
Differential Gene Expression in Late-Onset Friedreich Ataxia: A Comparative Transcriptomic Analysis Between Symptomatic and Asymptomatic Sisters


 , ,
, ,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
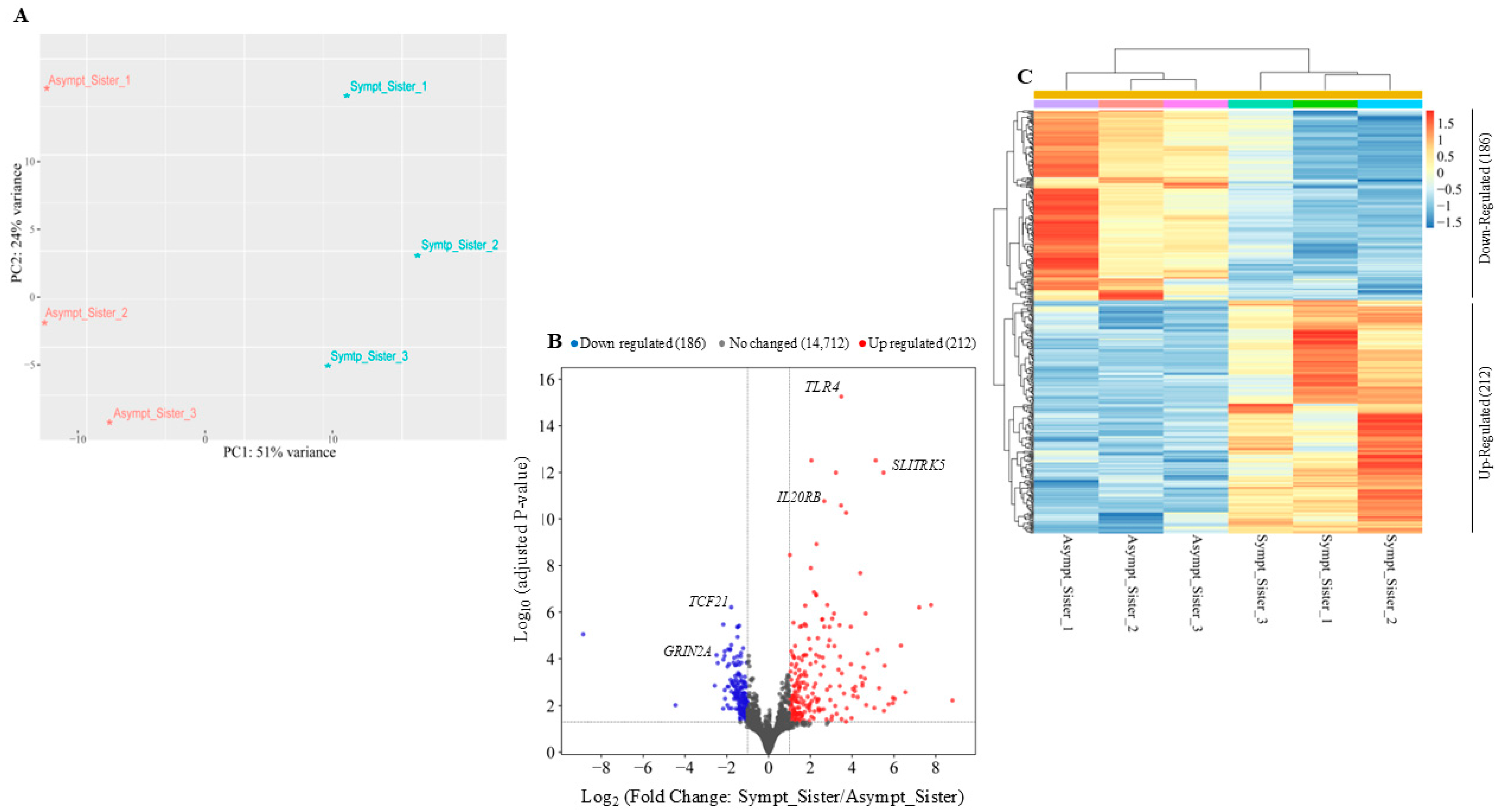
2.1. Transcriptomic Analysis
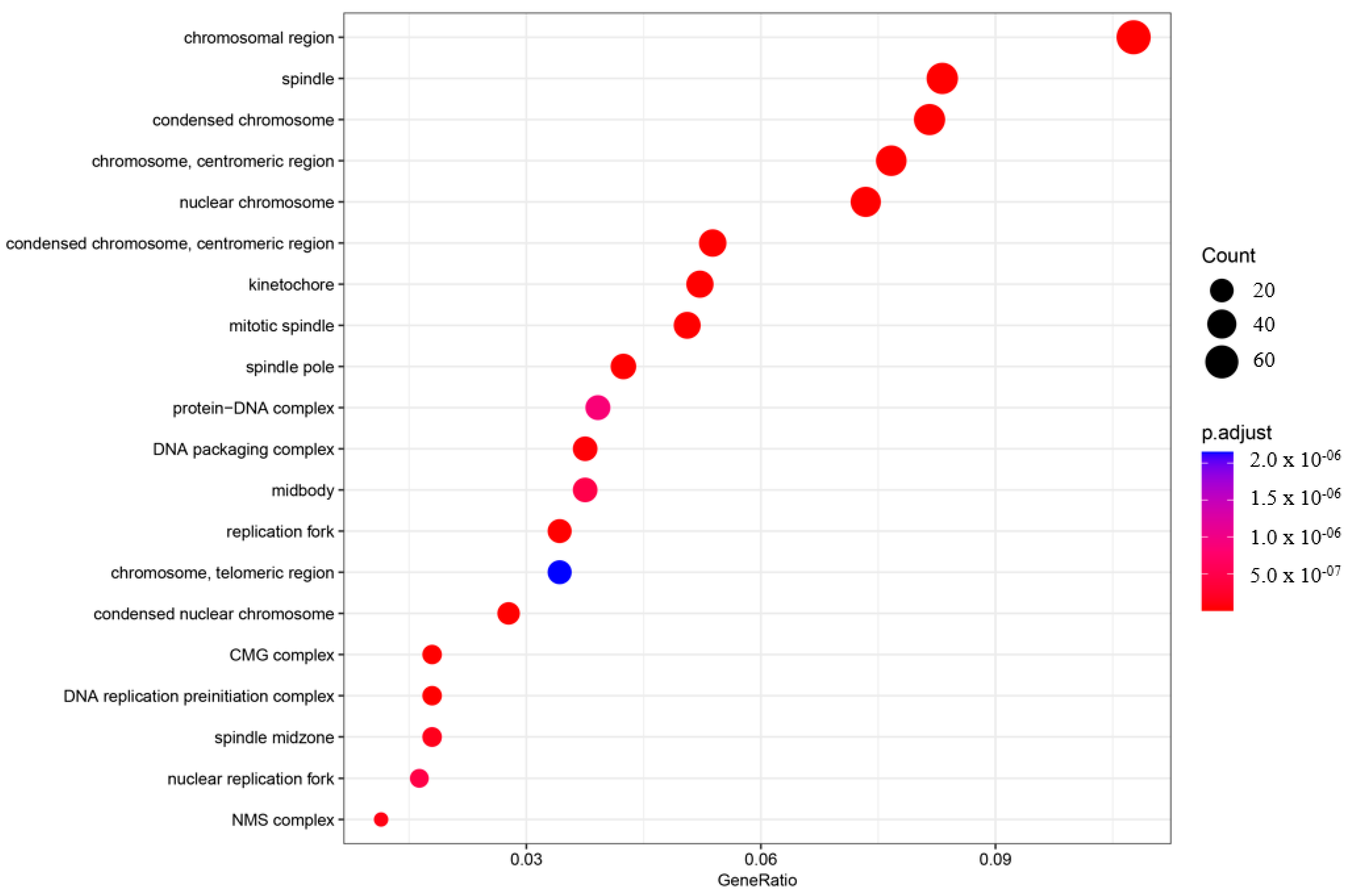
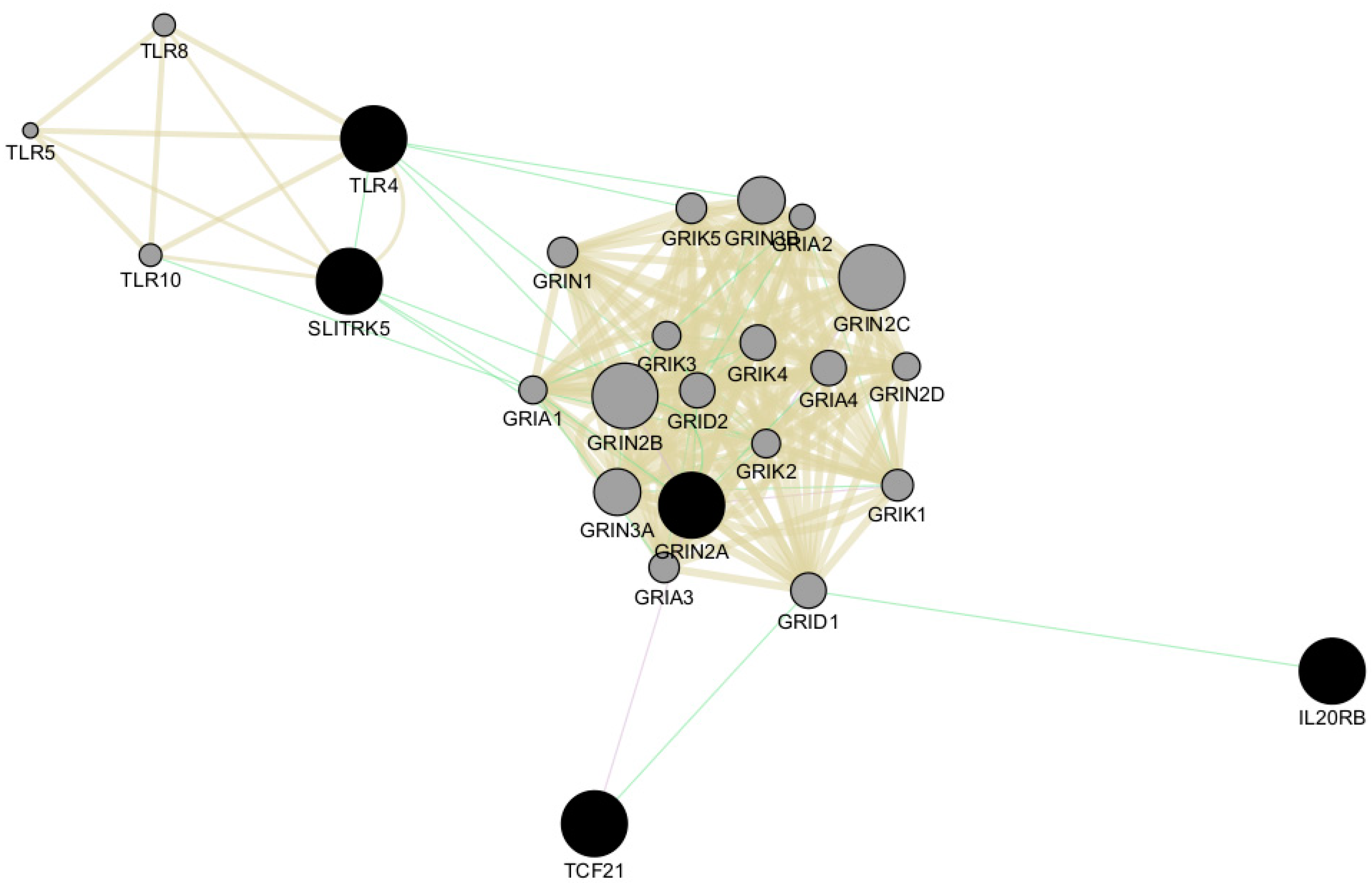
2.2. Gene Ontology (GO) and Pathway Analysis
3. Discussion
4. Materials and Methods
4.1. Subjects
4.2. Fibroblasts Cultures
4.3. RNA-Seq Analysis
4.4. Bioinformatic Analysis
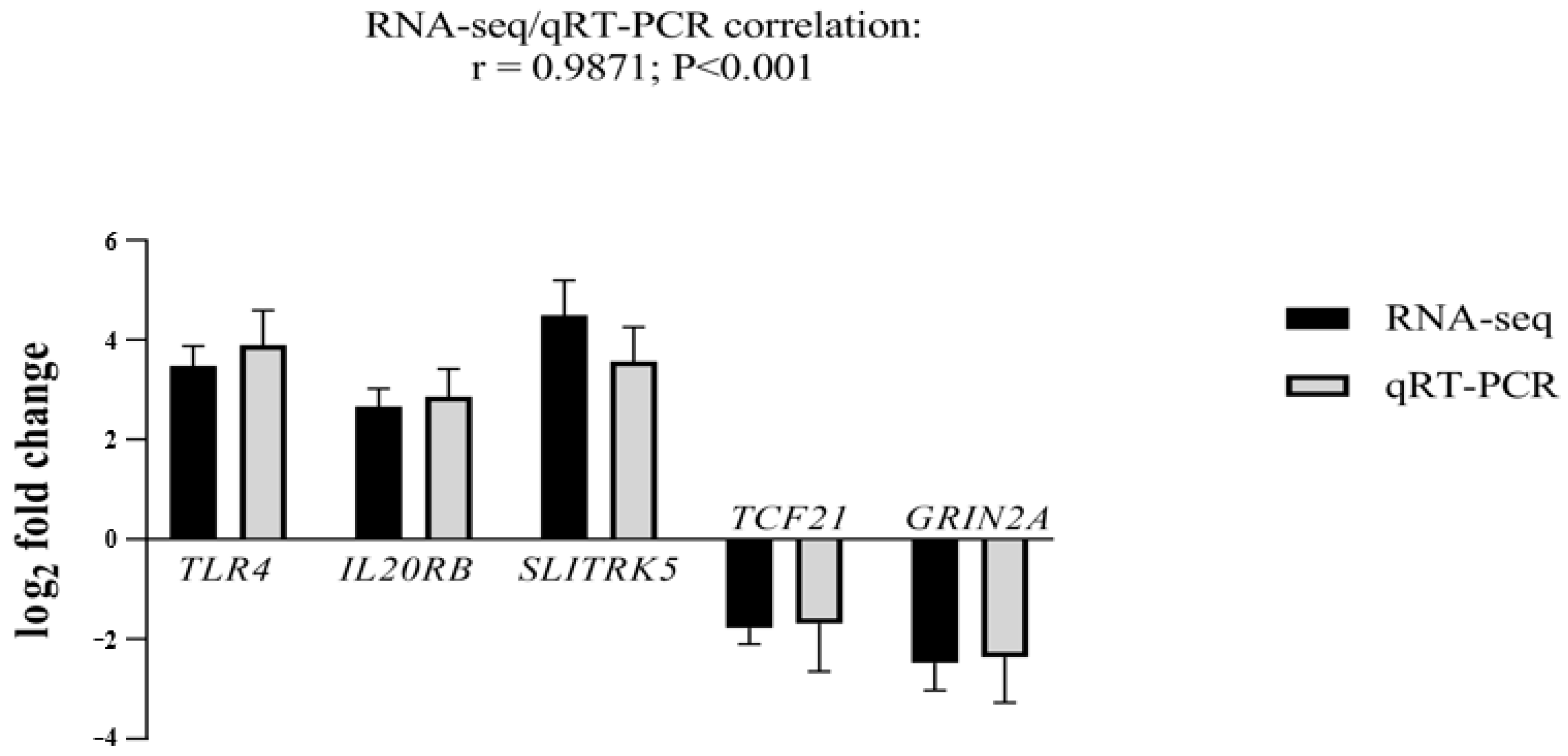
4.5. Quantitative Real-Time PCR (qRT-PCR) Validation
4.6. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Monfort, B.; Want, K.; Gervason, S.; D’Autréaux, B. Recent Advances in the Elucidation of Frataxin Biochemical Function Open Novel Perspectives for the Treatment of Friedreich’s Ataxia. Front. Neurosci. 2022, 16, 838335. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Keita, M.; McIntyre, K.; Rodden, L.N.; Schadt, K.; Lynch, D.R. Friedreich ataxia: Clinical features and new developments. Neurodegener. Dis. Manag. 2022, 12, 267–283. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Grander, M.; Haschka, D.; Indelicato, E.; Kremser, C.; Amprosi, M.; Nachbauer, W.; Henninger, B.; Stefani, A.; Högl, B.; Fischer, C.; et al. Genetic Determined Iron Starvation Signature in Friedreich’s Ataxia. Mov. Disord. 2024, 39, 1088–1098. [Google Scholar] [CrossRef] [PubMed]
- Gavriilaki, M.; Chatzikyriakou, E.; Moschou, M.; Arnaoutoglou, M.; Sakellari, I.; Kimiskidis, V.K. Therapeutic Biomarkers in Friedreich’s Ataxia: A Systematic Review and Meta-analysis. Cerebellum 2024, 23, 1184–1203. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cossée, M.; Puccio, H.; Gansmuller, A.; Koutnikova, H.; Dierich, A.; LeMeur, M.; Fischbeck, K.; Dollé, P.; Kœnig, M. Inactivation of the Friedreich ataxia mouse gene leads to early embryonic lethality without iron accumulation. Hum. Mol. Genet. 2000, 9, 1219–1226. [Google Scholar] [CrossRef]
- Becker, E.M.; Greer, J.M.; Ponka, P.; Richardson, D.R. Erythroid differentiation and protoporphyrin IX down-regulate frataxin expression in Friend cells: Characterization of frataxin expression compared to molecules involved in iron metabolism and hemoglobinization. Blood 2002, 99, 3813–3822. [Google Scholar] [CrossRef]
