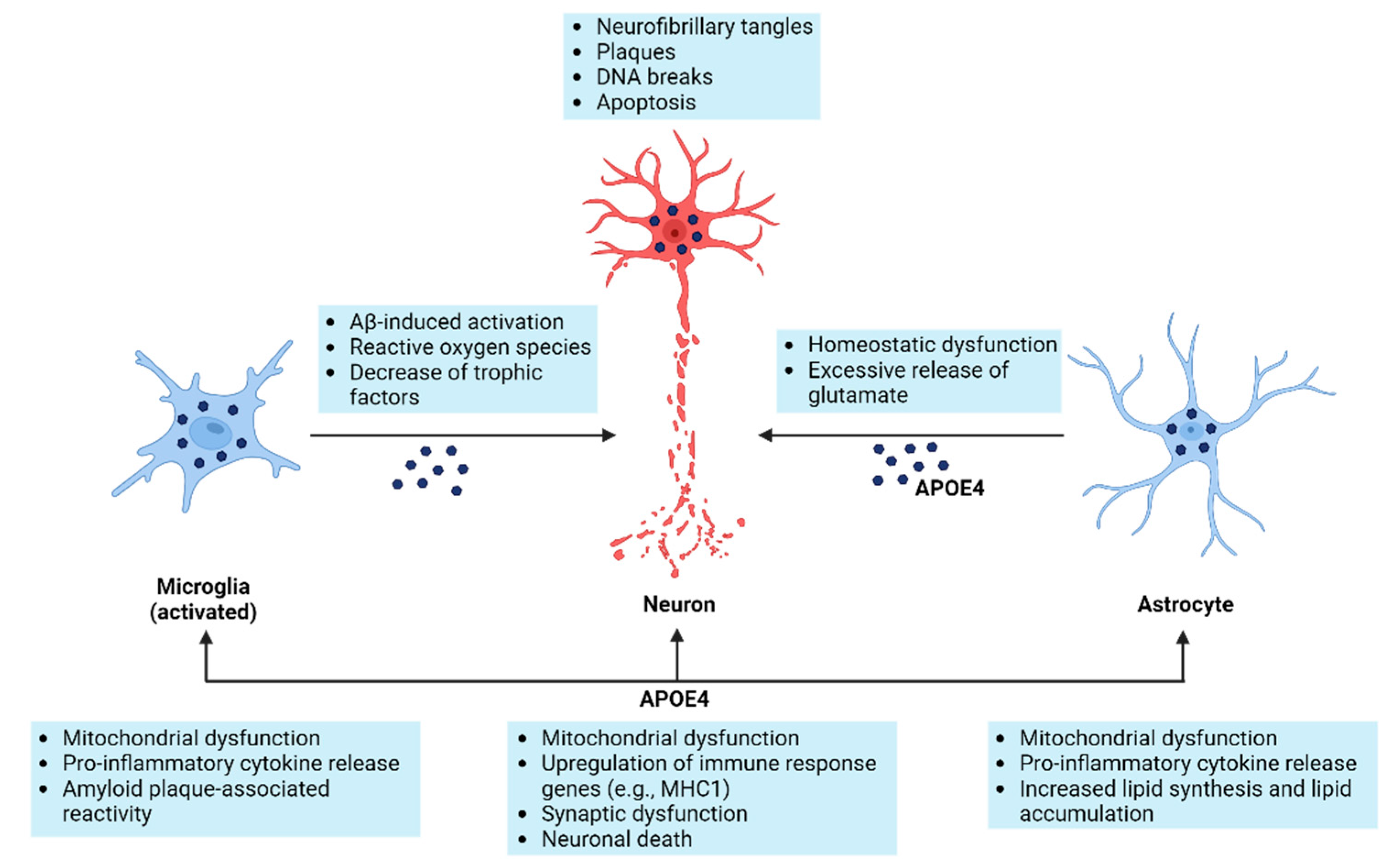
5. Cells Involved in Alzheimer’s Pathology
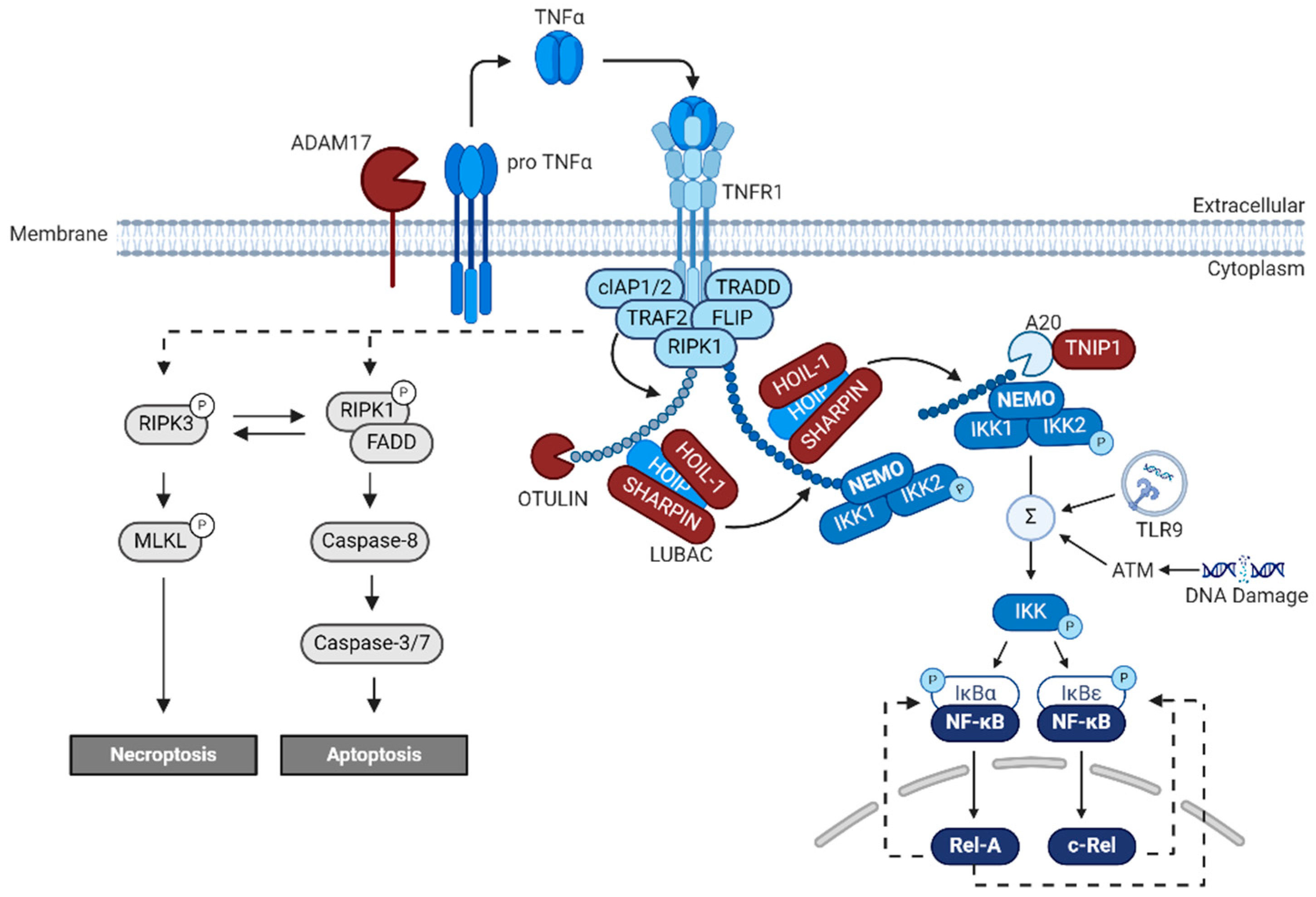
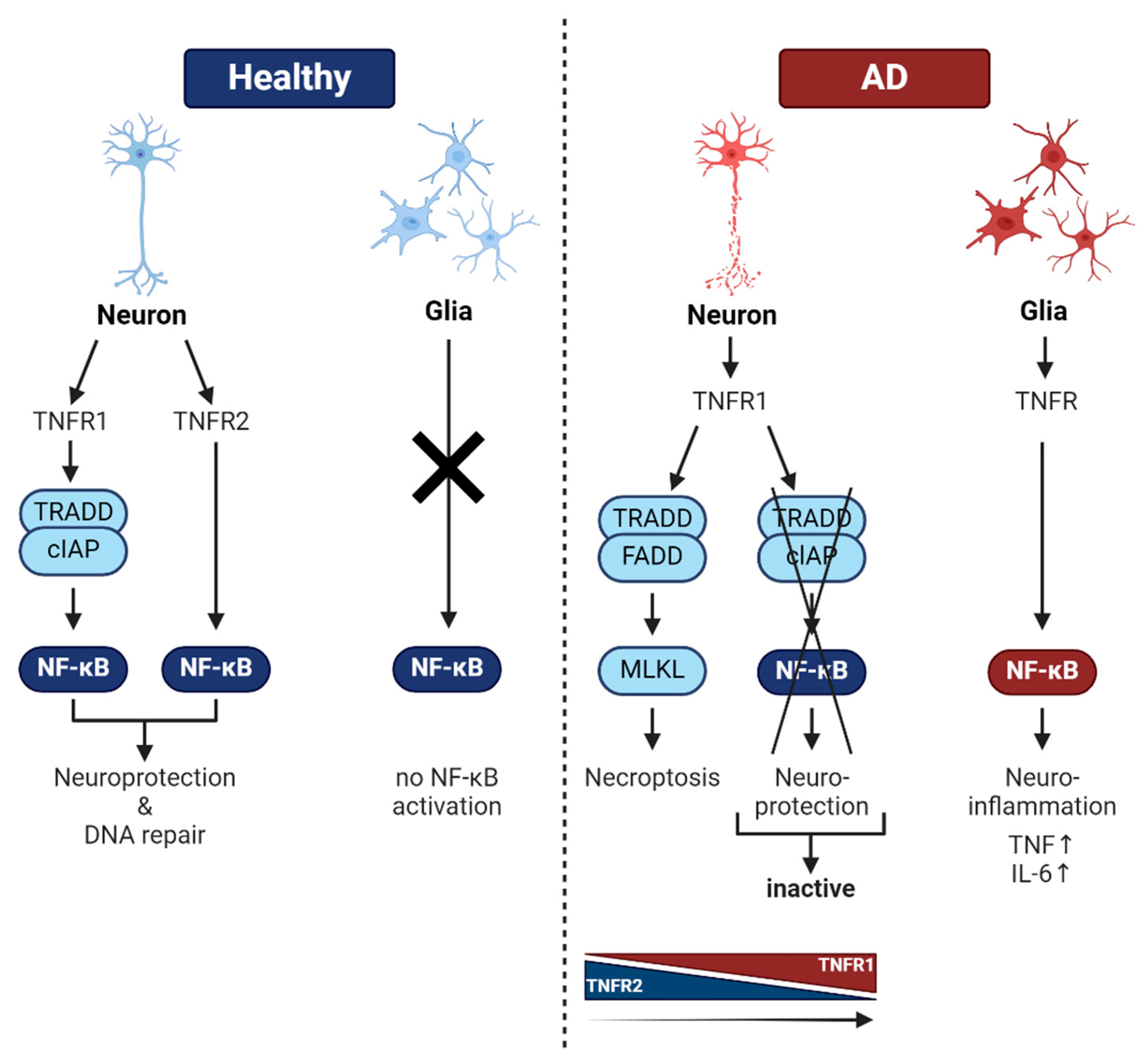
6. NF-κB
7. Small Molecule Drugs and Biologicals
7.1. Small Molecule Drugs
7.2. Biologicals
8. Mouse Models for NF-κB Function in Learning and Memory and AD
9. DNA Double-Strand Breaks in AD
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Alzheimer, A. über eigenartige Krankheitsfälle des späteren Alters. Z Für Gesamte Neurol. Psychiatr. 1911, 4, 356–385. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological Alterations in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Arnsten, A.F.T.; Datta, D.; Del Tredici, K.; Braak, H. Hypothesis: Tau pathology is an initiating factor in sporadic Alzheimer’s disease. Alzheimers Dement. J. Alzheimers Assoc. 2021, 17, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.R.; Rüb, U.; Orantes, M.; Braak, H. Phases of Aβ-deposition in the human brain and its relevance for the development of AD. Neurology 2002, 58, 1791–1800. [Google Scholar] [CrossRef]
- Brown, G.C.; Heneka, M.T. The endotoxin hypothesis of Alzheimer’s disease. Mol. Neurodegener. 2024, 19, 30. [Google Scholar] [CrossRef]
- Zhan, X.; Stamova, B.; Jin, L.-W.; DeCarli, C.; Phinney, B.; Sharp, F.R. Gram-negative bacterial molecules associate with Alzheimer disease pathology. Neurology 2016, 87, 2324–2332. [Google Scholar] [CrossRef]
- Klunk, W.E.; Engler, H.; Nordberg, A.; Wang, Y.; Blomqvist, G.; Holt, D.P.; Bergström, M.; Savitcheva, I.; Huang, G.; Estrada, S.; et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann. Neurol. 2004, 55, 306–319. [Google Scholar] [CrossRef]
- Palmqvist, S.; Tideman, P.; Mattsson-Carlgren, N.; Schindler, S.E.; Smith, R.; Ossenkoppele, R.; Calling, S.; West, T.; Monane, M.; Verghese, P.B.; et al. Blood Biomarkers to Detect Alzheimer Disease in Primary Care and Secondary Care. JAMA 2024, 332, 1245–1257. [Google Scholar] [CrossRef]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef]
- Piguet, O.; Double, K.L.; Kril, J.J.; Harasty, J.; Macdonald, V.; McRitchie, D.A.; Halliday, G.M. White matter loss in healthy ageing: A postmortem analysis. Neurobiol. Aging 2009, 30, 1288–1295. [Google Scholar] [CrossRef]
- Dickerson, B.C.; Stoub, T.R.; Shah, R.C.; Sperling, R.A.; Killiany, R.J.; Albert, M.S.; Hyman, B.T.; Blacker, D.; deToledo-Morrell, L. Alzheimer-signature MRI biomarker predicts AD dementia in cognitively normal adults. Neurology 2011, 76, 1395–1402. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K. Neuroanatomy and pathology of sporadic Alzheimer’s disease. In Advances in Anatomy, Embryology and Cell Biology; Springer: New York, NY, USA, 2015; Volume 215, pp. 1–162. [Google Scholar]
- Thal, D.R.; Walter, J.; Saido, T.C.; Fändrich, M. Neuropathology and biochemistry of Aβ and its aggregates in Alzheimer’s disease. Acta Neuropathol. 2015, 129, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Yang, D.-S.; Goulbourne, C.N.; Im, E.; Stavrides, P.; Pensalfini, A.; Chan, H.; Bouchet-Marquis, C.; Bleiwas, C.; Berg, M.J.; et al. Faulty autolysosome acidification in Alzheimer’s disease mouse models induces autophagic build-up of Aβ in neurons, yielding senile plaques. Nat. Neurosci. 2022, 25, 688–701. [Google Scholar] [CrossRef]
