

Physiopathological Roles of White Adiposity and Gut Functions in Neuroinflammation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
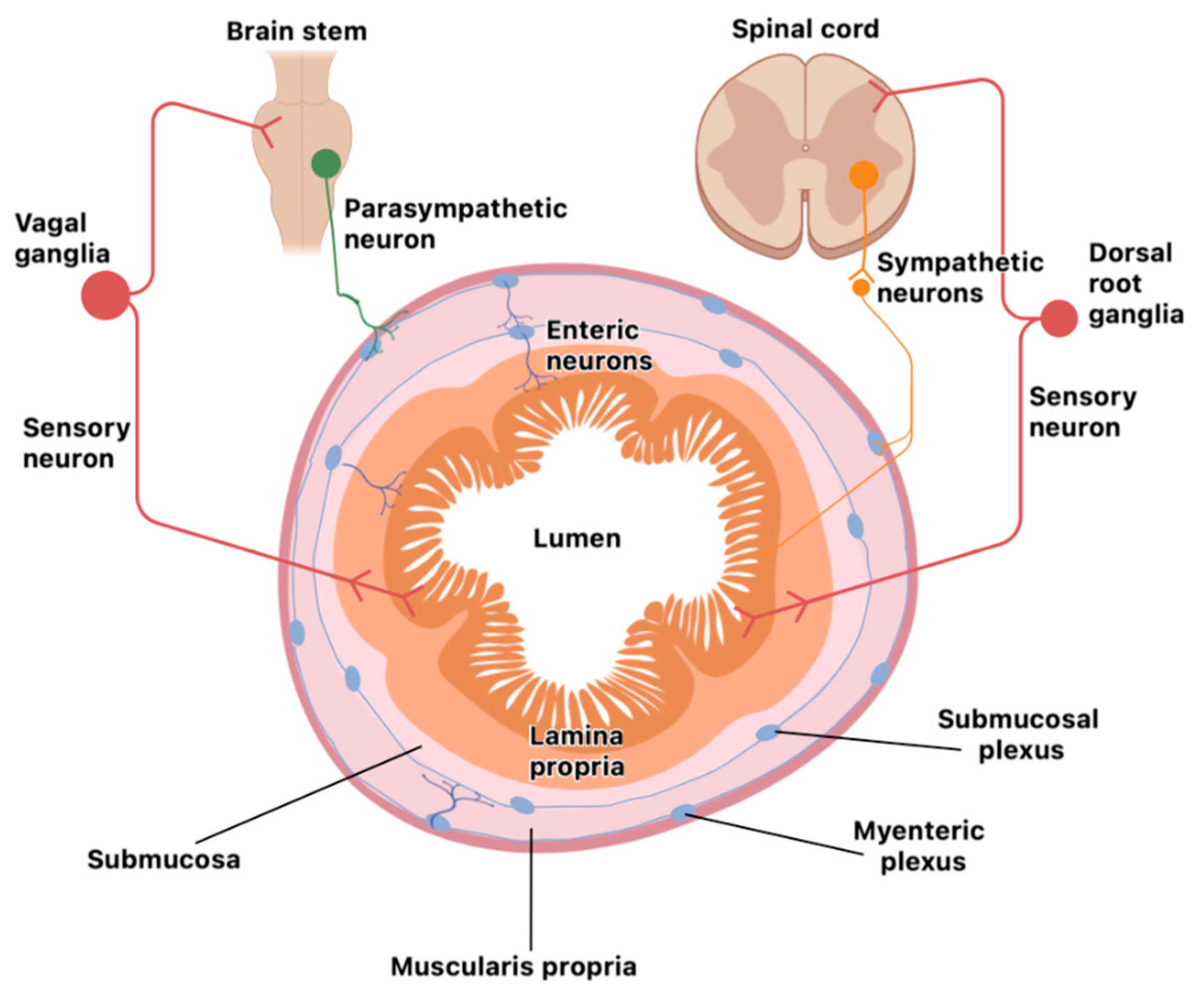
2. Relevance of WAT and Gastrointestinal Tract Functions in Neuroinflammation
Organization of the Nervous System in the Gut
3. Hypertrophic White-Adiposity-Induced Insulin Resistance (IR) as a Main Instigator of Neuroinflammation
3.1. Sympathetic Innervation of Adipose Tissue
3.2. Insulin-Resistant WAT Cells and Metainflammation Precede Neuroinflammation Development
Visceral Fat Depots of Large-Size WAT Adipocytes Characterize Obese Phenotypes
3.3. Hypothalamic-Mediated Mechanisms Involved in Neuroinflammation
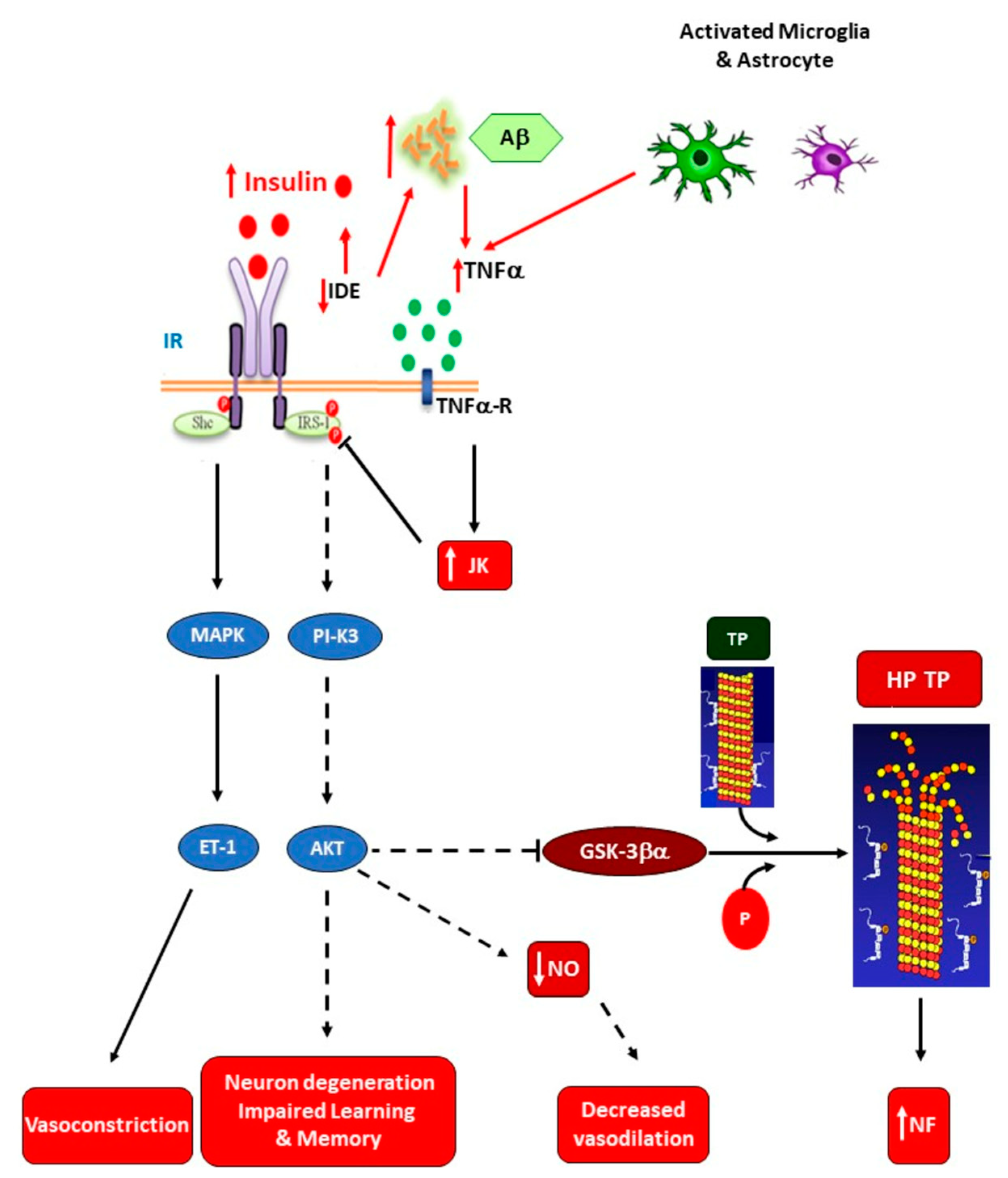
3.4. Insulin-Resistance-Related Neuroinflammation in Neurodegenerative Diseases
4. Gut and Neuroinflammation
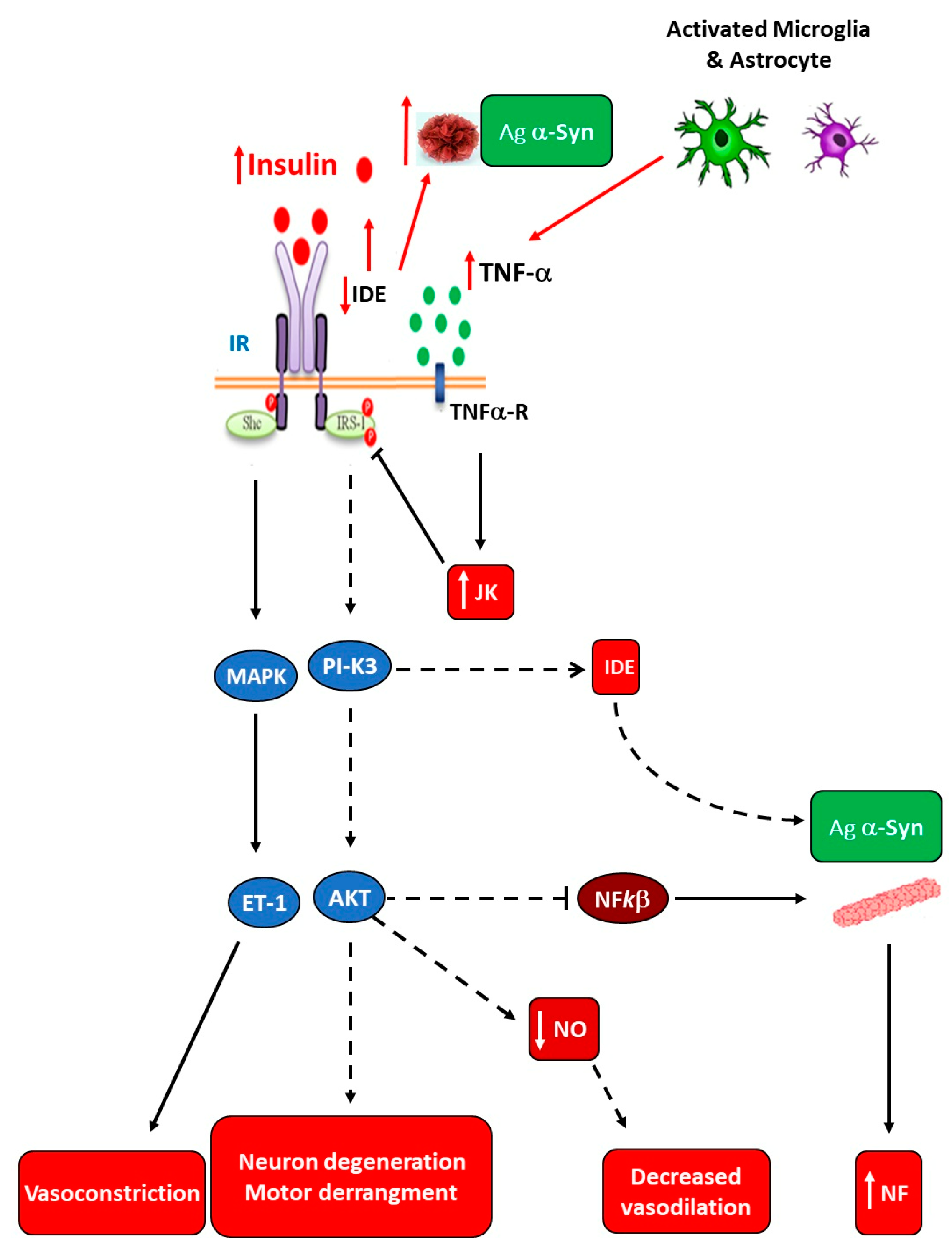
4.1. Role of Microbiota in Neurodegenerative Diseases
4.2. The Gut–Brain Axis: Role of the Hypothalamus
4.3. The Nervous System and Gut: Evidence of Cell Interactions
4.4. Role of the Microbiota in Tissue Barriers
4.5. Crosstalk Between the Intestinal Microbiome and Nervous System
4.6. The Role of Diet in Microbiota Composition and Neuroinflammation
5. Relationship Between Brain, Gut, and White Adiposity Processes
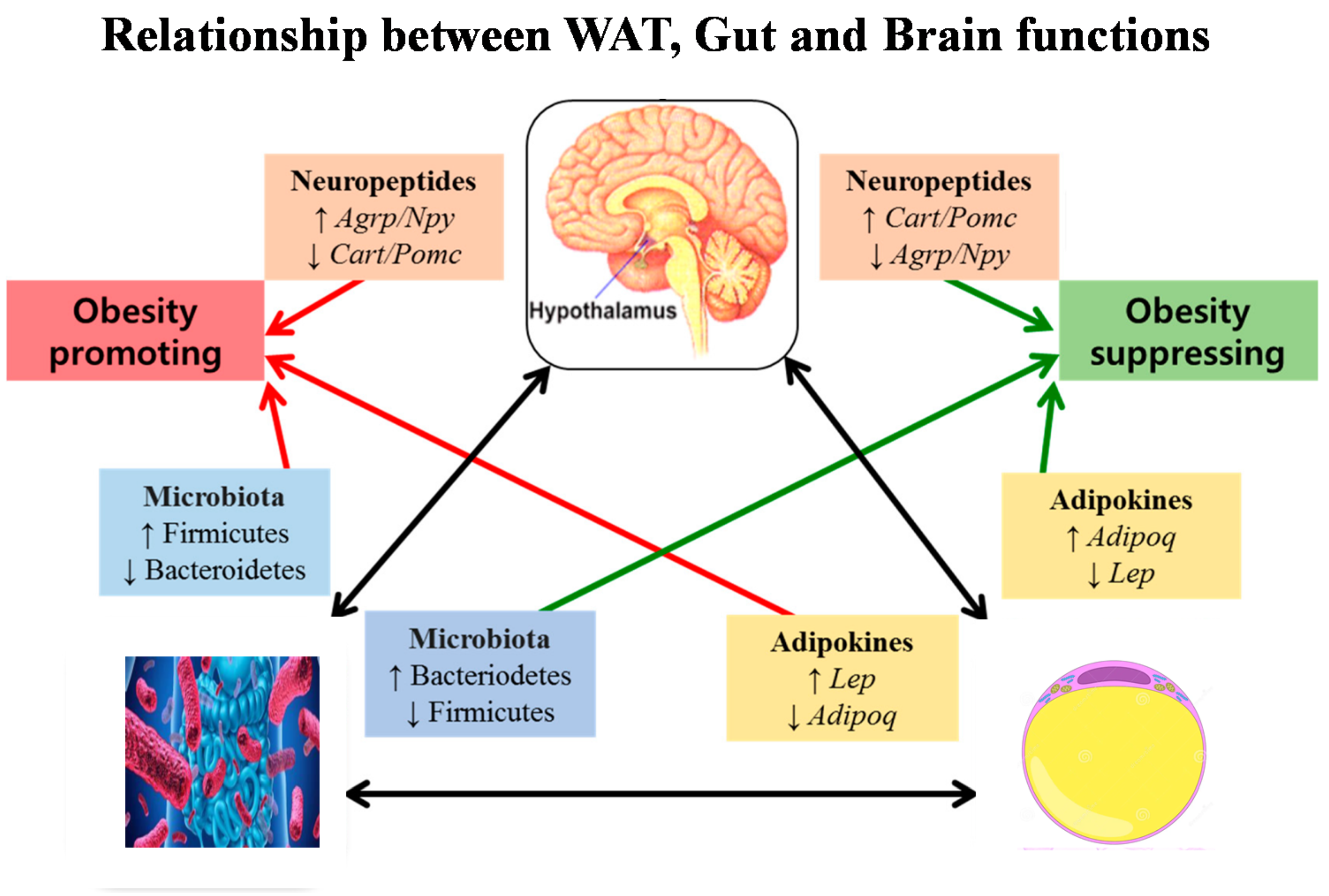
6. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Ab | Antibody |
| Aβ | Amyloid beta |
| Ach | Acetylcholine |
| AD | Alzheimer’s disease |
| AgRP | Aguti-related protein |
| AKT | Serine/threonine kinase |
| ALS | Amyotrophic lateral sclerosis |
| ANS | Autonomic nervous system |
| Arg1 | Arginase 1 |
| ASO | α-Syn-overexpressing |
| α-Syn | α-Synuclein |
| ATP | Adenosine triphosphate |
| ATRM | Adipose tissue-resident macrophage |
| BBB | Blood–brain barrier |
| BMP2 | Bone morphogenetic protein 2 |
| BMPR | Bone morphogenetic protein receptor |
| CART | Cocaine–amphetamine-related transcript |
| CCKR | Cholecystokinin receptor |
| CGRP | Calcitonin-gene-related peptide |
| CNS | Central nervous system |
| CpG ODN | CpG oligodeoxynucleotide |
| C-RP | C-reactive protein |
| CSF1 | Colony-stimulating factor 1 |
| CX3CL1 | Chemokine fractalkine |
| CX3CR1 | Fractalkine receptor 1 |
| DA | Dopamine |
| DAMP | Damage-associated molecular pattern |
| DC | Dendritic cells |
| DIO | Diet-induced obesity |
| DOPA | Dihydroxy phenyl alanine |
| ENS | Enteric nervous system |
| ER | Endoplasmic reticulum |
| FFA | Free fatty acid |
| FFAR | Free fatty acid receptor |
| GBA | Gut–brain axis |
| GC | Glucocorticoid |
| GLU | Glucose |
| GM | Gut microbiota |
| GR | Glucocorticoid receptor |
| GRE | Glucocorticoid response element |
| GSK3β | Glycogen synthase kinase 3β |
| GVB | Gut–vascular barrier |
| HD | Huntington’s disease |
| HFD | High-fat diet |
| HPA | Hypotalamo-pituitary–adrenal |
| IAPP | Islet amyloid polypeptide |
| IAPP | Islet amyloid polypeptide |
| IBD | Inflammatory bowel disease |
| IDE | Insulin-degrading enzyme |
| IFN-γ | Interferon-gamma |
| IKKβ | Inhibitory kappa beta kinase |
| IL-1 | Interleukin-1 |
| IL-6 | Interleukin-6 |
| ILC | Innate lymphoid cell |
| Ins | Insulin |
| IR | Insulin resistance |
| IRS-1 | Insulin receptor susbtrate-1 |
| JAK-STAT | Janus kinase–signal transducer and activator of transcription |
| L2-3 | Spinal lumbar segments 2 and 3 |
| LEP-R | Leptin receptor |
| LPM | Lamina propria macrophage |
| LPS | Lipopolysaccharide |
| MAOA | Monoamine oxidase A |
| microRNA | Micro ribonucleic acid |
| MM | Muscularis macrophage |
| MMP | Matrix metalloproteinases |
| MR | Mineralocorticoid receptor |
| mTOR | Mechanistic target of rapamycin |
| ND | Neurodegenerative disease |
| NE | Norepinephrine |
| NF | Neuroinflammation |
| NF-κB | Nuclear factor-kappa B |
| NGF | Nerve growth factor |
| NO | Nitric oxide |
| NPY | Neuropeptide Y |
| Ob-Rb | Leptin receptor form b |
| OS | Oxidative stress |
| PAI-1 | Plasminogen activator inhibitor type 1 |
| PAMP | Pathogen-associated molecular pattern |
| PD | Parkinson’s disease |
| PI3K | Phosphatidylinositol 3-kinase |
| PNS | Parasympathetic nervous system |
| POMC | Pro-opiomelanocortin |
| PRR | Pattern-recognition receptors |
| RA | Rheumatoid arthritis |
| Ramp1 | Receptor-activity-modifying protein 1 |
| ROS | Reactive oxygen substances |
| SAM | Sympathetic neuro-associated macrophage |
| SCFA | Short-chain fatty acid |
| SLC6A2 | Solute carrier family 6 member-2 |
| SNS | Sympathetic nervous system |
| SOCS3 | Suppressor of cytokine signaling 3 |
| SPF | Specific-pathogen-free |
| T13 | Spinal thoracic segment 13 |
| T2DM | Type 2 diabetes mellitus |
| Th | T helper lymphocytes |
| TLR7 | Toll-like receptor |
| TNF-α | Tumor necrosis-alpha |
| TOX01 | Forkhead box protein 01 |
| Trp | Tryptophan |
| UCP-1 | Uncoupling protein-1 |
| VIP | Vasoactive intestinal peptide |
| WAT | White adipose tissue |
| WHO | World Health Organization |
References
- Niranjan, R. Recent advances in the mechanisms of neuroinflammation and their roles in neurodegeneration. Neurochem. Int. 2018, 120, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Lyman, M.; Lloyd, D.G.; Ji, X.; Vizcaychipi, M.P.; Ma, D. Neuroinflammation: The role and consequences. J. Neuroscience. Research. 2014, 79, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Subhramanyam, C.S.; Wang, C.; Hu, Q.; Dheen, S.T. Microglia-mediated neuroinflammation in neurodegenerative diseases. J. Semin. Cell Dev. Biol. 2019, 94, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.C.; Chiu, K.; Ho, Y.S.; So, K.F. Modulation of neuroimmune responses on glia in the central nervous system: Implication in therapeutic intervention against neuroinflammation. J. Cell Mol. Immunol. 2009, 6, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Lyons, A.; McQuillan, K.; Deighan, B.F.; O’Reilly, J.A.; Downer, E.J.; Murphy, A.C.; Watson, M.; Piazza, A.; O’Connell, F.; Griffin, R.; et al. Decreased neuronal CD200 expression in IL-4-deficient mice results in increased neuroinflammation in response to lipopolysaccharide. J. Brain. Behavior. Immunity 2009, 23, 1020–1027. [Google Scholar] [CrossRef]
- Bessis, A.; Biber, K.; Bilbo, S.; Blurton-Jones, M.; Boddeke, E.; Brites, D.; Brône, B.; Brown, G.C.; Butovsky, O.; Carsonet, M.J.; et al. Microglia states and nomenclature: A field at its crossroads. Neuron. 2022, 110, 3458–3483. [Google Scholar]
- Woodburn, S.C.; Bollinger, J.L.; Wohleb, E.S. The semantics of microglia activation: Neuroinflammation, homeostasis, and stress. J. Neuroinflammation 2021, 18, 258. [Google Scholar] [CrossRef]
- Bazoukis, G.; Stavrakis, S.; Armoundas, A.A. Vagus Nerve Stimulation and Inflammation in Cardiovascular Disease: A State-of-the-Art Review. J. Am. Heart Assoc. 2023, 2, e030539. [Google Scholar] [CrossRef]
- Sun, Z.; Wang, X.; Feng, S.; Xie, C.; Xing, Y.; Guo, L.; Zhao, J.; Ji, C. A review of neuroendocrine immune system abnormalities in IBS based on the brain–gut axis and research progress of acupuncture intervention. Front. Neurosci. 2023, 17, 934341. [Google Scholar] [CrossRef]
