

MAD2L2 Dimerization Is Not Essential for Mitotic Regulation

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
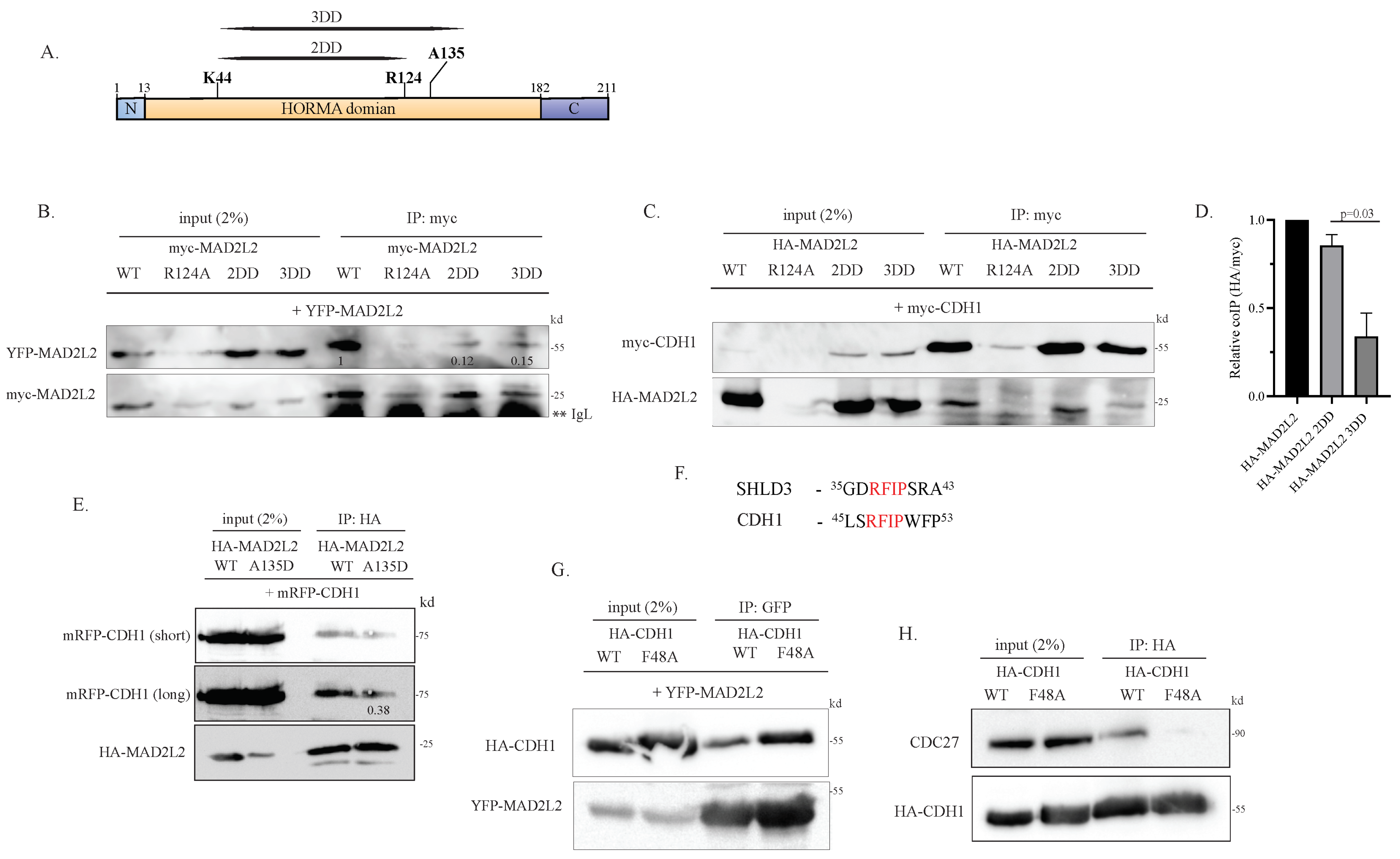
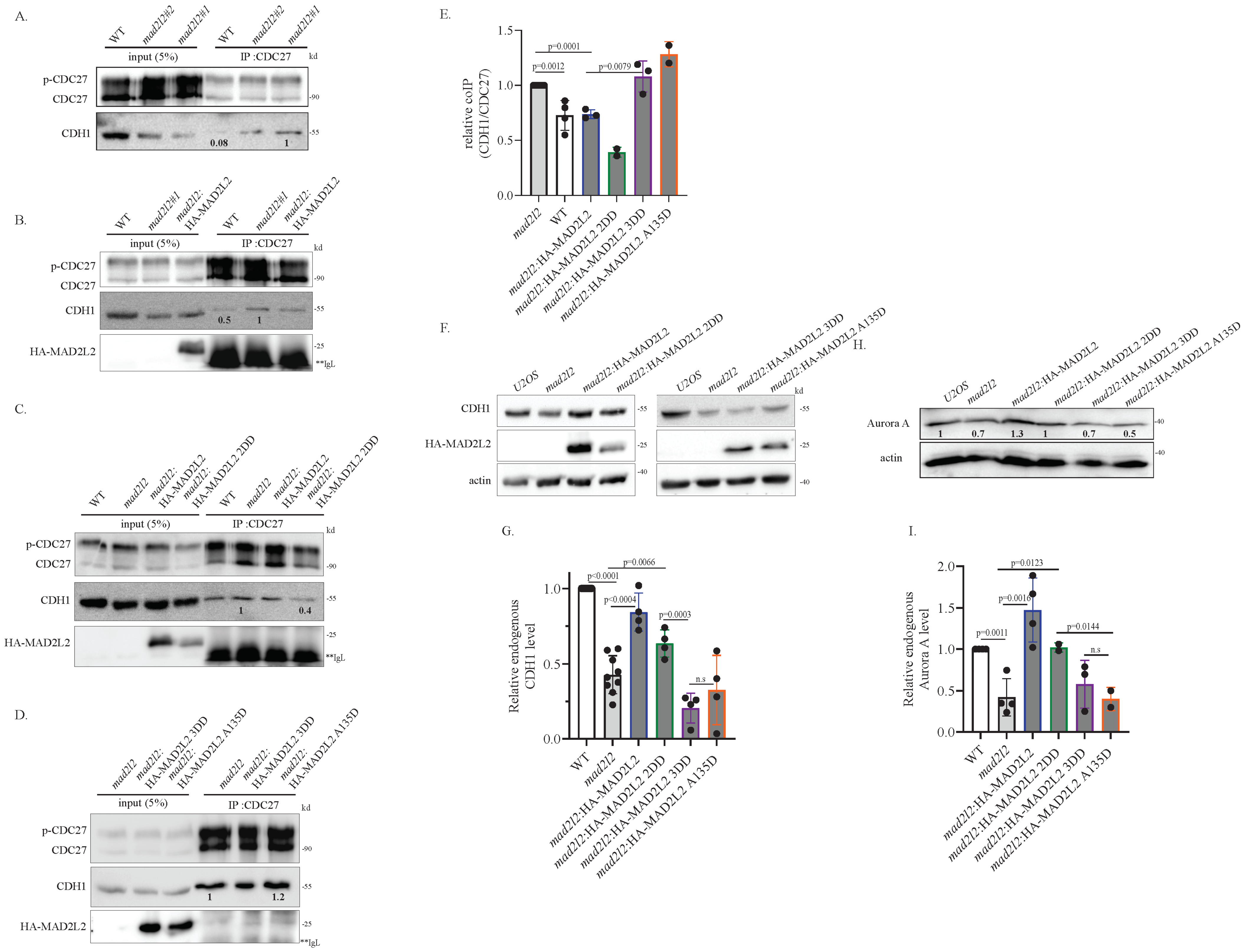
2.1. Monomeric MAD2L2 Can Bind to CDH1
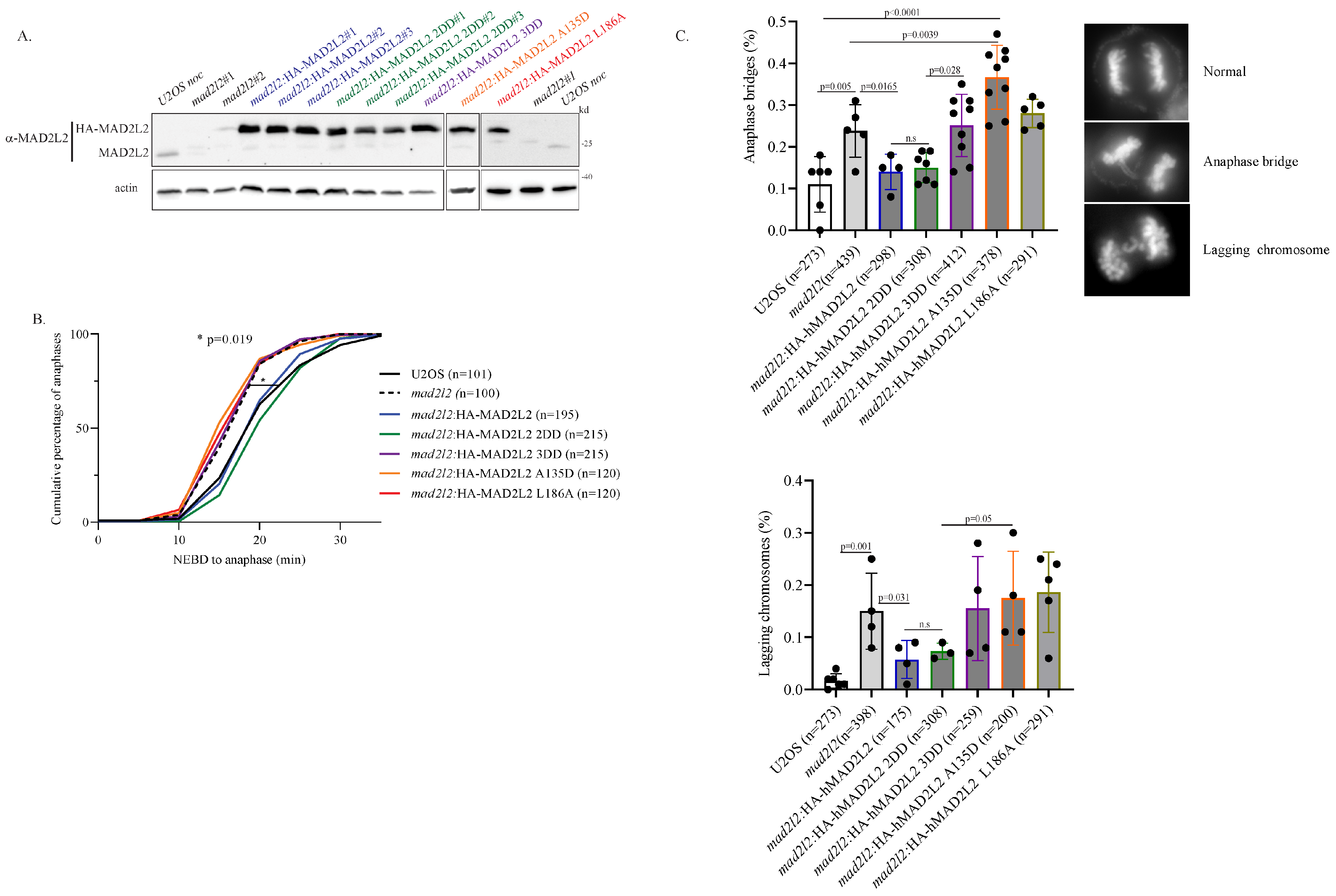
2.2. Monomeric MAD2L2 Can Properly Regulate Mitotic Entry
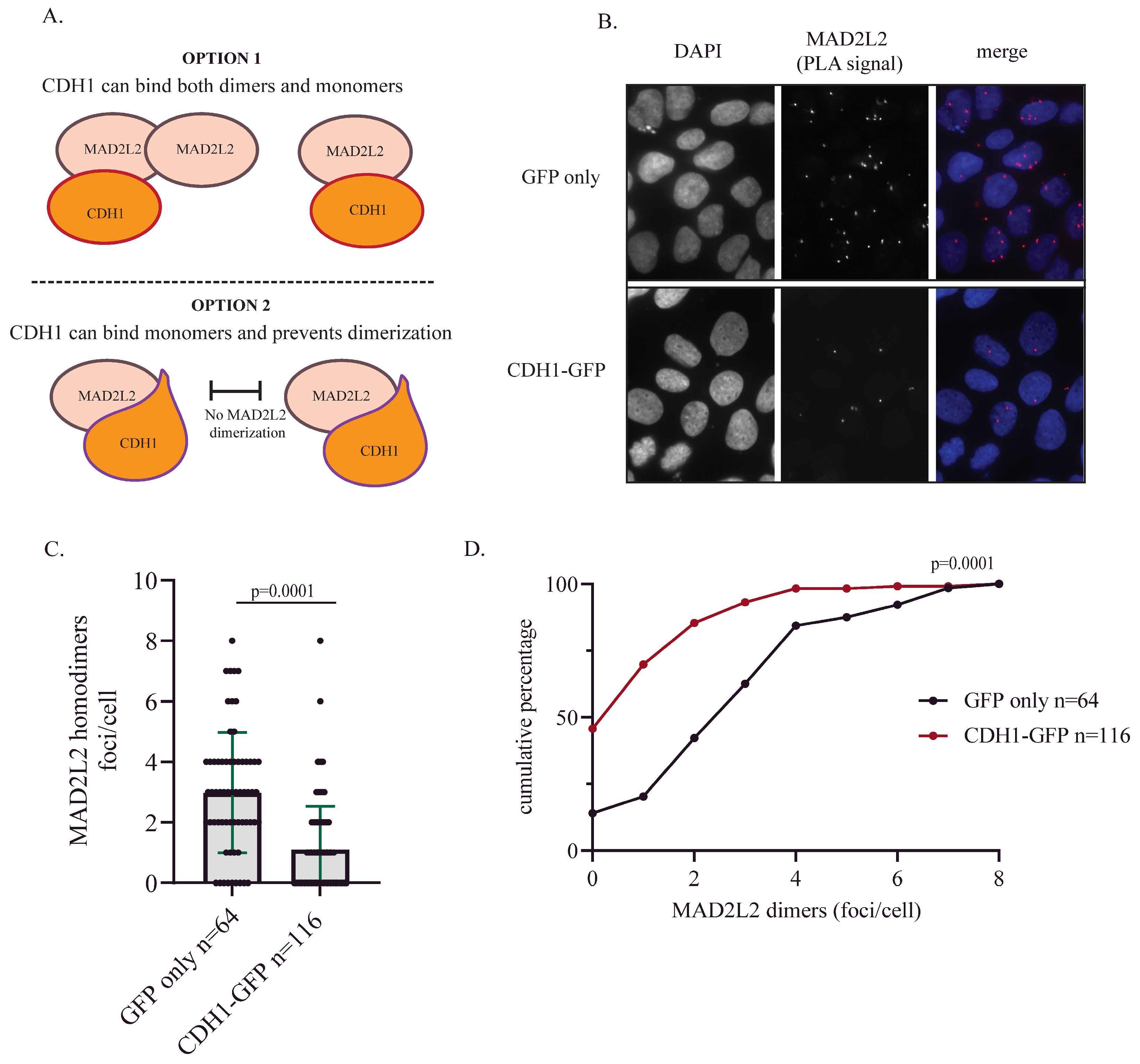
2.3. CDH1 Overexpression Reduces MAD2L2 Homodimerization
3. Discussion
4. Materials and Methods
4.1. Expression Vectors and Site-Directed Mutagenesis
4.2. Cell Culture, Transfection, and Synchronization
4.3. CRISPR/Cas9 Knockdown
4.4. Stable Complementation
4.5. Western Blot, Immunoprecipitation, and Antibodies
4.6. Time-Lapse Imaging and Analysis
4.7. Chromosome Spreads
4.8. Proximity Ligation Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Muniyappa, K.; Kshirsagar, R.; Ghodke, I. The HORMA domain: An evolutionarily conserved domain discovered in chromatin-associated proteins, has unanticipated diverse functions. Gene 2014, 545, 194–197. [Google Scholar] [CrossRef] [PubMed]
- de Krijger, I.; Boersma, V.; Jacobs, J.J. REV7: Jack of many trades. Trends Cell Biol. 2021, 31, 686–701. [Google Scholar] [CrossRef]
- Wang, X.; Pernicone, N.; Pertz, L.; Hua, D.; Zhang, T.; Listovsky, T.; Xie, W. REV7 has a dynamic adaptor region to accommodate small GTPase RAN/Shigella IpaB ligands, and its activity is regulated by the RanGTP/GDP switch. J. Biol. Chem. 2019, 294, 15733–15742. [Google Scholar] [CrossRef]