- Terai, K.; Matsuo, A.; McGeer, E.G.; McGeer, P.L. Enhancement of immunoreactivity for NF-kappa B in human cerebral infarctions. Brain Res. 1996, 739, 343–349. [Google Scholar] [CrossRef]
- Kaltschmidt, B.; Uherek, M.; Volk, B.; Baeuerle, P.A.; Kaltschmidt, C. Transcription factor NF-kappaB is activated in primary neurons by amyloid beta peptides and in neurons surrounding early plaques from patients with Alzheimer disease. Proc. Natl. Acad. Sci. USA 1997, 94, 2642–2647. [Google Scholar] [CrossRef]
- Boissière, F.; Hunot, S.; Faucheux, B.; Duyckaerts, C.; Hauw, J.-J.; Agid, Y.; Hirsch, E.C. Nuclear translocation of NF-κB in cholinergic neurons of patients with Alzheimer’s disease. NeuroReport 1997, 8, 2849–2852. [Google Scholar] [CrossRef]
- Kaltschmidt, B.; Uherek, M.; Wellmann, H.; Volk, B.; Kaltschmidt, C. Inhibition of NF-κB potentiates amyloid β-mediated neuronal apoptosis. Proc. Natl. Acad. Sci. USA 1999, 96, 9409–9414. [Google Scholar] [CrossRef]
- Yoshiyama, Y.; Arai, K.; Hattori, T. Enhanced expression of I-kappaB with neurofibrillary pathology in Alzheimer’s disease. NeuroReport 2001, 12, 2641–2645. [Google Scholar] [CrossRef]
- Jong Huat, T.; Camats-Perna, J.; Newcombe, E.A.; Onraet, T.; Campbell, D.; Sucic, J.T.; Martini, A.; Forner, S.; Mirzaei, M.; Poon, W.; et al. The impact of astrocytic NF-κB on healthy and Alzheimer’s disease brains. Sci. Rep. 2024, 14, 14305. [Google Scholar] [CrossRef]
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Lemaire, H.G.; Unterbeck, A.; Salbaum, J.M.; Masters, C.L.; Grzeschik, K.H.; Multhaup, G.; Beyreuther, K.; Müller-Hill, B. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 1987, 325, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Nichols, E.; Steinmetz, J.D.; Vollset, S.E.; Fukutaki, K.; Chalek, J.; Abd-Allah, F.; Abdoli, A.; Abualhasan, A.; Abu-Gharbieh, E.; Akram, T.T.; et al. Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: An analysis for the Global Burden of Disease Study 2019. Lancet Public Health 2022, 7, e105–e125. [Google Scholar] [CrossRef]
- Bellenguez, C.; Küçükali, F.; Jansen, I.E.; Kleineidam, L.; Moreno-Grau, S.; Amin, N.; Naj, A.C.; Campos-Martin, R.; Grenier-Boley, B.; Andrade, V.; et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 2022, 54, 412–436. [Google Scholar] [CrossRef] [PubMed]
- Campion, D.; Dumanchin, C.; Hannequin, D.; Dubois, B.; Belliard, S.; Puel, M.; Thomas-Anterion, C.; Michon, A.; Martin, C.; Charbonnier, F.; et al. Early-onset autosomal dominant Alzheimer disease: Prevalence, genetic heterogeneity, and mutation spectrum. Am. J. Hum. Genet. 1999, 65, 664–670. [Google Scholar] [CrossRef]
- Wiseman, F.K.; Al-Janabi, T.; Hardy, J.; Karmiloff-Smith, A.; Nizetic, D.; Tybulewicz, V.L.J.; Fisher, E.M.C.; Strydom, A. A genetic cause of Alzheimer disease: Mechanistic insights from Down syndrome. Nat. Rev. Neurosci. 2015, 16, 564–574. [Google Scholar] [CrossRef]