- Muhlenhoff, U.; Richhardt, N.; Ristow, M.; Kispal, G.; Lill, R. The yeast frataxin homolog Yfh1p plays a specific role in the maturation of cellular Fe/S proteins. Hum. Mol. Genet. 2002, 11, 2025–2036. [Google Scholar] [CrossRef]
- Seznec, H.; Simon, D.; Bouton, C.; Reutenauer, L.; Hertzog, A.; Golik, P.; Procaccio, V.; Patel, M.; Drapier, J.C.; Koenig, M.; et al. Friedreich ataxia: The oxidative stress paradox. Hum. Mol. Genet. 2005, 14, 463–474. [Google Scholar] [CrossRef]
- Steinkellner, H.; Singh, H.N.; Muckenthaler, M.U.; Goldenberg, H.; Moganty, R.R.; Scheiber-Mojdehkar, B.; Sturm, B. No changes in heme synthesis in human Friedreich s ataxia erythroid progenitor cells. Gene 2017, 621, 5–11. [Google Scholar] [CrossRef]
- Abeti, R.; Baccaro, A.; Esteras, N.; Giunti, P. Novel Nrf2-Inducer Prevents Mitochondrial Defects and Oxidative Stress in Friedreich’s Ataxia Models. Front. Cell. Neurosci. 2018, 12, 188. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- La Rosa, P.; Russo, M.; D’Amico, J.; Petrillo, S.; Aquilano, K.; Lettieri-Barbato, D. Nrf2 Induction Re-establishes a Proper Neuronal Differentiation Program in Friedreich’s Ataxia Neural Stem Cells. Front. Cell. Neurosci. 2019, 13, 356. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- La Rosa, P.; Petrillo, S.; Fiorenza, M.T.; Bertini, E.S.; Piemonte, F. Ferroptosis in Friedreich’s Ataxia: A Metal-Induced Neurodegenerative Disease. Biomolecules 2020, 10, 1551. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- La Rosa, P.; Petrillo, S.; Turchi, R.; Berardinelli, F.; Schirinzi, T.; Vasco, G.; Lettieri-Barbato, D.; Fiorenza, M.T.; Bertini, E.S.; Aquilano, K.; et al. The Nrf2 induction prevents ferroptosis in Friedreich’s Ataxia. Redox Biol. 2021, 38, 101791. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Petrillo, S.; Piermarini, E.; Pastore, A.; Vasco, G.; Schirinzi, T.; Carrozzo, R.; Bertini, E.; Piemonte, F. Nrf2-Inducers Counteract Neurodegeneration in Frataxin-Silenced Motor Neurons: Disclosing New Therapeutic Targets for Friedreich’s Ataxia. Int. J. Mol. Sci. 2017, 18, 2173. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Petrillo, S.; D’Amico, J.; La Rosa, P.; Bertini, E.S.; Piemonte, F. Targeting NRF2 for the Treatment of Friedreich’s Ataxia: A Comparison among Drugs. Int. J. Mol. Sci. 2019, 20, 5211. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Petrillo, S.; Santoro, M.; La Rosa, P.; Perna, A.; Gallo, M.G.; Bertini, E.S.; Silvestri, G.; Piemonte, F. Nuclear Factor Erythroid 2-Related Factor 2 Activation Might Mitigate Clinical Symptoms in Friedreich’s Ataxia: Clues of an Out-Brain Origin of the Disease From a Family Study. Front. Neurosci. 2021, 15, 638810. [Google Scholar] [CrossRef]
- Alsina, D.; Purroy, R.; Ros, J.; Tamarit, J. Iron in Friedreich Ataxia: A central role in the pathophysiology or an epiphenomenon? Pharmaceuticals 2018, 11, 89. [Google Scholar] [CrossRef]
- Cotticelli, M.G.; Xia, S.; Truitt, R.; Doliba, N.M.; Rozo, A.V.; Tobias, J.W.; Lee, T.; Chen, J.; Napierala, J.S.; Napierala, M.; et al. Acute frataxin knockdown in induced pluripotent stem cell-derived cardiomyocytes activates a type I interferon response. Dis. Model. Mech. 2023, 16, dmm049497. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Angulo, M.B.; Bertalovitz, A.; Argenziano, M.A.; Yang, J.; Patel, A.; Zesiewicz, T.; McDonald, T.V. Frataxin deficiency alters gene expression in Friedreich ataxia derived IPSC-neurons and cardiomyocytes. Mol. Genet. Genom. Med. 2023, 11, e2093. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Indelicato, E.; Kirchmair, A.; Amprosi, M.; Steixner, S.; Nachbauer, W.; Eigentler, A.; Wahl, N.; Apostolova, G.; Krogsdam, A.; Schneider, R.; et al. Skeletal muscle transcriptomics dissects the pathogenesis of Friedreich’s ataxia. Hum. Mol. Genet. 2023, 32, 2241–2250. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Indelicato, E.; Faserl, K.; Amprosi, M.; Nachbauer, W.; Schneider, R.; Wanschitz, J.; Sarg, B.; Boesch, S. Skeletal muscle proteome analysis underpins multifaceted mitochondrial dysfunction in Friedreich’s ataxia. Front. Neurosci. 2023, 17, 1289027. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Indelicato, E.; Wanschitz, J.; Löscher, W.; Boesch, S. Skeletal Muscle Involvement in Friedreich Ataxia. Int. J. Mol. Sci. 2024, 25, 9915. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cotticelli, M.G.; Xia, S.; Lin, D.; Lee, T.; Terrab, L.; Wipf, P.; Huryn, D.M.; Wilson, R.B. Ferroptosis as a Novel Therapeutic Target for Friedreich’s Ataxia. J. Pharmacol. Exp. Ther. 2019, 369, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Turchi, R.; Tortolici, F.; Guidobaldi, G.; Iacovelli, F.; Falconi, M.; Rufini, S.; Faraonio, R.; Casagrande, V.; Federici, M.; De Angelis, L.; et al. Frataxin deficiency induces lipid accumulation and affects thermogenesis in brown adipose tissue. Cell Death Dis. 2020, 11, 51–65, Erratum in: Cell Death Dis. 2020, 11, 165. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, L.; Fang, X.; Ling, B.; Wang, F.; Xia, Y.; Zhang, W.; Zhong, T.; Wang, X. Research progress on ferroptosis in the pathogenesis and treatment of neurodegenerative diseases. Front. Cell. Neurosci. 2024, 18, 1359453. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Santoro, M.; Perna, A.; La Rosa, P.; Petrillo, S.; Piemonte, F.; Rossi, S.; Riso, V.; Nicoletti, T.F.; Modoni, A.; Pomponi, M.G.; et al. Compound heterozygosity for an expanded (GAA) and a (GAAGGA) repeat at FXN locus: From a diagnostic pitfall to potential clues to the pathogenesis of Friedreich ataxia. Neurogenetics 2020, 21, 279–287. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sanchez, N.; Chapdelaine, P.; Rousseau, J.; Raymond, F.; Corbeil, J.; Tremblay, J.P. Characterization of frataxin gene network in Friedreich’s ataxia fibroblasts using the RNA-Seq technique. Mitochondrion 2016, 30, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Nachun, D.; Gao, F.; Isaacs, C.; Strawser, C.; Yang, Z.; Dokuru, D.; Van Berlo, V.; Sears, R.; Farmer, J.; Perlman, S.; et al. Peripheral blood gene expression reveals an inflammatory transcriptomic signature in Friedreich’s ataxia patients. Hum. Mol. Genet. 2018, 27, 2965–2977. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dionisi, C.; Chazalon, M.; Rai, M.; Keime, C.; Imbault, V.; Communi, D.; Puccio, H.; Schiffmann, S.N.; Pandolfo, M. Proprioceptors-enriched neuronal cultures from induced pluripotent stem cells from Friedreich ataxia patients show altered transcriptomic and proteomic profiles, abnormal neurite extension, and impaired electrophysiological properties. Brain Commun. 