- Choe, S.S.; Huh, J.Y.; Hwang, I.J.; Kim, J.I.; Kim, J.B. Adipose Tissue Remodeling: Its Role in energy Metabolism and Metabolic Disorders. Front. Endocrinol. 2016, 7, 30. [Google Scholar] [CrossRef]
- Candelario-Jalil, E.; Dijkhuizen, R.M.; Magnus, T. Neuroinflammation, Stroke, Blood-Brain Barrier: Dysfunction, and Imaging Modalities. Stroke 2022, 53, 1473–1486. [Google Scholar] [CrossRef] [PubMed]
- Maldonado-Ruiz, R.; Fuentes-Mera, L.; Camacho, A. Central Modulation of Neuroinflammation by Neuropeptides and Energy-Sensing Hormones during Obesity. Biomed. Res. Int. 2017, 2017, 7949582. [Google Scholar] [CrossRef] [PubMed]
- Bertocchi, A.; Carloni, S.; Ravenda, P.S.V.; Bertalot, G.; Spadoni, I.; Lo Cascio, A.; Gandini, S.; Lizier, M.; Braga, D.; Asnicar, F.; et al. Gut vascular barrier impairment leads to intestinal bacteria dissemination and colorectal cancer metastasis to liver. Cancer Cell 2021, 39, 708–724. [Google Scholar] [CrossRef] [PubMed]
- Carloni, S.; Bertocchi, A.; Mancinelli, S.; Bellini, M.; Erreni, M.; Borreca, A.; Braga, D.; Giugliano, S.; Mozzarelli, A.M.; Manganaro, D.; et al. Identification of a choroid plexus vascular barrier closing during intestinal inflammation. Science 2021, 374, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Fehervari, Z. A gut vascular barrier. Nat. Immunol. 2016, 17, 47. [Google Scholar] [CrossRef]
- Bilski, J.; Mazur-Bialy, A.; Wojcik, D.; Surmiak, M.; Magierowski, M.; Sliwowski, Z.; Pajdo, R.; Kwiecien, S.; Danielak, A.; Ptak-Belowska, A.; et al. Role of Obesity, Mesenteric Adipose Tissue, and Adipokines in Inflammatory Bowel Diseases. Biomolecules. 2019, 9, 780. [Google Scholar] [CrossRef]
- Rautmann, A.W.; de La Serre, C.B. Microbiota’s Role in Diet-Driven Alterations in Food Intake: Satiety, Energy Balance, and Reward. Nutrients 2021, 13, 3067. [Google Scholar] [CrossRef]
- Sartor, R.B. Mechanisms of disease: Pathogenesis of Crohn’s disease and ulcerative colitis. Nat. Clin. Pract. Gastroenterol. Hepatol. 2006, 3, 390–407. [Google Scholar] [CrossRef]
- Więckowska-Gacek, A.; Mietelska-Porowska, A.; Wydrych, M.; Wojda, U. Western diet as a trigger of Alzheimer’s disease: From metabolic syndrome and systemic inflammation to neuroinflammation and neurodegeneration. Ageing. Res. Rev. 2021, 70, 101397. [Google Scholar] [CrossRef]
- Spinedi, E.; Cardinali, D.P. Neuroendocrine-Metabolic Dysfunction and Sleep Disturbances in Neurodegenerative Disorders: Focus on Alzheimer’s Disease and Melatonin. Neuroendocrinology 2019, 108, 354–364. [Google Scholar] [CrossRef]
- Ordovas-Montanes, J.; Rakoff-Nahoum, S.; Huang, S.; Riol-Blanco, L.; Barreiro, O.; von Andrian, U.H. The Regulation of Immunological Processes by Peripheral Neurons in Homeostasis and Disease. Trends. Immunol. 2015, 36, 578–604. [Google Scholar] [CrossRef] [PubMed]
- Godinho-Silva, C.; Cardoso, F.; Veiga-Fernandes, H. Neuro-Immune Cell Units: A New Paradigm in Physiology. Ann. Rev. Immunol. 2019, 26, 19–46. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, A.J.; Beresford, L.J.; Bell, E.B.; Miyan, J.A. Mobilisation of specific T cells from lymph nodes in contact sensitivity requires substance P. J. Neuroimmunol. 2005, 164, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Veiga-Fernandes, H.; Pachnis, V. Neuroimmune regulation during intestinal development and homeostasis. Nat. Immunol. 2017, 18, 116–122. [Google Scholar] [CrossRef]
- Furness, J.B.; Kunze, W.A.; Clerc, N. Nutrient tasting and signaling mechanisms in the gut. II. The intestine as a sensory organ: Neural, endocrine, and immune responses. Am. J. Physiol. 1999, 277, G922–G928. [Google Scholar]
- Neunlist, M.; Van Landeghem, L.; Mahé, M.M.; Derkinderen, P.; des Varannes, S.B.; Rolli-Derkinderen, M. The digestive neuronal-glial-epithelial unit: A new actor in gut health and disease. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 90–100. [Google Scholar] [CrossRef]
- Liu, Y.A.; Chung, Y.C.; Pan, S.T.; Shen, M.Y.; Hou, Y.C.; Peng, S.J.; Pasricha, P.J.; Tang, S.C. 3-D imaging, illustration, and quantitation of enteric glial network in transparent human colon mucosa. Neurogastroenterol. Motil. 2013, 25, e324–e338. [Google Scholar] [CrossRef]
- Meisel, J.D.; Panda, O.; Mahanti, P.; Schroeder, F.C.; Kim, D.H. Chemosensation of bacterial secondary metabolites modulates neuroendocrine signaling and behavior of C. elegans. Cell 2014, 159, 267–280. [Google Scholar] [CrossRef]
- Makhijani, K.; Alexander, B.; Rao, D.; Petraki, S.; Herboso, L.; Kukar, K.; Batool, I.; Wachner, S.; Gold, K.S.; Wong, C.; et al. Regulation of Drosophila hematopoietic sites by Activin-β from active sensory neurons. Nat. Commun. 2017, 8, 15990. [Google Scholar] [CrossRef]
- Zeng, W.; Pirzgalska, R.M.; Pereira, M.M.; Kubasova, N.; Barateiro, A.; Seixas, E.; Lu, Y.H.; Kozlova, A.; Voss, H.; Martins, G.G.; et al. Sympathetic neuro-adipose connections mediate leptin-driven lipolysis. Cell 2015, 163, 84–94. [Google Scholar] [CrossRef]
- Fishman, R.B.; Dark, J. Sensory innervation of white adipose tissue. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1987, 253, R942–R944. [Google Scholar] [CrossRef] [PubMed]
- Song, C.K.; Schwartz, G.J.; Bartness, T.J. Anterograde transneuronal viral tract tracing reveals central sensory circuits from white adipose tissue. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, R501–R511. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.T.; Schwartz, G.J.; Nguyen, N.L.T.; Mendez, J.M.; Ryu, V.; Bartness, T.J. Leptin-sensitive sensory nerves innervate white fat. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E1338–E1347. [Google Scholar] [CrossRef] [PubMed]
- Garretson, J.T.; Szymanski, L.A.; Schwartz, G.J.; Xue, B.; Ryu, V.; Bartness, T.J. Lipolysis sensation by white fat afferent nerves triggers brown fat thermogenesis. Mol. Metab. 2016, 5, 626–634. [Google Scholar] [PubMed]
- Youngstrom, T.G.; Bartness, T.J. Catecholaminergic innervation of white adipose tissue in Siberian hamsters. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1995, 268, R744–R751. [Google Scholar] [CrossRef]
- Bartness, T.J.; Liu, Y.; Shrestha, Y.B.; Ryu, V. Neural innervation of white adipose tissue and the control of lipolysis. Front. Neuroendocrinol. 2014, 35, 473–493. [Google Scholar] [CrossRef]
- Bamshad, M.; Aoki, V.T.; Adkison, M.G.; Warren, W.S.; Bartness, T.J. Central nervous system origins of the sympathetic nervous system outflow to white adipose tissue. Am. J. Physiol. 1998, 275, R291–R299. [Google Scholar] [CrossRef]
- Harris, R.B.S. Denervation as a tool for testing sympathetic control of white adipose tissue. Physiol. Behav. 2018, 190, 3–10. [Google Scholar] [CrossRef]
- Shi, H.; Song, C.K.; Giordano, A.; Cinti, S.; Bartness, T.J. Sensory or sympathetic white adipose tissue denervation differentially affects depot growth and cellularity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R1028–R1037. [Google Scholar]