- Kaltschmidt, B.; Helweg, L.P.; Greiner, J.F.W.; Kaltschmidt, C. NF-κB in neurodegenerative diseases: Recent evidence from human genetics. Front. Mol. Neurosci. 2022, 15, 954541. [Google Scholar] [CrossRef]
- Jansen, I.E.; Savage, J.E.; Watanabe, K.; Bryois, J.; Williams, D.M.; Steinberg, S.; Sealock, J.; Karlsson, I.K.; Hägg, S.; Athanasiu, L.; et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 2019, 51, 404–413. [Google Scholar] [CrossRef]
- Duchateau, L.; Wawrzyniak, N.; Sleegers, K. The ABC’s of Alzheimer risk gene ABCA7. Alzheimers Dement. 2024, 20, 3629–3648. [Google Scholar] [CrossRef]
- Aikawa, T.; Ren, Y.; Yamazaki, Y.; Tachibana, M.; Johnson, M.R.; Anderson, C.T.; Martens, Y.A.; Holm, M.-L.; Asmann, Y.W.; Saito, T.; et al. ABCA7 haplodeficiency disturbs microglial immune responses in the mouse brain. Proc. Natl. Acad. Sci. USA 2019, 116, 23790–23796. [Google Scholar] [CrossRef]
- Huang, J.; Su, B.; Karhunen, V.; Gill, D.; Zuber, V.; Ahola-Olli, A.; Palaniswamy, S.; Auvinen, J.; Herzig, K.-H.; Keinänen-Kiukaanniemi, S.; et al. Inflammatory Diseases, Inflammatory Biomarkers, and Alzheimer Disease. Neurology 2023, 100, e568–e581. [Google Scholar] [CrossRef] [PubMed]
- Wang, T. TNF-alpha G308A Polymorphism and the Susceptibility to Alzheimer’s Disease: An Updated Meta-analysis. Arch. Med. Res. 2015, 46, 24–30.e1. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, A.; Cacabelos, R.; Sanpedro, C.; García-Fantini, M.; Aleixandre, M. Serum TNF-alpha levels are increased and correlate negatively with free IGF-I in Alzheimer disease. Neurobiol. Aging 2007, 28, 533–536. [Google Scholar] [CrossRef] [PubMed]
- Tobinick, E.; Gross, H.; Weinberger, A.; Cohen, H. TNF-alpha modulation for treatment of Alzheimer’s disease: A 6-month pilot study. MedGenMed Medscape Gen. Med. 2006, 8, 25. [Google Scholar]
- Diniz, B.S.; Teixeira, A.L.; Ojopi, E.B.; Talib, L.L.; Mendonça, V.A.; Gattaz, W.F.; Forlenza, O.V. Higher serum sTNFR1 level predicts conversion from mild cognitive impairment to Alzheimer’s disease. J. Alzheimers Dis. JAD 2010, 22, 1305–1311. [Google Scholar] [CrossRef]
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; van der Lee, S.J.; Amlie-Wolf, A.; et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 2019, 51, 414–430. [Google Scholar] [CrossRef]
- Arnaud, L.; Benech, P.; Greetham, L.; Stephan, D.; Jimenez, A.; Jullien, N.; García-González, L.; Tsvetkov, P.O.; Devred, F.; Sancho-Martinez, I.; et al. APOE4 drives inflammation in human astrocytes via TAGLN3 repression and NF-κB activation. Cell Rep. 2022, 40, 111200. [Google Scholar] [CrossRef]
- Song, W.; Lahiri, D.K. Functional identification of the promoter of the gene encoding the Rhesus monkey β-amyloid precursor protein. Gene 1998, 217, 165–176. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Blumenfeld, J.; Yip, O.; Kim, M.J.; Huang, Y. Cell type-specific roles of APOE4 in Alzheimer disease. Nat. Rev. Neurosci. 2024, 25, 91–110. [Google Scholar] [CrossRef]