2023, 5, fcad007. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lai, J.I.; Nachun, D.; Petrosyan, L.; Throesch, B.; Campau, E.; Gao, F.; Baldwin, K.K.; Coppola, G.; Gottesfeld, J.M.; Soragni, E. Transcriptional profiling of isogenic Friedreich ataxia neurons and effect of an HDAC inhibitor on disease signatures. J. Biol. Chem. 2019, 294, 1846–1859. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chen, G.; Li, L.; Tao, H. Bioinformatics Identification of Ferroptosis-Related Biomarkers and Therapeutic Compounds in Ischemic Stroke. Front. Neurol. 2021, 12, 745240. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, J.Z.; Fan, B.Y.; Sun, T.; Wang, X.X.; Li, J.J.; Zhang, J.P.; Gu, G.J.; Shen, W.Y.; Liu, D.R.; Wei, Z.J.; et al. Bioinformatics analysis of ferroptosis in spinal cord injury. Neural Regen. Res. 2023, 18, 626–633. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sharma, V.; Sharma, P.; Singh, T.G. Mechanistic insights on TLR-4 mediated inflammatory pathway in neurodegenerative diseases. Pharmacol. Rep. 2024, 76, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef]
- Manoharan, R.R.; Prasad, A.; Pospíšil, P.; Kzhyshkowska, J. ROS signaling in innate immunity via oxidative protein modifications. Front. Immunol. 2024, 15, 1359600. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Van der Horst, D.; Carter-Timofte, M.E.; van Grevenynghe, J.; Laguette, N.; Dinkova-Kostova, A.T.; Olagnier, D. Regulation of innate immunity by Nrf2. Curr. Opin. Immunol. 2022, 78, 102247. [Google Scholar] [CrossRef]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting molecular cross-talk between Nrf2 and NF-κB response pathways. Biochem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [CrossRef]
- Apolloni, S.; Milani, M.; D’Ambrosi, N. Neuroinflammation in Friedreich’s Ataxia. Int. J. Mol. Sci. 2022, 23, 6297. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Khan, W.; Corben, L.A.; Bilal, H.; Vivash, L.; Delatycki, M.B.; Egan, G.F.; Harding, I.H. Neuroinflammation in the Cerebellum and Brainstem in Friedreich Ataxia: An [18F]-FEMPA PET Study. Mov. Disord. 2022, 37, 218–224. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, Y.; Ji, M.; Wu, C.; Zhang, Y.; Ji, S. Targeting ferroptosis in neuroimmune and neurodegenerative disorders for the development of novel therapeutics. Biomed. Pharmacother. 2024, 176, 116777. [Google Scholar] [CrossRef]