- Liang, L.; Feng, J.; Zuo, P.; Yang, J.; Lu, Y.; Yin, Y. Molecular basis for assembly of the shieldin complex and its implications for NHEJ. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Sale, J.E.; Lehmann, A.R.; Woodgate, R. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat. Rev. Mol. Cell Biol. 2012, 13, 141–152. [Google Scholar] [CrossRef]
- Vaisman, A.; Woodgate, R. Translesion DNA polymerases in eukaryotes: What makes them tick? Crit. Rev. Biochem. Mol. Biol. 2017, 52, 274–303. [Google Scholar] [CrossRef]
- Livneh, Z.; Z, O.; Shachar, S. Multiple two-polymerase mechanisms in mammalian translesion DNA synthesis. Cell Cycle 2010, 9, 729–735. [Google Scholar] [CrossRef]
- Lee, Y.-S.; Gregory, M.T.; Yang, W. Human Pol ζ purified with accessory subunits is active in translesion DNA synthesis and complements Pol η in cisplatin bypass. Proc. Natl. Acad. Sci. 2014, 111, 2954–2959. [Google Scholar] [CrossRef]
- Hara, K.; Hashimoto, H.; Murakumo, Y.; Kobayashi, S.; Kogame, T.; Unzai, S.; Akashi, S.; Takeda, S.; Shimizu, T.; Sato, M. Crystal Structure of Human REV7 in Complex with a Human REV3 Fragment and Structural Implication of the Interaction between DNA Polymerase ζ and REV1. J. Biol. Chem. 2010, 285, 12299–12307. [Google Scholar] [CrossRef]