- Micheau, O.; Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Piras, S.; Furfaro, A.L.; Domenicotti, C.; Traverso, N.; Marinari, U.M.; Pronzato, M.A.; Nitti, M. RAGE Expression and ROS Generation in Neurons: Differentiation versus Damage. Oxid. Med. Cell. Longev. 2016, 2016, 9348651. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, K.; Mattson, M.P. The transcription factor NF-kappaB mediates increases in calcium currents and decreases in NMDA- and AMPA/kainate-induced currents induced by tumor necrosis factor-alpha in hippocampal neurons. J. Neurochem. 1998, 70, 1876–1886. [Google Scholar] [CrossRef] [PubMed]
- Guerrini, L.; Blasi, F.; Denis-Donini, S. Synaptic activation of NF-kappa B by glutamate in cerebellar granule neurons in vitro. Proc. Natl. Acad. Sci. USA 1995, 92, 9077–9081. [Google Scholar] [CrossRef] [PubMed]
- Kaltschmidt, C.; Kaltschmidt, B.; Baeuerle, P.A. Stimulation of ionotropic glutamate receptors activates transcription factor NF-kappa B in primary neurons. Proc. Natl. Acad. Sci. USA 1995, 92, 9618–9622. [Google Scholar] [CrossRef]
- Meffert, M.K.; Chang, J.M.; Wiltgen, B.J.; Fanselow, M.S.; Baltimore, D. NF-kappa B functions in synaptic signaling and behavior. Nat. Neurosci. 2003, 6, 1072–1078. [Google Scholar] [CrossRef]
- Ghosh, G.; van Duyne, G.; Ghosh, S.; Sigler, P.B. Structure of NF-kappa B p50 homodimer bound to a kappa B site. Nature 1995, 373, 303–310. [Google Scholar] [CrossRef]
- Kaltschmidt, B.; Greiner, J.F.W.; Kadhim, H.M.; Kaltschmidt, C. Subunit-Specific Role of NF-κB in Cancer. Biomedicines 2018, 6, 44. [Google Scholar] [CrossRef]
- Zhao, M.; Chauhan, P.; Sherman, C.A.; Singh, A.; Kaileh, M.; Mazan-Mamczarz, K.; Ji, H.; Joy, J.; Nandi, S.; De, S.; et al. NF-κB subunits direct kinetically distinct transcriptional cascades in antigen receptor-activated B cells. Nat. Immunol. 2023, 24, 1552–1564. [Google Scholar] [CrossRef]
- Fricke, F.; Malkusch, S.; Wangorsch, G.; Greiner, J.F.; Kaltschmidt, B.; Kaltschmidt, C.; Widera, D.; Dandekar, T.; Heilemann, M. Quantitative single-molecule localization microscopy combined with rule-based modeling reveals ligand-induced TNF-R1 reorganization toward higher-order oligomers. Histochem. Cell Biol. 2014, 142, 91–101. [Google Scholar] [CrossRef]
- Haas, T.L.; Emmerich, C.H.; Gerlach, B.; Schmukle, A.C.; Cordier, S.M.; Rieser, E.; Feltham, R.; Vince, J.; Warnken, U.; Wenger, T.; et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol. Cell 2009, 36, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Iwai, K. LUBAC-mediated linear ubiquitination in tissue homeostasis and disease. J. Biochem. 2023, 174, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Oda, H.; Manthiram, K.; Chavan, P.P.; Rieser, E.; Veli, Ö.; Kaya, Ö.; Rauch, C.; Nakabo, S.; Kuehn, H.S.; Swart, M.; et al. Biallelic human SHARPIN loss of function induces autoinflammation and immunodeficiency. Nat. Immunol. 2024, 25, 764–777. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, F.; Nishimasu, H.; Ishitani, R.; Goto, E.; Noguchi, T.; Mio, K.; Kamei, K.; Ma, A.; Iwai, K.; Nureki, O. Specific recognition of linear polyubiquitin by A20 zinc finger 7 is involved in NF-κB regulation. EMBO J. 2012, 31, 3856–3870. [Google Scholar] [CrossRef]