- Foster, M.T.; Bartness, T.J. Sympathetic but not sensory denervation stimulates white adipocyte proliferation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 291, R1630–R1637. [Google Scholar] [CrossRef]
- Herradon, G.; Ramos-Alvarez, M.P.; Gramage, E. Connecting Metainflammation and Neuroinflammation Through the PTN-MK-RPTPβ/ζ Axis: Relevance in Therapeutic Development. Front Pharmacol. 2019, 10, 377. [Google Scholar]
- Pagano, E.S.; Spinedi, E.; Gagliardino, J.J. White Adipose Tissue and Circadian Rhythm Dysfunctions in Obesity: Pathogenesis and Available Therapies. Neuroendocrinology. 2017, 104, 347–363. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, S.E.; Gee, L.L.; Wachtel, M.S.; Frezza, E.E. Adipose tissue: The new endocrine organ? Dig. Dis. Sci. 2009, 54, 1847–1856. [Google Scholar] [CrossRef] [PubMed]
- Lelliot, C.; Vidal Puig, A.J. Lipotoxicity, an imbalance between lipogenesis de novo and fatty acid-oxidation. Int. J. Obesity 2004, 28, S22–S28. [Google Scholar] [CrossRef]
- Olefsky, J.M. Decreased insulin binding to adipocytes and circulating monocytes from obese subjects. J. Clin. Investig. 1976, 57, 1165–1172. [Google Scholar] [CrossRef]
- Molina, J.M.; Ciaraldi, T.P.; Brady, D.; Olefsky, J.M. Decreased activation rate of insulin-stimulated glucose transport in adipocytes from obese subjects. Diabetes 1989, 38, 991–995. [Google Scholar] [CrossRef]
- Carnie, J.A.; Smith, D.G.; Mavris-Vavayannis, M. Effects of insulin on lipolysis and lipogenesis in adipocytes from genetically obese (ob/ob) mice. Biochem. J. 1979, 184, 107–112. [Google Scholar] [CrossRef]
- Pocai, A.; Lam, T.K.; Gutierrez-Juarez, R.; Obici, S.; Schwartz, G.J.; Bryan, J.; Aguilar-Bryan, L.; Rossetti, L. Hypothalamic K(ATP) channels control hepatic glucose production. Nature 2005, 434, 1026–1031. [Google Scholar] [CrossRef]
- Prodi, E.; Obici, S. Minireview: The brain as a molecular target for diabetic therapy. Endocrinology. 2006, 147, 2664–2669. [Google Scholar] [CrossRef]
- Frühbeck, G.; Gómez-Ambrosi, J.; Muruzábal, F.J.; Burrell, M.A. The adipocyte: A model for integration of endocrine and metabolic signaling in energy metabolism regulation. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E827–E84747. [Google Scholar] [CrossRef]
- Banks, A.S.; Davis, S.M.; Bates, S.H.; Myers, M.G., Jr. Activation of downstream signals by the long form of the leptin receptor. J. Biol. Chem. 2000, 275, 14563–14572. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, A.; Murakami, T.; Otani, S.; Kuwajima, M.; Shima, K. Leptin affects pancreatic endocrine functions through the sympathetic nervous system. Endocrinology 1998, 139, 3863–3870. [Google Scholar] [CrossRef] [PubMed]
- Huan, J.N.; Li, J.; Han, Y.; Chen, K.; Wu, N.; Zhao, A.Z. Adipocyte-selective reduction of the leptin receptors induced by antisense RNA leads to increased adiposity, dyslipidemia, and insulin resistance. J. Biol. Chem. 2003, 278, 45638–45650. [Google Scholar] [CrossRef] [PubMed]
- Kern, P.A.; Saghizadeh, M.; Ong, J.M.; Bosch, R.J.; Deem, R.; Simsolo, R.B. The expression of tumor necrosis factor in human adipose tissue. Regulation by obesity, weight loss, and relationship to lipoprotein lipase. J. Clin. Investig. 1995, 95, 2111–2119. [Google Scholar] [CrossRef]
- Fain, J.N.; Madan, A.K.; Hiler, M.L.; Cheema, P.; Bahouth, S.W. Comparison of the release of adipokines by adipose tissue, adipose tissue matrix, and adipocytes from visceral and subcutaneous abdominal adipose tissues of obese humans. Endocrinology 2004, 145, 2273–2282. [Google Scholar] [CrossRef]
- Okamoto, Y.; Kihara, S.; Funahashi, T.; Matsuzawa, Y.; Libby, P. Adiponectin: A key adipocytokine in metabolic syndrome. Clin. Sci. 2006, 110, 267–278. [Google Scholar] [CrossRef]
- Niswender, K.D.; Schwartz, M.W. Insulin and leptin revisited: Adiposity signals with overlapping physiological and intracellular signaling capabilities. Front. Neuroendocrinol. 2003, 24, 1–10. [Google Scholar] [CrossRef]
- Anubhuti Arora, S. Leptin and its metabolic interactions: An update. Diabetes. Obes. Metab. 2008, 10, 973–993. [Google Scholar] [CrossRef]
- Frühbeck, G. Intracellular signalling pathways activated by leptin. Biochem. J. 2006, 393, 7–20. [Google Scholar] [CrossRef]
- Uyama, N.; Geerts, A.; Reynaert, H. Neural connections between the hypothalamus and the liver. Anat. Rec. A. Discov. Mol. Cell Evol. Biol. 2004, 280, 808–820. [Google Scholar] [CrossRef]
- Flak, J.N.; Myers, M.G., Jr. CNS Mechanisms of Leptin Action. Mol. Endocrinol. 2016, 30, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Unger, R.H. Minireview: Weapons of lean body mass destruction: The role of ectopic lipids in the metabolic syndrome. Endocrinology 2003, 144, 5159–5165. [Google Scholar] [CrossRef] [PubMed]
- Morioka, T.; Asilmaz, E.; Hu, J.; Dishinger, J.F.; Kurpad, A.J.; Elias, C.F.; Li, H.; Elmquist, J.K.; Kennedy, R.T.; Kulkarni, R.N. Disruption of leptin receptor expression in the pancreas directly affects beta cell growth and function in mice. J. Clin. Investig. 2007, 117, 2860–2868. [Google Scholar] [CrossRef] [PubMed]
- Timper, K.; Bruning, J.C. Hypothalamic circuits regulating appetite and energy homeostasis: Pathways to obesity. Dis. Model. Mech. 2017, 10, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Thaler, J.P.; Yi, C.X.; Schur, E.A.; Guyenet, S.J.; Hwang, B.H.; Dietrich, M.O.; Zhao, X.; Sarruf, D.A.; Izgur, V.; Maravilla, K.R.; et al. Obesity is associated with hypothalamic injury in rodents and humans. J. Clin. Investig. 2012, 122, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Velloso, L.A.; Schwartz, M.W. Altered hypothalamic function in diet-induced obesity. Int. J. Obes. 2011, 35, 1455–1465. [Google Scholar] [CrossRef]
- Bergen, H.T.; Mizuno, T.; Taylor, J.; Mobbs, C.V. Resistance to diet-induced obesity is associated with increased proopiomelanocortin mRNA and decreased neuropeptide Y mRNA in the hypothalamus. Brain. Res. 1999, 851, 198–203. [Google Scholar] [CrossRef]
- Briggs, D.I.; Lemus, M.B.; Kua, E.; Andrews, Z.B. Diet-induced obesity attenuates fasting-induced hyperphagia. J. Neuroendocrinol. 2011, 23, 620–626. [Google Scholar] [CrossRef]
- Milanski, M.; Degasperi, G.; Coope, A.; Morari, J.; Denis, R.; Cintra, D.E.; Tsukumo, D.M.; Anhe, G.; Amaral, M.E.; Takahashi, H.K.; et al. Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: Implications for the pathogenesis of obesity. J. Neurosci. 2009, 29, 359–370. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, G.; Zhang, H.; Karin, M.; Bai, H.; Cai, D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell 2008, 135, 61–73. [Google Scholar] [CrossRef]
- Mendes, N.F.; Gaspar, J.M.; Lima-Junior, J.C.; Donato, J., Jr.; Velloso, L.A.; Araujo, E.P. TGF-beta1 down-regulation in the mediobasal hypothalamus attenuates hypothalamic inflammation and protects against diet-induced obesity. Metabolism 2018, 85, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Kim, K.K.; Park, B.S.; Kim, D.H.; Jeong, B.; Kang, D.; Lee, T.H.; Park, J.W.; Kim, J.G.; Lee, B.J. Function of astrocyte MyD88 in high-fat-diet-induced hypothalamic inflammation. J. Neuroinflamm. 2020, 17, 195. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, J.M.; Mendes, N.F.; Correa-da-Silva, F.; Lima-Junior, J.C.; Gaspar, R.C.; Ropelle, E.R.; Araujo, E.P.; Carvalho, H.M.; Velloso, L.A. Downregulation of HIF complex in the hypothalamus exacerbates diet-induced obesity. Brain. Behav. Immun. 2018, 73, 550–561. [Google Scholar] [CrossRef] [PubMed]