- Rizzo, A.A.; Vassel, F.-M.; Chatterjee, N.; D’souza, S.; Li, Y.; Hao, B.; Hemann, M.T.; Walker, G.C.; Korzhnev, D.M. Rev7 dimerization is important for assembly and function of the Rev1/Polζ translesion synthesis complex. Proc. Natl. Acad. Sci. USA 2018, 115, EB191–EB200. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, A.A.; Korzhnev, D.M. The Rev1-Polζ translesion synthesis mutasome: Structure, interactions and inhibition. Enzymes 2019, 45, 139–181. [Google Scholar]
- McPherson, K.S.; Rizzo, A.A.; Erlandsen, H.; Chatterjee, N.; Walker, G.C.; Korzhnev, D.M. Evolution of Rev7 interactions in eukaryotic TLS DNA polymerase Polζ. J. Biol. Chem. 2022, 299, 102859. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, S.; Hara, K.; Shimizu, T.; Sato, M.; Hashimoto, H. Structural Basis of Recruitment of DNA Polymerase ζ by Interaction between REV1 and REV7 Proteins. J. Biol. Chem. 2012, 287, 33847–33852. [Google Scholar] [CrossRef]
- Pernicone, N.; Elias, M.; Onn, I.; Tobi, D.; Listovsky, T. Disrupting the MAD2L2-Rev1 Complex Enhances Cell Death upon DNA Damage. Molecules 2022, 27, 636. [Google Scholar] [CrossRef]
- Wojtaszek, J.L.; Chatterjee, N.; Najeeb, J.; Ramos, A.; Lee, M.; Bian, K.; Xue, J.Y.; Fenton, B.A.; Park, H.; Li, D.; et al. A Small Molecule Targeting Mutagenic Translesion Synthesis Improves Chemotherapy. Cell 2019, 178, 152–159.e11. [Google Scholar] [CrossRef]
- Doles, J.; Oliver, T.G.; Cameron, E.R.; Hsu, G.; Jacks, T.; Walker, G.C.; Hemann, M.T. Suppression of Rev3, the catalytic subunit of Polζ, sensitizes drug-resistant lung tumors to chemotherapy. Proc. Natl. Acad. Sci. USA 2010, 107, 20786–20791. [Google Scholar] [CrossRef]