- Zhou, J.; Wu, R.; High, A.A.; Slaughter, C.A.; Finkelstein, D.; Rehg, J.E.; Redecke, V.; Häcker, H. A20-binding inhibitor of NF-κB (ABIN1) controls Toll-like receptor-mediated CCAAT/enhancer-binding protein β activation and protects from inflammatory disease. Proc. Natl. Acad. Sci. USA 2011, 108, E998-1006. [Google Scholar] [CrossRef]
- Wajant, H.; Siegmund, D. TNFR1 and TNFR2 in the Control of the Life and Death Balance of Macrophages. Front. Cell Dev. Biol. 2019, 7, 91. [Google Scholar] [CrossRef]
- Asimakidou, E.; Reynolds, R.; Barron, A.M.; Lo, C.H. Autolysosomal acidification impairment as a mediator for TNFR1 induced neuronal necroptosis in Alzheimer’s disease. Neural Regen. Res. 2024, 19, 1869–1870. [Google Scholar] [CrossRef]
- Jayaraman, A.; Htike, T.T.; James, R.; Picon, C.; Reynolds, R. TNF-mediated neuroinflammation is linked to neuronal necroptosis in Alzheimer’s disease hippocampus. Acta Neuropathol. Commun. 2021, 9, 159. [Google Scholar] [CrossRef]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 years of NF-κB: A blossoming of relevance to human pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef]
- Kaltschmidt, B.; Witte, K.E.; Greiner, J.F.W.; Weissinger, F.; Kaltschmidt, C. Targeting NF-κB Signaling in Cancer Stem Cells: A Narrative Review. Biomedicines 2022, 10, 261. [Google Scholar] [CrossRef]
- Shorey, C.L.; Mulla, R.T.; Mielke, J.G. The effects of synthetic glucocorticoid treatment for inflammatory disease on brain structure, function, and dementia outcomes: A systematic review. Brain Res. 2023, 1798, 148157. [Google Scholar] [CrossRef] [PubMed]
- Nerius, M.; Haenisch, B.; Gomm, W.; Doblhammer, G.; Schneider, A. Glucocorticoid Therapy is Associated with a Lower Risk of Dementia. J. Alzheimers Dis. 2020, 73, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Lam, Y.A.; Pickart, C.M.; Alban, A.; Landon, M.; Jamieson, C.; Ramage, R.; Mayer, R.J.; Layfield, R. Inhibition of the ubiquitin-proteasome system in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 9902–9906. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.-C.; Augur, Z.M.; Arbery, M.; Ashour, N.; Barrett, K.; Pearse, R.V., II; Tio, E.S.; Duong, D.M.; Felsky, D.; De Jager, P.L.; et al. Person-specific differences in ubiquitin-proteasome mediated proteostasis in human neurons. Alzheimers Dement. 2024, 20, 2952–2967. [Google Scholar] [CrossRef] [PubMed]
- Decourt, B.; Drumm-Gurnee, D.; Wilson, J.; Jacobson, S.; Belden, C.; Sirrel, S.; Ahmadi, M.; Shill, H.; Powell, J.; Walker, A.; et al. Poor Safety and Tolerability Hamper Reaching a Potentially Therapeutic Dose in the Use of Thalidomide for Alzheimer’s Disease: Results from a Double-Blind, Placebo-Controlled Trial. Curr. Alzheimer Res. 2017, 14, 403–411. [Google Scholar] [CrossRef]
- Jordan, F.; Quinn, T.J.; McGuinness, B.; Passmore, P.; Kelly, J.P.; Smith, C.T.; Murphy, K.; Devane, D. Aspirin and Other Non-Steroidal Anti-Inflammatory Drugs for the Prevention of Dementia; Jordan, F., Ed.; Cochrane Library: London, UK, 2020. [Google Scholar]