- De Souza, C.T.; Araujo, E.P.; Bordin, S.; Ashimine, R.; Zollner, R.L.; Boschero, A.C.; Saad, M.J.; Velloso, L.A. Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology 2005, 146, 4192–4199. [Google Scholar] [CrossRef]
- de Paula, G.C.; Brunetta, H.S.; Engel, D.F.; Gaspar, J.M.; Velloso, L.A.; Engblom, D.; de Oliveira, J.; de Bem, A.F. Hippocampal Function Is Impaired by a Short-Term High-Fat Diet in Mice: Increased Blood-Brain Barrier Permeability and Neuroinflammation as Triggering Events. Front. Neurosci. 2021, 15, 734158. [Google Scholar] [CrossRef]
- Hsu, T.M.; Konanur, V.R.; Taing, L.; Usui, R.; Kayser, B.D.; Goran, M.I.; Kanoski, S.E. Effects of sucrose and high fructose corn syrup consumption on spatial memory function and hippocampal neuroinflammation in adolescent rats. Hippocampus 2015, 25, 227–239. [Google Scholar] [CrossRef]
- Carraro, R.S.; Souza, G.F.; Solon, C.; Razolli, D.S.; Chausse, B.; Barbizan, R.; Victorio, S.C.; Velloso, L.A. Hypothalamic mitochondrial abnormalities occur downstream of inflammation in diet-induced obesity. Mol. Cell Endocrinol. 2017, 460, 238–245. [Google Scholar] [CrossRef]
- Wardzinski, E.K.; Kistenmacher, A.; Melchert, U.H.; Jauch-Chara, K.; Oltmanns, K.M. Impaired brain energy gain upon a glucose load in obesity. Metabolism. 2018, 85, 90–96. [Google Scholar] [CrossRef]
- Valdearcos, M.; Douglass, J.D.; Robblee, M.M.; Dorfman, M.D.; Stifler, D.R.; Bennett, M.L.; Gerritse, I.; Fasnacht, R.; Barres, B.A.; Thaler, J.P.; et al. Microglial Inflammatory Signaling Orchestrates the Hypothalamic Immune Response to Dietary Excess and Mediates Obesity Susceptibility. Cell Metab. 2017, 26, 185–197. [Google Scholar] [CrossRef]
- Davidson, T.L.; Hargrave, S.L.; Swithers, S.E.; Sample, C.H.; Fu, X.; Kinzig, K.P.; Zheng, W. Inter-relationships among diet, obesity and hippocampal-dependent cognitive function. Neuroscience 2013, 253, 110–122. [Google Scholar] [CrossRef]
- Kratz, M.; Baars, T.; Guyenet, S. The relationship between high-fat dairy consumption and obesity, cardiovascular, and metabolic disease. Eur. J. Nutr. 2013, 52, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Zhang, H.; Yin, Y.; Li, J.; Tang, Y.; Purkayastha, S.; Li, L.; Cai, D. Obesity- and aging-induced excess of central transforming growth factor-beta potentiates diabetic development via an RNA stress response. Nat. Med. 2014, 20, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Yuan, L.; Yu, H.; Xi, Y.; Xiao, R. Mitochondrial dysfunction and oxidative damage in the brain of diet-induced obese rats but not in diet-resistant rats. Life. Sci. 2014, 110, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Martín, M.F.; Alzamendi, A.; Harnichar, A.E.; Castrogiovanni, D.; Zubiría, M.G.; Spinedi, E.; Giovambattista, A. Role of glucocorticoid receptor (GR) in white adipose tissue beiging. Life. Sci. 2023, 322, 121681. [Google Scholar] [CrossRef] [PubMed]
- Houstis, N.; Rosen, E.D.; Lander, E.S. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 2006, 440, 944–948. [Google Scholar] [CrossRef]
- Zhang, L.; Ebenezer, P.J.; Dasuri, K.; Fernandez-Kim, S.O.; Francis, J.; Mariappan, N.; Gao, Z.; Ye, J.; Bruce-Keller, A.J.; Keller, J.N. Aging is associated with hypoxia and oxidative stress in adipose tissue: Implications for adipose function. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E599–E607. [Google Scholar] [CrossRef]
- Salgado-Somoza, A.; Teijeira-Fernández, E.; Fernández, A.L.; GonzálezJuanatey, J.R.; Eiras, S. 2010 Proteomic analysis of epicardial and subcutaneous adipose tissue reveals differences in proteins involved in oxidative stress. Am. J. Physiol. Heart. Circ. Physiol. 2010, 299, H202–H209. [Google Scholar] [CrossRef]
- Long, E.K.; Olson, D.M.; Bernlohr, D.A. High-fat diet induces changes in adipose tissue trans-4-oxo-2-nonenal and trans-4-hydroxy-2-nonenal levels in a depot-specific manner. Free. Radic. Biol. Med. 2013, 63, 390–398. [Google Scholar] [CrossRef]
- Hauck, A.K.; Zhou, T.; Hahn, W.; Petegrosso, R.; Kuang, R.; Chen, Y.; Bernlohr, D.A. Obesity-induced protein carbonylation in murine adipose tissue regulates the DNA-binding domain of nuclear zinc finger proteins. J. Biol. Chem. 2018, 293, 13464–13476. [Google Scholar] [CrossRef]
- Stanley, M.; MacAuley, S.L.; Holtzman, D.M. Changes in insulin and insulin signaling in Alzheimer’s disease: Cause or consequence? J. Exp. Med. 2016, 213, 1375–1385. [Google Scholar] [CrossRef]
- Janson, J.; Laedtke, T.; Parisi, J.E.; O’Brien, P.; Petersen, R.C.; Butler, P.C. Increased risk of type 2 diabetes in alzheimer disease. Diabetes. 2004, 53, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Talbot, K.; Wang, H.Y.; Kazi, H.; Han, L.Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef] [PubMed]
- Grillo, C.A.; Piroli, G.G.; Lawrence, R.C.; Wrighten, S.A.; Green, A.J.; Wilson, S.P.; Sakai, R.R.; Kelly, S.J.; Wilson, M.A.; Mott, D.D.; et al. Hippocampal insulin resistance impairs spatial learning and synaptic plasticity. Diabetes 2015, 64, 3927–3936. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.R.; Lyra, E.; Silva, N.M.; Figueiredo, C.P.; Frozza, R.L.; Ledo, J.H.; Beckman, D.; Katashima, C.K.; Razolli, D.; Carvalho, B.M.; et al. Alzheimer-associated oligomers impact the central nervous system to induce peripheral metabolic deregulation. Mol. Med. 2015, 7, 190–210. [Google Scholar] [CrossRef]
- Yarchoan, M.; Toledo, J.B.; Lee, E.B.; Arvanitakis, Z.; Kazi, H.; Han, L.Y.; Louneva, N.; Lee, V.M.; Kim, S.F.; Trojanowski, J.Q.; et al. Abnormal serine phosphorylation of insulin receptor substrate 1 is associated with tau pathology in Alzheimer’s disease and tauopathies. Acta Neuropathol. 2014, 128, 679–689. [Google Scholar] [CrossRef]
- Zhao, W.Q.; De Felice, F.G.; Fernandez, S.; Chen, H.; Lambert, M.P.; Quon, M.J.; Krafft, G.A.; Klein, W.L. Amyloid b oligomers induce impairment of neuronal insulin receptors. FASEB J. 2008, 22, 246–260. [Google Scholar] [CrossRef]
- Lourenco, M.V.; Ferreira, S.T.; De Felice, F.G. Neuronal stress signaling and eIF2a phosphorylation as molecular links between Alzheimer’s disease and diabetes. Prog. Neurobiol. 2015, 129, 37–57. [Google Scholar] [CrossRef]
- Aviles-Olmos, I.; Limousin, P.; Lees, A.; Foltynie, T. Parkinson’s disease, insulin resistance and novel agents of neuroprotection. Brain 2013, 136, 374–384. [Google Scholar] [CrossRef]
- Perry, V.H. The influence of systemic inflammation on inflammation in the brain: Implications for chronic neurodegenerative disease. Brain. Behav. Immun. 2004, 18, 407–413. [Google Scholar] [CrossRef]