- Ghezraoui, H.; Oliveira, C.; Becker, J.R.; Bilham, K.; Moralli, D.; Anzilotti, C.; Fischer, R.; Deobagkar-Lele, M.; Sanchiz-Calvo, M.; Fueyo-Marcos, E.; et al. 53BP1 cooperation with the REV7–shieldin complex underpins DNA structure-specific NHEJ. Nature 2018, 560, 122–127. [Google Scholar] [CrossRef]
- Xu, G.; Chapman, J.R.; Brandsma, I.; Yuan, J.; Mistrik, M.; Bouwman, P.; Bartkova, J.; Gogola, E.; Warmerdam, D.; Barazas, M.; et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature 2015, 521, 541–544. [Google Scholar] [CrossRef]
- Boersma, V.; Moatti, N.; Segura-Bayona, S.; Peuscher, M.H.; van der Torre, J.; Wevers, B.A.; Orthwein, A.; Durocher, D.; Jacobs, J.J.L. MAD2L2 controls DNA repair at telomeres and DNA breaks by inhibiting 5′ end resection. Nature 2015, 521, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Noordermeer, S.M.; Adam, S.; Setiaputra, D.; Barazas, M.; Pettitt, S.J.; Ling, A.K.; Olivieri, M.; Álvarez-Quilón, A.; Moatti, N.; Zimmermann, M.; et al. The shieldin complex mediates 53BP1-dependent DNA repair. Nature 2018, 560, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Setiaputra, D.; Durocher, D. Shieldin – the protector of DNA ends. Embo Rep. 2019, 20. [Google Scholar] [CrossRef]
- de Krijger, I.; Föhr, B.; Pérez, S.H.; Vincendeau, E.; Serrat, J.; Thouin, A.M.; Susvirkar, V.; Lescale, C.; Paniagua, I.; Hoekman, L.; et al. MAD2L2 dimerization and TRIP13 control shieldin activity in DNA repair. Nat. Commun. 2021, 12, 1–15. [Google Scholar] [CrossRef]