- Sivamaruthi, B.S.; Raghani, N.; Chorawala, M.; Bhattacharya, S.; Prajapati, B.G.; Elossaily, G.M.; Chaiyasut, C. NF-κB Pathway and Its Inhibitors: A Promising Frontier in the Management of Alzheimer’s Disease. Biomedicines 2023, 11, 2587. [Google Scholar] [CrossRef]
- Kobayashi, K.; Imagama, S.; Ohgomori, T.; Hirano, K.; Uchimura, K.; Sakamoto, K.; Hirakawa, A.; Takeuchi, H.; Suzumura, A.; Ishiguro, N.; et al. Minocycline selectively inhibits M1 polarization of microglia. Cell Death Dis. 2013, 4, e525. [Google Scholar] [CrossRef]
- Howard, R.; Zubko, O.; Gray, R.; Bradley, R.; Harper, E.; Kelly, L.; Pank, L.; O’Brien, J.; Fox, C.; Tabet, N.; et al. Minocycline 200 Mg or 400 Mg Versus Placebo for Mild Alzheimer’s Disease: The MADE Phase II, Three-Arm RCT. In Efficacy and Mechanism Evaluation; NIHR Journals Library: Southampton, UK, 2020. [Google Scholar]
- Torres-Acosta, N.; O’Keefe, J.H.; O’Keefe, E.L.; Isaacson, R.; Small, G. Therapeutic Potential of TNF- α Inhibition for Alzheimer’s Disease Prevention. J. Alzheimers Dis. 2020, 78, 619–626. [Google Scholar] [CrossRef]
- Kaltschmidt, B.; Ndiaye, D.; Korte, M.; Pothion, S.; Arbibe, L.; Prüllage, M.; Pfeiffer, J.; Lindecke, A.; Staiger, V.; Israël, A.; et al. NF-κB Regulates Spatial Memory Formation and Synaptic Plasticity through Protein Kinase A/CREB Signaling. Mol. Cell. Biol. 2006, 26, 2936–2946. [Google Scholar] [CrossRef]
- O’Mahony, A.; Raber, J.; Montano, M.; Foehr, E.; Han, V.; Lu, S.; Kwon, H.; LeFevour, A.; Chakraborty-Sett, S.; Greene, W.C. NF-κB/Rel Regulates Inhibitory and Excitatory Neuronal Function and Synaptic Plasticity. Mol. Cell. Biol. 2006, 26, 7283–7298. [Google Scholar] [CrossRef]
- Schmeisser, M.J.; Baumann, B.; Johannsen, S.; Vindedal, G.F.; Jensen, V.; Hvalby, Ø.C.; Sprengel, R.; Seither, J.; Maqbool, A.; Magnutzki, A.; et al. IκB Kinase/Nuclear Factor κB-Dependent Insulin-Like Growth Factor 2 (Igf2) Expression Regulates Synapse Formation and Spine Maturation via Igf2 Receptor Signaling. J. Neurosci. 2012, 32, 5688–5703. [Google Scholar] [CrossRef] [PubMed]
- Maqbool, A.; Lattke, M.; Wirth, T.; Baumann, B. Sustained, neuron-specific IKK/NF-κB activation generates a selective neuroinflammatory response promoting local neurodegeneration with aging. Mol. Neurodegener. 2013, 8, 40. [Google Scholar] [CrossRef] [PubMed]
- Schnöder, L.; Quan, W.; Yu, Y.; Tomic, I.; Luo, Q.; Hao, W.; Peng, G.; Li, D.; Fassbender, K.; Liu, Y. Deficiency of IKKβ in neurons ameliorates Alzheimer’s disease pathology in APP- and tau-transgenic mice. FASEB J. 2023, 37, e22778. [Google Scholar] [CrossRef] [PubMed]
- Cruz, J.C.; Tseng, H.-C.; Goldman, J.A.; Shih, H.; Tsai, L.-H. Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron 2003, 40, 471–483. [Google Scholar] [CrossRef]
- Welch, G.M.; Boix, C.A.; Schmauch, E.; Davila-Velderrain, J.; Victor, M.B.; Dileep, V.; Bozzelli, P.L.; Su, Q.; Cheng, J.D.; Lee, A.; et al. Neurons burdened by DNA double-strand breaks incite microglia activation through antiviral-like signaling in neurodegeneration. Sci. Adv. 2022, 8, eabo4662. [Google Scholar] [CrossRef] [PubMed]