- Koeppen, A.H.; Michael, S.C.; Knutson, M.D.; Haile, D.J.; Qian, J.; Levi, S.; Santambrogio, P.; Garrick, M.D.; Lamarche, J.B. The Dentate Nucleus in Friedreich’s Ataxia: The Role of Iron-Responsive Proteins. Acta Neuropathol. 2007, 114, 163–173. [Google Scholar] [CrossRef]
- Franco, C.; Genis, L.; Navarro, J.A.; Pérez-Domper, P.; Fernandez, A.M.; Schneuwly, S.; Alemán, I.T. A Role for Astrocytes in Cerebellar Deficits in Frataxin Deficiency: Protection by Insulin-like Growth Factor I. Mol. Cell Neurosci. 2017, 80, 100–110. [Google Scholar] [CrossRef]
- Zeitlberger, A.M.; Thomas-Black, G.; Garcia-Moreno, H.; Foiani, M.; Heslegrave, A.J.; Zetterberg, H.; Giunti, P. Plasma Markers of Neurodegeneration Are Raised in Friedreich’s Ataxia. Front. Cell Neurosci. 2018, 12, 366. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Harding, I.H.; Lynch, D.R.; Koeppen, A.H.; Pandolfo, M. Central Nervous System Therapeutic Targets in Friedreich Ataxia. Hum. Gene Ther. 2020, 31, 1226–1236. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Schoenfeld, R.; Shan, Y.; Tsai, H.-J.; Hammock, B.; Cortopassi, G. Frataxin Deficiency Induces Schwann Cell Inflammation and Death. Biochim. Biophys. Acta 2009, 1792, 1052–1061. [Google Scholar] [CrossRef]
- Blumberg, H.; Conklin, D.; Xu, W.; Grossmann, A.; Brender, T.; Carollo, S.; Eagan, M.; Foster, D.; Haldeman, B.A.; Hammond, A.; et al. Interleukin 20: Discovery, receptor identification, and role in epidermal function. Cell 2001, 104, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Cornut, M.; Bourdonnay, E.; Henry, T. Transcriptional Regulation of Inflammasomes. Int. J. Mol. Sci. 2020, 21, 8087. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chiriac, M.T.; Hracsko, Z.; Günther, C.; Gonzalez-Acera, M.; Atreya, R.; Stolzer, I.; Wittner, L.; Dressel, A.; Schickedanz, L.; Gamez-Belmonte, R.; et al. IL-20 controls resolution of experimental colitis by regulating epithelial IFN/STAT2 signalling. Gut 2024, 73, 282–297. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Olagnier, D.; Brandtoft, A.M.; Gunderstofte, C.; Villadsen, N.L.; Krapp, C.; Thielke, A.L.; Laustsen, A.; Peri, S.; Hansen, A.L.; Bonefeld, L.; et al. Nrf2 negatively regulates STING indicating a link between antiviral sensing and metabolic reprogramming. Nat. Commun. 2018, 9, 3506–35018. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, L.; Mei, R.; Ai, M.; Pang, R.; Xia, D.; Chen, L.; Zhong, L. The Role of SLITRK5 in Central Nervous System. Biomed Res. Int. 2022, 2022, 4678026. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, Y.; Zhang, L.; Ai, M.; Xia, D.; Chen, H.; Pang, R.; Mei, R.; Zhong, L.; Chen, L. Upregulation of SLITRK5 in patients with epilepsy and in a rat model. Synapse 2023, 77, e22266. [Google Scholar] [CrossRef] [PubMed]
- Cardis, R.; Cabungcal, J.H.; Dwir, D.; Do, K.Q.; Steullet, P. A lack of GluN2A-containing NMDA receptors confers a vulnerability to redox dysregulation: Consequences on parvalbumin interneurons, and their perineuronal nets. Neurobiol. Dis. 2018, 109, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Camp, C.R.; Vlachos, A.; Klöckner, C.; Krey, I.; Banke, T.G.; Shariatzadeh, N.; Ruggiero, S.M.; Galer, P.; Park, K.L.; Caccavano, A.; et al. Loss of Grin2a causes a transient delay in the electrophysiological maturation of hippocampal parvalbumin interneurons. Commun. Biol. 2023, 6, 952. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Strehlow, V.; Myers, K.A.; Morgan, A.T.; Scheffer, I.E.; Lemke, J.R. GRIN2A-Related Disorders. In GeneReviews® [Internet]; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993–2024. [Google Scholar] [PubMed]
- Ao, X.; Ding, W.; Zhang, Y.; Ding, D.; Liu, Y. TCF21: A critical transcription factor in health and cancer. J. Mol. Med. 2020, 98, 1055–1068. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; He, F.; Zhang, Y.; Li, S.; Lu, R.; Wei, X.; Huang, J. Transcription Factor 21: A Transcription Factor That Plays an Important Role in Cardiovascular Disease. Pharmacology 2024, 109, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Gambardella, L.; McManus, S.A.; Moignard, V.; Sebukhan, D.; Delaune, A.; Andrews, S.; Bernard, W.G.; Morrison, M.A.; Riley, P.R.; Göttgens, B.; et al. BNC1 regulates cell heterogeneity in human pluripotent stem cell derived-epicardium. Development 2019, 146, dev174441. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Wang, L.; Wang, S.; Li, W. RSeQC: Quality control of RNA-seq experiments. Bioinformatics 2012, 28, 2184–2185. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.; Ibrahim, J.G.; Love, M.I. Heavy-tailed prior distributions for sequence count data: Removing the noise and preserving large differences. Bioinformatics 2019, 35, 2084–2092. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Chen, M.; Huang, X.; Zhang, G.; Zeng, L.; Zhang, G.; Wu, S.; Wang, Y. SRplot: A free online platform for data visualization and graphing. PLoS ONE 2023, 18, e0294236. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE Guidelines—Minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrillo, S.; Perna, A.; Quatrana, A.; Silvestri, G.; Bertini, E.; Piemonte, F.; Santoro, M. Differential Gene Expression in Late-Onset Friedreich Ataxia: A Comparative Transcriptomic Analysis Between Symptomatic and Asymptomatic Sisters. Int. J. Mol. Sci. 2024, 25, 11615. https://doi.org/10.3390/ijms252111615
Petrillo S, Perna A, Quatrana A, Silvestri G, Bertini E, Piemonte F, Santoro M. Differential Gene Expression in Late-Onset Friedreich Ataxia: A Comparative Transcriptomic Analysis Between Symptomatic and Asymptomatic Sisters. International Journal of Molecular Sciences. 2024; 25(21):11615. https://doi.org/10.3390/ijms252111615
Chicago/Turabian StylePetrillo, Sara, Alessia Perna, Andrea Quatrana, Gabriella Silvestri, Enrico Bertini, Fiorella Piemonte, and Massimo Santoro. 2024. "Differential Gene Expression in Late-Onset Friedreich Ataxia: A Comparative Transcriptomic Analysis Between Symptomatic and Asymptomatic Sisters" International Journal of Molecular Sciences 25, no. 21: 11615. https://doi.org/10.3390/ijms252111615
APA StylePetrillo, S., Perna, A., Quatrana, A., Silvestri, G., Bertini, E., Piemonte, F., & Santoro, M. (2024). Differential Gene Expression in Late-Onset Friedreich Ataxia: A Comparative Transcriptomic Analysis Between Symptomatic and Asymptomatic Sisters. International Journal of Molecular Sciences, 25(21), 11615. https://doi.org/10.3390/ijms252111615