- Stark, R.; Roden, M. ESCI Award 2006. Mitochondrial function and endocrine diseases. Eur. J. Clin. Investig. 2007, 37, 236–248. [Google Scholar] [CrossRef]
- Rantham Prabhakara, J.P.; Feist, G.; Thomasson, S.; Thompson, A.; Schommer, E.; Ghribi, O. Differential effects of 24-hydroxycholesterol and 27-hydroxycholesterol on tyrosine hydroxylase and alpha-synuclein in human neuroblastoma SH-SY5Y cells. J. Neurochem. 2008, 107, 1722–1729. [Google Scholar] [CrossRef] [PubMed]
- Li, L.-Y.; Liu, S.-F.; Zhuang, J.L.; Li, M.M.; Huang, Z.-P.; Chen, Y.H.; Chen, X.-R.; Chen, C.-N.; Li-Chao Ye, S.L. Recent research progress on metabolic syndrome and risk of Parkinson’s disease. Rev. Neurosci. 2022, 34, 719–735. [Google Scholar] [CrossRef] [PubMed]
- Miraglia, F.; Colla, E. Microbiome, Parkinson’s disease and molecular mimicry. Cells 2019, 8, 222. [Google Scholar] [CrossRef] [PubMed]
- Erny, D.; Hrabě de Angelis, A.L.; Jaitin, D.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T.; et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 2015, 18, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Liao, J.; Xia, Y.; Liu, X.; Jones, R.; Haran, J.; McCormick, B.; Sampson, T.R.; Alam, A.; Ye, K. Gut microbiota regulate Alzheimer’s disease pathologies and cognitive disorders via PUFA-associated neuroinflammation. Gut 2022, 71, 2233–2252. [Google Scholar] [CrossRef]
- Warnecke, T.; Schäfer, K.-H.; Claus, I.; Del Tredici, K.; Jost, W.H. Gastrointestinal involvement in Parkinson’s disease: Pathophysiology, diagnosis, and management. NPJ. Parkinsons. Dis. 2022, 8, 31. [Google Scholar] [CrossRef]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell 2016, 167, 1469–1480. [Google Scholar] [CrossRef]
- Seo, D.-o.; Holtzman, D.M. Current understanding of the Alzheimer’s disease-associated microbiome and therapeutic strategies. Exp. Mol. Med. 2024, 56, 86–94. [Google Scholar] [CrossRef]
- van de Pavert, S.; Oliver, B.J.; Goverse, G.; Vondenhoff, M.F.; Greuter, M.; Beke, P.; Kusser, K.; Höpken, U.E.; Lipp, M.; Niederreither, K.; et al. Chemokine CXCL13 is essential for lymph node initiation and is induced by retinoic acid and neuronal stimulation. Nat. Immunol. 2009, 10, 1193–1199. [Google Scholar] [CrossRef]
- Patel, A.; Harker, N.; Moreira-Santos, L.; Ferreira, M.; Alden, K.; Timmis, J.; Foster, K.; Garefalaki, A.; Pachnis, P.; Andrews, P.; et al. Differential RET signaling pathways drive development of the enteric lymphoid and nervous systems. Sci. Signal. 2012, 5, ra55. [Google Scholar] [CrossRef]
- van de Pavert, S.A.; Ferreira, M.; Domingues, R.G.; Ribeiro, H.; Molenaar, R.; Moreira-Santos, L.; Almeida, F.F.; Ibiza, S.; Barbosa, I.; Goverse, G. Maternal retinoids control type 3 innate lymphoid cells and set the offspring immunity. Nature 2014, 508, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.; Artis, D.; Chiu, I.M. Neuro-immune Interactions in the Tissues. Immunity 2020, 52, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Veiga-Fernandes, H.; Mucida, D. Neuro-Immune Interactions at Barrier Surfaces. Cell 2016, 165, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Kouassi, E.; Li, S.Y.; Boukhris, W.; Millet, I.; Revillard, J.P. Opposite effects of the catecholamines dopamine and norepinephrine on murine polyclonal B-cell activation. Immunopharmacology 1988, 16, 125–137. [Google Scholar] [CrossRef]
- Kruszewska, B.; Felten, S.Y.; Moynihan, J.A. Alterations in cytokine and antibody production following chemical sympathectomy in two strains of mice. J. Immunol. 1995, 155, 4613–4620. [Google Scholar] [CrossRef]
- Veiga-Fernandes, H.; Coles, M.C.; Foster, K.E.; Patel, A.; Williams, A.; Natarajan, D.; Barlow, A.; Pachnis, V.; Kioussis, D. Tyrosine kinase receptor RET is a key regulator of Peyer’s patch organogenesis. Nature 2007, 446, 547–551. [Google Scholar] [CrossRef]
- Vulchanova, L.; Casey, M.A.; Crabb, G.W.; Kennedy, W.R.; Brown, D.R. Anatomical evidence for enteric neuroimmune interactions in Peyer’s patches. J. Neuroimmunol. 2007, 185, 64–74. [Google Scholar] [CrossRef]
- Ma, B.; von Wasielewski, R.; Lindenmaier, W.; Dittmar, K.E.J. Immmunohistochemical study of the blood and lymphatic vasculature and the innervation of mouse gut and gut-associated lymphoid tissue. Anat. Histol. Embryol. 2007, 36, 62–74. [Google Scholar] [CrossRef]
- Kasprowicz, D.J.; Kohm, A.P.; Berton, M.T.; Chruscinski, A.J.; Sharpe, A.; Sanders, M.V. Stimulation of the B Cell Receptor, CD86 (B7-2), and the β2-Adrenergic Receptor Intrinsically Modulates the Level of IgG1 and IgE Produced per B Cell. J. Immunol. 2000, 165, 680–690. [Google Scholar] [CrossRef]
- Straub, R.H.; Pongratz, G.; Weidler, C.; Linde, H.-J.; Kirschning, C.J.; Glück, T.; Schölmerich, J.; Falk, W. Ablation of the sympathetic nervous system decreases gram-negative and increases gram-positive bacterial dissemination: Key roles for tumor necrosis factor/phagocytes and interleukin-4/lymphocytes. J. Infect. Dis. 2005, 192, 560–572. [Google Scholar] [CrossRef]
- Cheadle, G.A.; Costantini, T.W.; Bansal, V.; Eliceiri, B.P.; Coimbra, R. Cholinergic signaling in the gut: A novel mechanism of barrier protection through activation of enteric glia cells. Surg. Infect. 2014, 15, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, D.; Miller, J.; Merad, M. Dendritic cell and macrophage heterogeneity in vivo. Immunity 2011, 35, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Rescigno, M.; Urbano, M.; Valzasina, B.; Francolini, M.; Rotta, G.; Bonasio, R.; Granucci, F.; Kraehenbuhl, J.P.; Ricciardi-Castagnoli, P. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat. Immunol. 2001, 2, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Niess, J.H.; Brand, S.; Gu, X.; Landsman, L.; Jung, S.; McCormick, B.A.; Vyas, J.M.; Boes, M.; Ploegh, H.L.; Fox, J.G.; et al. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science 2005, 307, 254–258. [Google Scholar] [CrossRef]
- Mazzini, E.; Massimiliano, L.; Penna, G.; Rescigno, M. Oral tolerance can be established via gap junction transfer of fed antigens from CX3CR1 macrophages to CD103 dendritic cells. Immunity 2011, 35, 323–335. [Google Scholar] [CrossRef]
- Rescigno, M. Intestinal dendritic cells. Adv. Immunol. 2010, 107, 109–138. [Google Scholar]
- Scott, C.L.; Aumeunier, A.M.; Mowat, A.M. Intestinal CD103+ dendritic cells: Master regulators of tolerance? Trends. Immunol. 2011, 32, 412–419. [Google Scholar] [CrossRef]
- Hadis, U.; Wahl, B.; Schulz, O.; Hardtke-Wolenski, M.; Schippers, A.; Wagner, N.; Müller, W.; Sparwasser, T.; Förster, R.; Pabst, O. Intestinal tolerance requires gut homing and expansion of FoxP3+ regulatory T cells in the lamina propria. Immunity 2011, 34, 237–246. [Google Scholar] [CrossRef]
- Shakhar, G.; Kolesnikov, M. Intestinal macrophages and DCs close the gap o tolerance. Immunity 2014, 40, 171–173. [Google Scholar] [CrossRef]
- De Schepper, S.; Stakenborg, N.; Matteoli, G.; Verheijden, S.; Boeckxstaens, G.E. Muscularis macrophages: Key players in intestinal homeostasis and disease. Cell Immunol. 2018, 330, 142–150. [Google Scholar] [CrossRef]