- Clairmont, C.S.; Sarangi, P.; Ponnienselvan, K.; Galli, L.D.; Csete, I.; Moreau, L.; Adelmant, G.; Chowdhury, D.; Marto, J.A.; D’andrea, A.D. TRIP13 regulates DNA repair pathway choice through REV7 conformational change. Nat. Cell Biol. 2020, 22, 87–96. [Google Scholar] [CrossRef]
- Chen, J.; Fang, G. MAD2B is an inhibitor of the anaphase-promoting complex. Genes Dev. 2001, 15, 1765–1770. [Google Scholar] [CrossRef]
- Pfleger, C.M.; Salic, A.; Lee, E.; Kirschner, M.W. Inhibition of Cdh1–APC by the MAD2-related protein MAD2L2: A novel mechanism for regulating Cdh1. Genes Dev. 2001, 15, 1759–1764. [Google Scholar] [CrossRef]
- Sudakin, V.; Ganoth, D.; Dahan, A.; Heller, H.; Hershko, J.; Luca, F.C.; Ruderman, J.V.; Hershko, A. The cyclosome, a large complex containing cyclin-selective ubiquitin ligase activity, targets cyclins for destruction at the end of mitosis. Mol. Biol. Cell 1995, 6, 185–197. [Google Scholar] [CrossRef]
- King, R.W.; Peters, J.-M.; Tugendreich, S.; Rolfe, M.; Hieter, P.; Kirschner, M.W. A 20s complex containing CDC27 and CDC16 catalyzes the mitosis-specific conjugation of ubiquitin to cyclin B. Cell 1995, 81, 279–288. [Google Scholar] [CrossRef]
- Pines, J. Cubism and the cell cycle: The many faces of the APC/C. Nat. Rev. Mol. Cell Biol. 2011, 12, 427–438. [Google Scholar] [CrossRef]