- Seok, J.; Warren, H.S.; Cuenca, A.G.; Mindrinos, M.N.; Baker, H.V.; Xu, W.; Richards, D.R.; McDonald-Smith, G.P.; Gao, H.; Hennessy, L.; et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl. Acad. Sci. USA. 2013, 110, 3507–3512. [Google Scholar] [CrossRef]
- Tanaka, T.; Huang, X.; Halicka, H.D.; Zhao, H.; Traganos, F.; Albino, A.P.; Dai, W.; Darzynkiewicz, Z. Cytometry of ATM activation and histone H2AX phosphorylation to estimate extent of DNA damage induced by exogenous agents. Cytom. Part J. Int. Soc. Anal. Cytol. 2007, 71, 648–661. [Google Scholar] [CrossRef]
- Vilenchik, M.M.; Knudson, A.G. Endogenous DNA double-strand breaks: Production, fidelity of repair, and induction of cancer. Proc. Natl. Acad. Sci. USA 2003, 100, 12871–12876. [Google Scholar] [CrossRef]
- Takahashi, A.; Ohnishi, T. Does gammaH2AX foci formation depend on the presence of DNA double strand breaks? Cancer Lett. 2005, 229, 171–179. [Google Scholar] [CrossRef]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef]
- Kastan, M.B.; Lim, D.S. The many substrates and functions of ATM. Nat. Rev. Mol. Cell Biol. 2000, 1, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Madabhushi, R.; Pan, L.; Tsai, L.-H. DNA damage and its links to neurodegeneration. Neuron 2014, 83, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Welch, G.; Tsai, L.-H. Mechanisms of DNA damage-mediated neurotoxicity in neurodegenerative disease. EMBO Rep. 2022, 23, e54217. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, E.; Beach, T.; Shen, Y.; Li, R.; Chang, Y. Deficiency of the Mre11 DNA repair complex in Alzheimer’s disease brains. Brain Res. Mol. Brain Res. 2004, 128, 1–7. [Google Scholar] [CrossRef]
- Lovell, M.A.; Markesbery, W.R. Oxidative DNA damage in mild cognitive impairment and late-stage Alzheimer’s disease. Nucleic Acids Res. 2007, 35, 7497–7504. [Google Scholar] [CrossRef]
- Lu, T.; Pan, Y.; Kao, S.-Y.; Li, C.; Kohane, I.; Chan, J.; Yankner, B.A. Gene regulation and DNA damage in the ageing human brain. Nature 2004, 429, 883–891. [Google Scholar] [CrossRef]
- Shanbhag, N.M.; Evans, M.D.; Mao, W.; Nana, A.L.; Seeley, W.W.; Adame, A.; Rissman, R.A.; Masliah, E.; Mucke, L. Early neuronal accumulation of DNA double strand breaks in Alzheimer’s disease. Acta Neuropathol. Commun. 2019, 7, 77. [Google Scholar] [CrossRef]
- Jovasevic, V.; Wood, E.M.; Cicvaric, A.; Zhang, H.; Petrovic, Z.; Carboncino, A.; Parker, K.K.; Bassett, T.E.; Moltesen, M.; Yamawaki, N.; et al. Formation of memory assemblies through the DNA-sensing TLR9 pathway. Nature 2024, 628, 145–153. [Google Scholar] [CrossRef]
- Adamec, E.; Vonsattel, J.P.; Nixon, R.A. DNA strand breaks in Alzheimer’s disease. Brain Res. 1999, 849, 67–77. [Google Scholar] [CrossRef]
- Shen, X.; Chen, J.; Li, J.; Kofler, J.; Herrup, K. Neurons in Vulnerable Regions of the Alzheimer’s Disease Brain Display Reduced ATM Signaling. eNeuro 2016, 3, 1–18. [Google Scholar] [CrossRef]
- Mertens, J.; Herdy, J.R.; Traxler, L.; Schafer, S.T.; Schlachetzki, J.C.M.; Böhnke, L.; Reid, D.A.; Lee, H.; Zangwill, D.; Fernandes, D.P.; et al. Age-dependent instability of mature neuronal fate in induced neurons from Alzheimer’s patients. Cell Stem Cell 2021, 28, 1533–1548.e6. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Finding | Antibody | Case Number | References |