- Gabanyi, I.; Muller, P.A.; Feighery, L.; Oliveira, T.Y.; Costa-Pinto, F.A.; Mucida, D. Neuro-immune Interactions Drive Tissue Programming in Intestinal Macrophages. Cell 2016, 164, 378–391. [Google Scholar] [CrossRef] [PubMed]
- Estévez, A.G.; Sahawneh, M.A.; Lange, P.S.; Bae, N.; Egea, M.; Ratan, R.R. Arginase 1 regulation of nitric oxide production is key to survival of trophic factor-deprived motor neurons. J. Neurosci. 2006, 26, 8512–8516. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, B.E.; Midtvedt, T.; Strandberg, K. Effects of microbial contamination on the cecum enlargement of germfree rats. Scand. J. Gastroenterol. 1970, 5, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Koscsó, B.; Rajani, G.M.; Stevanovic, K.; Berres, M.L.; Hashimoto, D.; Mortha, A.; Leboeuf, M.; Li, X.-M.; Mucida, D.; et al. Crosstalk between muscularis macrophages andentericneurons regulates gastrointestinal motility. Cell 2014, 158, 300–313. [Google Scholar] [CrossRef]
- Stead, R.H.; Dixon, M.F.; Bramwell, N.H.; Riddell, R.H.; Bienenstock, J. Mast cells are closely apposed to nerves in the human gastrointestinal mucosa. Gastroenterology 1989, 97, 575–585. [Google Scholar] [CrossRef]
- Wu, J.J.; Rothman, T.P.; Gershon, M.D. Development of the interstitial cell of Cajal: Origin, kit dependence and neuronal and nonneuronal sources of kit ligand. J. Neurosci. Res. 2000, 59, 384–401. [Google Scholar] [CrossRef]
- Brandt, E.B.; Strait, R.T.; Hershko, D.; Wang, Q.; Muntel, E.E.; Scribner, T.A.; Zimmermann, N.; Finkelman, F.D.; Rothenberg, M.E. Mast cells are required for experimental oral allergen-induced diarrhea. J. Clin. Investig. 2003, 112, 1666–1677. [Google Scholar] [CrossRef]
- De Winter, B.Y.; van den Wijngaard, R.M.; de Jonge, W.J. Intestinal mast cells in gut inflammation and motility disturbances. Biochim. Biophys. Acta 2012, 1822, 66–73. [Google Scholar] [CrossRef]
- Barbara, G.; Wang, B.; Stanghellini, V.; de Giorgio, R.; Cremon, C.; Di Nardo, G.; Trevisani, M.; Campi, B.; Geppetti, P.; Tonini, M.; et al. Mast cell-dependent excitation of visceral-nociceptive sensory neurons in irritable bowel syndrome. Gastroenterology 2007, 132, 26–37. [Google Scholar] [CrossRef]
- Cheng, L.; Luo, Q.Q.; Chen, S.L. The role of intestinal mast cell infiltration in irritable bowel syndrome. J. Dig. Dis. 2021, 22, 143–151. [Google Scholar] [CrossRef]
- Nystrom, E.E.L.; Birchenough, G.M.H.; van der Post, S.; Arike, L.; Gruber, A.D.; Hansson, G.C.; Johansson, M.E.V. Calcium-activated Chloride Channel Regulator 1 (CLCA1) Controls Mucus Expansion in Colon by Proteolytic Activity. EBioMedicine 2018, 33, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Jacobson, A.; Meerschaert, K.A.; Sifakis, J.J.; Wu, M.; Chen, X.; Yang, T.; Zhou, Y.; Anekal, P.V.; Rucker, R.A.; et al. Nociceptor neurons direct goblet cells via a CGRP-RAMP1 axis to drive mucus production and gut barrier protection. Cell 2022, 185, 4190–4205.e25. [Google Scholar] [CrossRef] [PubMed]
- Bennstein, S.B.; Uhrberg, M. Biology and therapeutic potential of human innate lymphoid cells. FEBS J. 2022, 289, 3967–3981. [Google Scholar] [CrossRef] [PubMed]
- Klose, C.S.N.; Mahlakõiv, T.; Moeller, J.B.; Rankin, L.C.; Flamar, A.L.; Kabata, H.; Monticelli, L.A.; Moriyama, S.; Putzel, G.G.; Rakhilin, N.; et al. The neuropeptide neuromedin U stimulates innate lymphoid cells and type 2 inflammation. Nature 2017, 549, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Tracey, K.J. Physiology and immunology of the cholinergic antiinflammatory pathway. J. Clin. Investig. 2007, 117, 289–296. [Google Scholar] [CrossRef]
- Matteoli, G.; Boeckxstaens, G.E. The vagal innervation of the gut and immune homeostasis. Gut 2013, 62, 1214–1222. [Google Scholar] [CrossRef]
- Galle-Treger, L.; Suzuki, Y.; Patel, N.; Sankaranarayanan, I.; Aron, J.L.; Hadi Maazi, H.; Chen, L.; Akbari, O. Nicotinic acetylcholine receptor agonist attenuates ILC2-dependent airway hyperreactivity. Nat. Comm. 2016, 7, 13202. [Google Scholar] [CrossRef]
- Dalli, J.; Colas, R.A.; Arnardottir, H.; Serhan, C.N. Vagal Regulation of Group 3 Innate Lymphoid Cells and the Immunoresolvent PCTR1 Controls Infection Resolution. Immunity 2017, 46, 92–105. [Google Scholar] [CrossRef]
- Moriyama, S.; Brestoff, J.R.; Flamar, A.-L.; Moeller, J.B.; Klose, C.S.N.; Rankin, L.C.; Yudanin, L.A.; Monticelli, L.A.; Putzel, G.B.; Rodewald, H.-R.; et al. β2-adrenergic receptor-mediated negative regulation of group 2 innate lymphoid cell responses. Science 2018, 359, 1056–1061. [Google Scholar] [CrossRef]
- Wallrapp, A.; Riesenfeld, S.J.; Burkett, P.R.; Abdulnour, R.E.; Nyman, J.; Dionne, D.; Hofree, M.; Cuoco, M.S.; Rodman, C.; Farouq, D.; et al. The neuropeptide NMU amplifies ILC2-driven allergic lung inflammation. Nature 2017, 549, 351–356. [Google Scholar] [CrossRef]
- Quatrini, L.; Wieduwild, E.; Guia, S.; Bernat, C.; Glaichenhaus, N.; Vivier, E.; Ugolini, S. Host resistance to endotoxic shock requires the neuroendocrine regulation of group 1 innate lymphoid cells. J. Exp. Med. 2017, 214, 3531–3541. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.N.; Guo, Y.B.; Li, X.; Li, C.L.; Tan, W.P.; Fan, X.L.; Qin, Z.L.; Chen, D.; Wen, W.P.; Zheng, S.G.; et al. ILC2 frequency and activity are inhibited by glucocorticoid treatment via STAT pathway in patients with asthma. Allergy 2018, 73, 1860–1870. [Google Scholar] [CrossRef] [PubMed]
- Talbot, S.; Abdulnour, R.E.; Burkett, P.R.; Lee, S.; Cronin, S.J.; Pascal, M.A.; Laedermann, C.; Foster, S.L.; Tran, J.V.; Lai, N.; et al. Silencing Nociceptor Neurons Reduces Allergic Airway Inflammation. Neuron 2015, 87, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Seillet, C.; Luong, K.; Tellier, J.; Jacquelot, N.; Shen, R.D.; Hickey, P.; Wimmer, V.C.; Whitehead, L.; Rogers, K.; Smyth, G.K.; et al. The neuropeptide VIP confers anticipatory mucosal immunity by regulating ILC3 activity. Nat. Immunol. 2020, 21, 168–177. [Google Scholar] [CrossRef]
- Takenaka, M.C.; Araujo, L.P.; Maricato, J.T.; Nascimento, V.M.; Guereschi, M.G.; Rezende, R.M.; Quintana, F.J.; Basso, A.S. Norepinephrine Controls Effector T Cell Differentiation through beta2-Adrenergic Receptor-Mediated Inhibition of NF-kappaB and AP-1 in Dendritic Cells. J. Immunol. 2016, 196, 637–644. [Google Scholar] [CrossRef]
- Maestroni, G.J. Dendritic cell migration controlled by alpha 1b-adrenergic receptors. J. Immunol. 2000, 165, 6743–6747. [Google Scholar] [CrossRef]
- Delgado, M.; Chorny, A.; Gonzalez-Rey, E.; Ganea, D. Vasoactive intestinal peptide generates CD4+CD25+ regulatory T cells in vivo. J. Leukoc. Biol. 2005, 78, 1327–1338. [Google Scholar] [CrossRef]
- Padro, C.J.; Sanders, V.M. Neuroendocrine regulation of inflammation. Semin. Immunol. 2014, 26, 357–368. [Google Scholar] [CrossRef]
- Nakai, A.; Hayano, Y.; Furuta, F.; Noda, M.; Suzuki, K. Control of lymphocyte egress from lymph nodes through β2-adrenergic receptors. J. Exp. Med. 2014, 211, 2583–2598. [Google Scholar] [CrossRef]