- Kramer, E.R.; Scheuringer, N.; Podtelejnikov, A.V.; Mann, M.; Peters, J.-M. Mitotic Regulation of the APC Activator Proteins CDC20 and CDH1. Mol. Biol. Cell 2000, 11, 1555–1569. [Google Scholar] [CrossRef] [PubMed]
- Listovsky, T.; Oren, Y.S.; Yudkovsky, Y.; Mahbubani, H.M.; Weiss, A.M.; Lebendiker, M.; Brandeis, M. Mammalian Cdh1/Fzr mediates its own degradation. EMBO J. 2004, 23, 1619–1626. [Google Scholar] [CrossRef] [PubMed]
- Visintin, R.; Prinz, S.; Amon, A. CDC20 and CDH1: A Family of Substrate-Specific Activators of APC-Dependent Proteolysis. Science 1997, 278, 460–463. [Google Scholar] [CrossRef] [PubMed]
- Listovsky, T.; Sale, J.E. Sequestration of CDH1 by MAD2L2 prevents premature APC/C activation prior to anaphase onset. J. Cell Biol. 2013, 203, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Pernicone, N.; Grinshpon, S.; Listovsky, T. CDH1 binds MAD2L2 in a Rev1-like pattern. Biochem. Biophys. Res. Commun. 2020, 531, 566–572. [Google Scholar] [CrossRef]
- Vassel, F.M.; Laverty, D.J.; Bian, K.; Piett, C.G.; Hemann, M.T.; Walker, G.C.; Nagel, Z.D. REV7 Monomer Is Unable to Par-ticipate in Double Strand Break Repair and Translesion Synthesis but Suppresses Mitotic Errors. Int. J. Mol. Sci. 2023, 24, 15799. [Google Scholar] [CrossRef]
- Bhat, A.; Wu, Z.; Maher, V.M.; McCormick, J.J.; Xiao, W. Rev7/Mad2B plays a critical role in the assembly of a functional mitotic spindle. Cell Cycle 2015, 14, 3929–3938. [Google Scholar] [CrossRef]