|---|---|---|---|
| strong IR in neurons in AD, much lower in controls whole hippocampus formation extremely high and mainly nuclear in CA3 prominent staining in neuronal processes as well as neurofibrillary tangles and dystrophic neurites staining of astrocytes no microglial staining | anti-RELA | 7 non-AD 7 AD | [16] |
| 65% of plaques surrounded by cells with activated nuclear RELA pyramidal neurons and glia surrounding plaques, presumable astrocytes, not in activated microglia, strongest staining in primitive plaques | anti-RELA-NLS mAb | 4 AD | [17] |
| highly enriched nuclear RELA in cholinergic neurons of AD (nucleus basalis of Meynert) | RELA | [18] | |
| RELA in cells around plaques, graded loss of RELA IR during plaque maturation from primitive to classical plaques, lower IR in AD compared to controls in early plaque stages | anti-RELA-NLS mAb | [19] | |
| enhanced expression of IκBα in the cytoplasm of degenerating neurons with tangle formation REL nuclear in CA1 neurons with tangles (AT8 double pos.) southwestern blotting revealed strong binding of kB motif dsDNA to neuronal elements in AD but not in controls RELA mainly nuclear but p50 mainly cytoplasmic | anti-IκBα, anti-p50 anti-RELA anti-REL AT8 | [20] | |
| nuclear P-RELA highly enriched (>10-fold) in GFAP + astrocytes in AD compared to non-AD, p50 not nuclear | [21] |
| Gene | Function | Cell Type |
|---|---|---|
| APOE | lipid transporter | neurons and astrocytes |
| CR1 | complement receptor | macrophages and B-cells |
| BIN1 | adaptor protein | neurons |
| TREM2 | Aβ42 receptor | microglia |
| CLU | chaperone | astrocytes |
| PICALM | Phosphatidylinositol-binding clathrin assembly protein | oligodendrocytes |
| MS4A | Membrane-spanning 4-domains protein with no orthologous mouse protein | microglia |
| APP | Aβ precursor protein | neurons |
| Tau (MAPT) | microtubule-associated protein | neurons |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaltschmidt, B.; Czaniera, N.J.; Schulten, W.; Kaltschmidt, C. NF-κB in Alzheimer’s Disease: Friend or Foe? Opposite Functions in Neurons and Glial Cells. Int. J. Mol. Sci. 2024, 25, 11353. https://doi.org/10.3390/ijms252111353
Kaltschmidt B, Czaniera NJ, Schulten W, Kaltschmidt C. NF-κB in Alzheimer’s Disease: Friend or Foe? Opposite Functions in Neurons and Glial Cells. International Journal of Molecular Sciences. 2024; 25(21):11353. https://doi.org/10.3390/ijms252111353
Chicago/Turabian StyleKaltschmidt, Barbara, Nele Johanne Czaniera, Wiebke Schulten, and Christian Kaltschmidt. 2024. "NF-κB in Alzheimer’s Disease: Friend or Foe? Opposite Functions in Neurons and Glial Cells" International Journal of Molecular Sciences 25, no. 21: 11353. https://doi.org/10.3390/ijms252111353
APA StyleKaltschmidt, B., Czaniera, N. J., Schulten, W., & Kaltschmidt, C. (2024). NF-κB in Alzheimer’s Disease: Friend or Foe? Opposite Functions in Neurons and Glial Cells. International Journal of Molecular Sciences, 25(21), 11353. https://doi.org/10.3390/ijms252111353