- Bonaz, B.; Sinniger, V.; Hoffmann, D.; Clarençon, D.; Mathieu, N.; Dantzer, C.; Vercueil, L.; Picq, C.; Trocmé, C.; Faure, P.; et al. Chronic vagus nerve stimulation in Crohn’s disease: A 6-month follow-up pilot study. Neurogastroenterol. Motil. 2016, 28, 948–953. [Google Scholar] [CrossRef]
- Koopman, F.A.; Chavan, S.S.; Miljko, S.; Grazio, S.; Sokolovic, S.; Schuurman, P.R.; Mehta, A.D.; Levine, Y.A.; Faltys, M.; Zitnik, R.; et al. Vagus nerve stimulation inhibits cytokine production and attenuates disease severity in rheumatoid arthritis. Proc. Natl. Acad. Sci. USA 2016, 113, 8284–8289. [Google Scholar] [CrossRef] [PubMed]
- Guarini, S.; Altavilla, D.; Cainazzo, M.M.; Giuliani, D.; Bigiani, A.; Marini, H.; Squadrito, G.; Minutoli, L.; Bertolini, A.; Marini, R.; et al. Efferent vagal fibre stimulation blunts nuclear factor-kappaB activation and protects against hypovolemic hemorrhagic shock. Circulation 2003, 107, 1189–1194. [Google Scholar] [CrossRef] [PubMed]
- Levine, Y.A.; Koopman, F.A.; Faltys, M.; Caravaca, A.; Bendele, A.; Zitnik, R.; Margriet, J.; Vervoordeldonk, M.J.; Tak, P.P. Neurostimulation of the cholinergic anti-inflammatory pathway ameliorates disease in rat collagen-induced arthritis. PLoS ONE 2014, 9, e104530. [Google Scholar] [CrossRef] [PubMed]
- Carloni, S.; Rescigno, M. The gut-brain vascular axis in neuroinflammation. Semin. Immunol. 2023, 69, 101802. [Google Scholar] [CrossRef] [PubMed]
- Braniste, V.; Al-Asmakh, M.; Kowal, C.; Anuar, F.; Abbaspour, A.; Tóth, M.; Korecka, A.; Bakocevic, N.; Ng, L.G.; Kundu, P. The gut microbiota influences blood-brain barrier permeability in mice. Sci. Transl. Med. 2014, 19, 263ra158. [Google Scholar] [CrossRef]
- Mouries, J.P.; Silvestri, A.; Spadoni, I.; Sorribas, M.; Wiest, R.; Mileti, E.; Galbiati, M.P.; Adorini, L.; Penna, G.; Rescigno, M. Microbiota-driven gut vascular barrier disruption is a prerequisite for non-alcoholic steatohepatitis development. J. Hepatol. 2019, 71, 1216–1228. [Google Scholar] [CrossRef]
- Häuser, W.; Janke, K.-H.; Klump, B.; Hinz, A. Anxiety and depression in patients with inflammatory bowel disease: Comparisons with chronic liver disease patients and the general population. Inflamm. Bowel. Dis. 2011, 17, 621–632. [Google Scholar] [CrossRef]
- Panara, A.J.; Yarur, A.J.; Rieders, B.; Proksell, S.; Deshpande, A.R.; Abreu, M.T.; Sussman, D.A. The incidence and risk factors for developing depression after being diagnosed with inflammatory bowel disease: A cohort study. Aliment. Pharmacol. Ther. 2014, 39, 802–810. [Google Scholar] [CrossRef]
- Di Tommaso, N.; Santopaolo, F.; Gasbarrini, A.; Ponziani, F.R. The Gut-Vascular Barrier as a New Protagonist in Intestinal and Extraintestinal Diseases. Int. J. Mol. Sci. 2023, 24, 1470. [Google Scholar] [CrossRef]
- Obata, Y.; Castaño, A.; Boeing, S.; Bon-Frauches, A.C.; Fung, C.; Fallesen, T.; Gomez de Agüero, N.; Yilmaz, B.; Lopes, R.; Huseynova, A. Neuronal programming by microbiota regulates intestinal physiology. Nature 2020, 578, 284–289. [Google Scholar] [CrossRef]
- Sudo, N.; Chida, Y.; Aiba, Y.; Sonoda, J.; Oyama, N.; Yu, X.-N.; Kubo, C.; Koga, Y. Postnatal microbial colonization programs the hypothalamic-pituitary-adrenal system for stress response in mice. J. Physiol. 2004, 558, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Farzi, A.; Reichmann, F.; Meinitzer, A.; Mayerhofer, R.; Jain, P.; Hassan, A.M.; Fröhlich, E.E.; Wagner, K.E.; Rinner, B.; Holzer, P. Synergistic effects of NOD1 or NOD2 and TLR4 activation on mouse sickness behavior in relation to immune and brain activity markers. Brain. Behav. Immun. 2015, 44, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Savignac, H.M.; Tramullas, M.; Kiely, B.; Dinan, T.G.; Cryan, J.F. Bifidobacteria modulate cognitive processes in an anxious mouse strain. Behav. Brain. Res. 2015, 287, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, E.E.; Farzi, A.; Mayerhofer, R.; Reichmann, F.; Jačan, A.; Wagner, B.; Zinser, E.; Bordag, N.; Magnes, C.; Fröhlich, E.; et al. Cognitive impairment by antibiotic-induced gut dysbiosis: Analysis of gut microbiota-brain communication. Brain. Behav. Immun. 2016, 56, 140–155. [Google Scholar] [CrossRef]
- Fung, T.C.; Olson, C.A.; Hsiao, E.Y. Interactions between the microbiota, immune and nervous systems in health and disease. Nat. Neurosci. 2017, 20, 145–155. [Google Scholar] [CrossRef]
- Sherwin, E.; Dinan, T.G.; Cryan, J.F. Recent developments in understanding the role of the gut microbiota in brain health and disease. Ann. NY Acad. Sci. 2018, 1420, 5–25. [Google Scholar] [CrossRef]
- Forsythe, P.; Bienenstock, J.; Kunze, W.A. Vagal pathways for microbiome-brain-gut axis communication. Adv. Exp. Med. Biol. 2014, 817, 115–133. [Google Scholar]
- Lyte, M.; Li, W.; Opitz, N.; Gaykema, R.P.A.; Goehler, L.E. Induction of anxiety-like behavior in mice during the initial stages of infection with the agent of murine colonic hyperplasia Citrobacter rodentium. Physiol. Behav. 2006, 89, 350–357. [Google Scholar] [CrossRef]
- Tanida, M.; Yamano, T.; Maeda, K.; Okumura, N.; Fukushima, Y.; Nagai, K. Effects of intraduodenal injection of Lactobacillus johnsonii La1 on renal sympathetic nerve activity and blood pressure in urethane-anesthetized rats. Neurosci. Lett. 2005, 389, 109–114. [Google Scholar] [CrossRef]
- Takagi, H.; Kuruma, I. Effect of bacterial lipopolysaccharide on the content of serotonin and norepinephrine in rabbit brain. Jpn. J. Pharmacol. 1966, 16, 478–479. [Google Scholar] [CrossRef]
- Gaillard, P.J.; de Boer, A.B.G.; Breimer, D.D. Pharmacological investigations on lipopolysaccharide-induced permeability changes in the blood-brain barrier in vitro. Microvasc. Res. 2003, 65, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Hoyles, L.; Snelling, T.; Umlai, U.-K.; Jeremy, K.V.; Nicholson, J.K.; Carding, S.R.; Glem, R.C. Microbiome-host systems interactions: Protective effects of propionate upon the blood-brain barrier McArthur, S. Microbiome-host systems interactions: Protective effects of propionate upon the blood-brain barrier. Microbiome 2018, 6, 55. [Google Scholar] [CrossRef] [PubMed]
- Clarke, G.; Grenham, S.; Scully, P.; Fitzgerald, P.; Moloney, R.D.; Shanahan, F.; Dinan, T.G.; Cryan, J.F. The microbiome-gut-brain axis during early life regulates the hippocampal serotonergic system in a sex-dependent manner. Mol. Psychiatry 2013, 18, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Mikocka-Walus, A.; Knowles, S.R.; Keefer, L.; Graff, L. Controversies revisited: A systematic review of the comorbidity of depression and anxiety with inflammatory bowel diseases. Inflamm. Bowel. Dis. 2016, 22, 752–762. [Google Scholar] [CrossRef]
- Neuendorf, R.; Harding, A.; Stello, N.; Hanes, D.; Wahbeh, H. Depression and anxiety in patients with inflammatory bowel diseases: A systematic review. J. Psychosom. Res. 2016, 87, 70–80. [Google Scholar] [CrossRef]
- Nikolaus, S.; Schulte, B.; Al-Massad, N.; Thieme, F.; Schulte, D.M.; Bethge, J.; Rehman, A.; Tran, F.; Aden, K.; Häsler, R.; et al. Increased tryptophan metabolism is associated with activity of inflammatory bowel diseases. Gastroenterology 2017, 153, 1504–1516. [Google Scholar] [CrossRef]