- Brito, D.A.; Rieder, C.L. The ability to survive mitosis in the presence of microtubule poisons differs significantly between human nontransformed (RPE-1) and cancer (U2OS, HeLa) cells. Cell Motil. Cytoskelet. 2008, 66, 437–447. [Google Scholar] [CrossRef]
- Floyd, S.; Pines, J.; Lindon, C. APC/CCdh1 Targets Aurora Kinase to Control Reorganization of the Mitotic Spindle at Anaphase. Curr. Biol. 2008, 18, 1649–1658. [Google Scholar] [CrossRef]
- Clute, P.; Pines, J. Temporal and spatial control of cyclin B1 destruction in metaphase. Nat. Cell Biol. 1999, 1, 82–87. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barda, N.; Ayiku, P.J.; Bar-on, A.; Movshovitz, S.; Listovsky, T. MAD2L2 Dimerization Is Not Essential for Mitotic Regulation. Int. J. Mol. Sci. 2024, 25, 11485. https://doi.org/10.3390/ijms252111485
Barda N, Ayiku PJ, Bar-on A, Movshovitz S, Listovsky T. MAD2L2 Dimerization Is Not Essential for Mitotic Regulation. International Journal of Molecular Sciences. 2024; 25(21):11485. https://doi.org/10.3390/ijms252111485
Chicago/Turabian StyleBarda, Nomi, Philippa Jennifer Ayiku, Amit Bar-on, Sahar Movshovitz, and Tamar Listovsky. 2024. "MAD2L2 Dimerization Is Not Essential for Mitotic Regulation" International Journal of Molecular Sciences 25, no. 21: 11485. https://doi.org/10.3390/ijms252111485
APA StyleBarda, N., Ayiku, P. J., Bar-on, A., Movshovitz, S., & Listovsky, T. (2024). MAD2L2 Dimerization Is Not Essential for Mitotic Regulation. International Journal of Molecular Sciences, 25(21), 11485. https://doi.org/10.3390/ijms252111485