- Sobesky, J.L.; Barrientos, R.M.; De May, H.S.; Thompson, B.M.; Weber, M.D.; Watkins, L.R.; Maier, S.F. High-fat diet consumption disrupts memory and primes elevations in hippocampal IL-1β, an effect that can be prevented with dietary reversal or IL-1 receptor antagonism. Brain. Behav. Immun. 2014, 42, 22–32. [Google Scholar] [CrossRef]
- de La Serre, C.B.; Ellis, C.L.; Lee, J.; Hartman, A.L.; Rutledge, J.C.; Raybould, H.E. Propensity to high-fat diet-induced obesity in rats is associated with changes in the gut microbiota and gut inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G440–G448. [Google Scholar] [CrossRef]
- Hamilton, M.K.; Boudry, G.; Lemay, D.G.; Raybould, H.E. Changes in intestinal barrier function and gut microbiota in high-fat diet-fed rats are dynamic and region dependent. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G840–G851. [Google Scholar] [CrossRef]
- Vaughn, A.C.; Cooper, E.M.; DiLorenzo, P.M.; O’Loughlin, L.J.; Konkel, M.E.; Peters, J.H.; Hajnal, A.; Sen, T.; Lee, S.H.; de La Serre, C.B.; et al. Energy-dense diet triggers changes in gut microbiota, reorganization of gut-brain vagal communication and increases body fat accumulation. Acta Neurobiol. Exp. 2017, 77, 18–30. [Google Scholar] [CrossRef]
- Hildebrandt, M.A.; Hoffman, C.; Sherrill-Mix, S.A.; Keilbaugh, S.A.; Hamady, M.; Chen, Y.-Y.; Knight, R.; Ahima, R.S.; Bushman, F.; Wu, G.D. High Fat Diet Determines the composition of the murine gut microbiome independently of obesity. Gastroenterology 2009, 137, 1716–1724. [Google Scholar] [CrossRef]
- Kim, J.S.; Kirkland, R.A.; Lee, S.H.; Cawthon, C.R.; Rzepka, K.W.; Minaya, D.M.; De Lartigue, G.; Czaja, K.; de La Serre, C.B. Gut microbiota composition modulates inflammation and structure of the vagal afferent pathway. Physiol. Behav. 2020, 225, 113082. [Google Scholar] [CrossRef]
- Gannon, O.J.; Robison, L.S.; Salinero, A.E.; Abi-Ghanem, C.; Mansour, F.M.; Kelly, R.D.; Tyagi, A.; Brawley, R.R.; Ogg, J.D.; Zuloaga, K.L. High-fat diet exacerbates cognitive decline in mouse models of Alzheimer’s disease and mixed dementia in a sex-dependent manner. J. Neuroinflamm. 2022, 19, 110. [Google Scholar] [CrossRef]
- Jones, N.S.; Watson, K.Q.; Rebeck, G.W. High-fat diet increases gliosis and immediate early gene expression in APOE3 mice, but not APOE4 mice. J. Neuroinflamm. 2021, 18, 214. [Google Scholar] [CrossRef]
- Vega-Torres, J.D.; Ontiveros-Angel, P.; Stuffle, E.C.; Solak, S.; Terrones, E.; Tyner, E.; Oropeza, M.; Dela Peña, I.; Obenaus, A.; Ford, B.D.; et al. Short-term exposure to an obesogenic diet during adolescence elicits anxiety-related behavior and neuroinflammation: Modulatory effects of exogenous neuregulin-1. Transl. Psychiatry 2022, 12, 83. [Google Scholar] [CrossRef]
- Lee, B.; Choi, G.M.; Sur, B. Silibinin prevents depression-like behaviors in a single prolonged stress rat model: The possible role of serotonin. BMC Complement. Med. Ther. 2020, 20, 70. [Google Scholar] [CrossRef]
- O’Mahony, S.M.; Clarke, G.; Borre, Y.E.; Dinan, T.G.; Cryan, J.F. Serotonin, tryptophan metabolism and the brain-gut-microbiome axis. Behav. Brain Res. 2015, 277, 32–48. [Google Scholar] [CrossRef]
- Pirzgalska, R.M.; Seixas, E.; Seidman, J.S.; Link, V.M.; Sánchez, N.M.; Mahú, I.; Mendes, R.; Gres, V.; Kubasova, N.; Morris, I.; et al. Sympathetic neuron associated macrophages contribute to obesity by importing and metabolizing norepinephrine. Nat. Med. 2017, 23, 1309–1318. [Google Scholar] [CrossRef]
- Fischer, K.; Ruiz, H.H.; Jhun, K.; Finan, B.; Oberlin, D.J.; van der Heide, V.; Kalinovich, A.V.; Petrovic, N.; Wolf, Y.; Clemmensen, C. Alternatively activated macrophages do not synthesize catecholamines or contribute to adipose tissue adaptive thermogenesis. Nat. Med. 2017, 23, 623. [Google Scholar] [CrossRef]
- Gaskell, W.H. On the structure, distribution and function of the nerves which innervate the visceral and vascular systems. J. Physiol. 1886, 7, 1–80. [Google Scholar] [CrossRef]
- Berthoud, H.; Carlson, N.; Powley, T. Topography of efferent vagal innervation of the rat gastrointestinal tract. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1991, 260, R200–R207. [Google Scholar] [CrossRef] [PubMed]
- Bi, S.; Chen, J.; Behles, R.R.; Hyun, J.; Kopin, A.S.; Moran, T.H. Differential body weight and feeding responses to high-fat diets in rats and mice lacking cholecystokinin 1 receptors. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R55–R63. [Google Scholar] [CrossRef] [PubMed]
- Paulino, G.; Barbier de la Serre, C.; Knotts, T.A.; Oort, P.J.; Newman, J.W.; Adams, S.H.; Raybould, H.E. Increased expression of receptors for orexigenic factors in nodose ganglion of diet-induced obese rats. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E898–E903. [Google Scholar] [CrossRef] [PubMed]
- de Lartigue, G.; Ronveaux, C.C.; Raybould, H.E. Deletion of leptin signaling in vagal afferent neurons results in hyperphagia and obesity. Mol. Metab. 2014, 3, 595–607. [Google Scholar] [CrossRef]
- Minnone, G.; De Benedetti, F.; Bracci-Laudiero, L. NGF and its receptors in the regulation of inflammatory response. IJMS 2017, 18, 1028. [Google Scholar] [CrossRef]
- Reinshagen, M.; Rom, H.; Steinkamp, M.; Lieb, K.; Geerling, I.; Von Herbay, A.; Flamig, G.; Eysselein, V.E.; Adler, G. Protective role of neurotrophins in experimental inflammation of the rat gut. Gastroenterology 2000, 119, 368–376. [Google Scholar] [CrossRef]
- Harrington, A.M.; Brierley, S.M.; Isaacs, N.; Hughes, P.A.; Castro, J.; Blackshaw, L.A. Sprouting of colonic afferent central terminals and increased spinal mitogen-activated protein kinase expression in a mouse model of chronic visceral hypersensitivity. J. Comp. Neurol. 2012, 520, 2241–2255. [Google Scholar] [CrossRef]
- Ansari, A.Z.; Bose, S.; Yadav, M.K.; Wang, J.H.; Song, Y.-K.; Ko, S.-G.; Kim, H. CST, an Herbal Formula, Exerts Anti-Obesity Effects through Brain-Gut-Adipose Tissue Axis Modulation in High-Fat Diet Fed Mice. Molecules. 2016, 21, 1522. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spinedi, E.; Docena, G.H. Physiopathological Roles of White Adiposity and Gut Functions in Neuroinflammation. Int. J. Mol. Sci. 2024, 25, 11741. https://doi.org/10.3390/ijms252111741
Spinedi E, Docena GH. Physiopathological Roles of White Adiposity and Gut Functions in Neuroinflammation. International Journal of Molecular Sciences. 2024; 25(21):11741. https://doi.org/10.3390/ijms252111741
Chicago/Turabian StyleSpinedi, Eduardo, and Guillermo Horacio Docena. 2024. "Physiopathological Roles of White Adiposity and Gut Functions in Neuroinflammation" International Journal of Molecular Sciences 25, no. 21: 11741. https://doi.org/10.3390/ijms252111741
APA StyleSpinedi, E., & Docena, G. H. (2024). Physiopathological Roles of White Adiposity and Gut Functions in Neuroinflammation. International Journal of Molecular Sciences, 25(21), 11741. https://doi.org/10.3390/ijms252111741