
Molecular Dynamics Simulations of Nucleosomes Containing Histone Variant H2A.J

 , ,
, ,
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
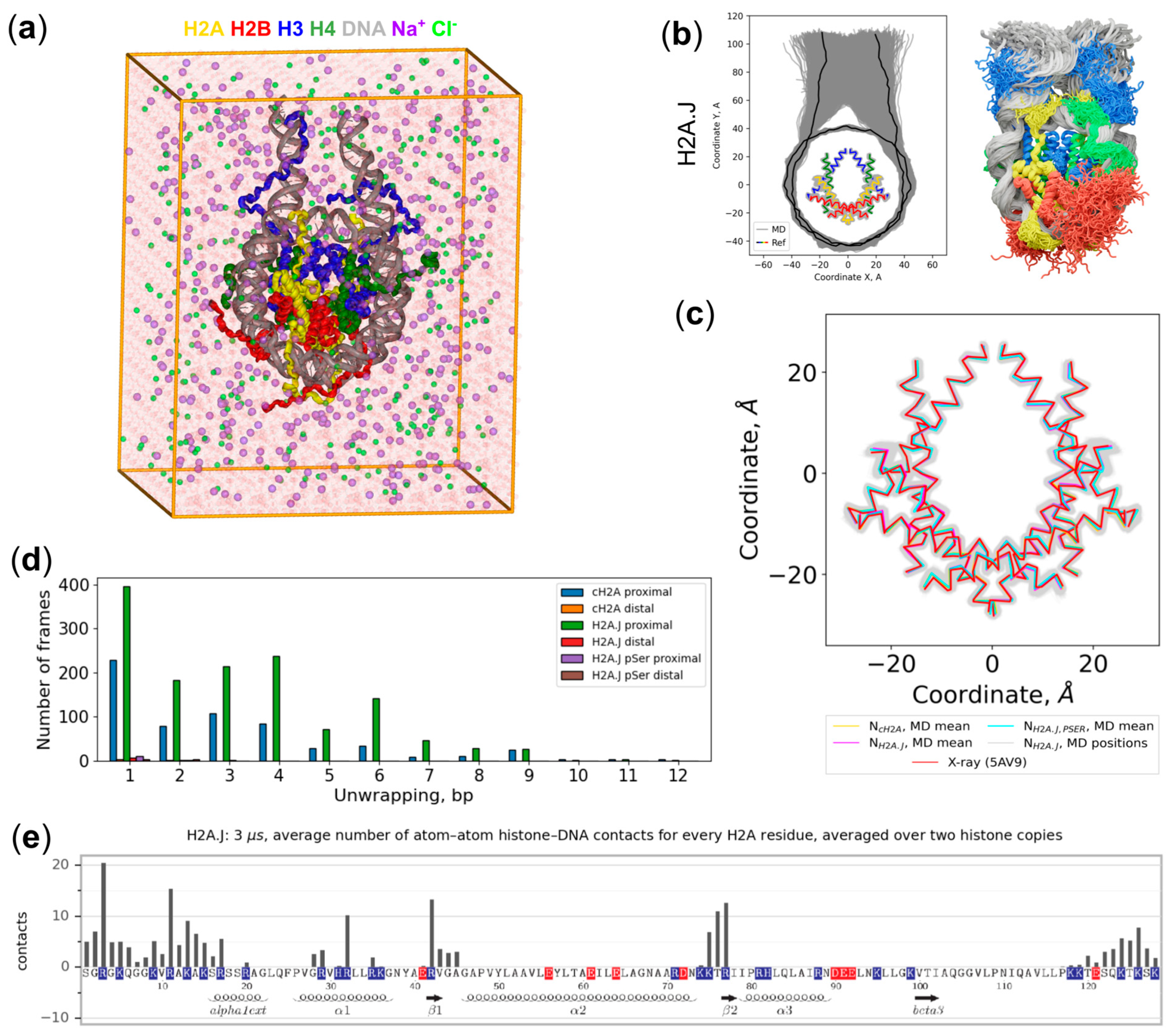
2.1. Overview of MD Simulations
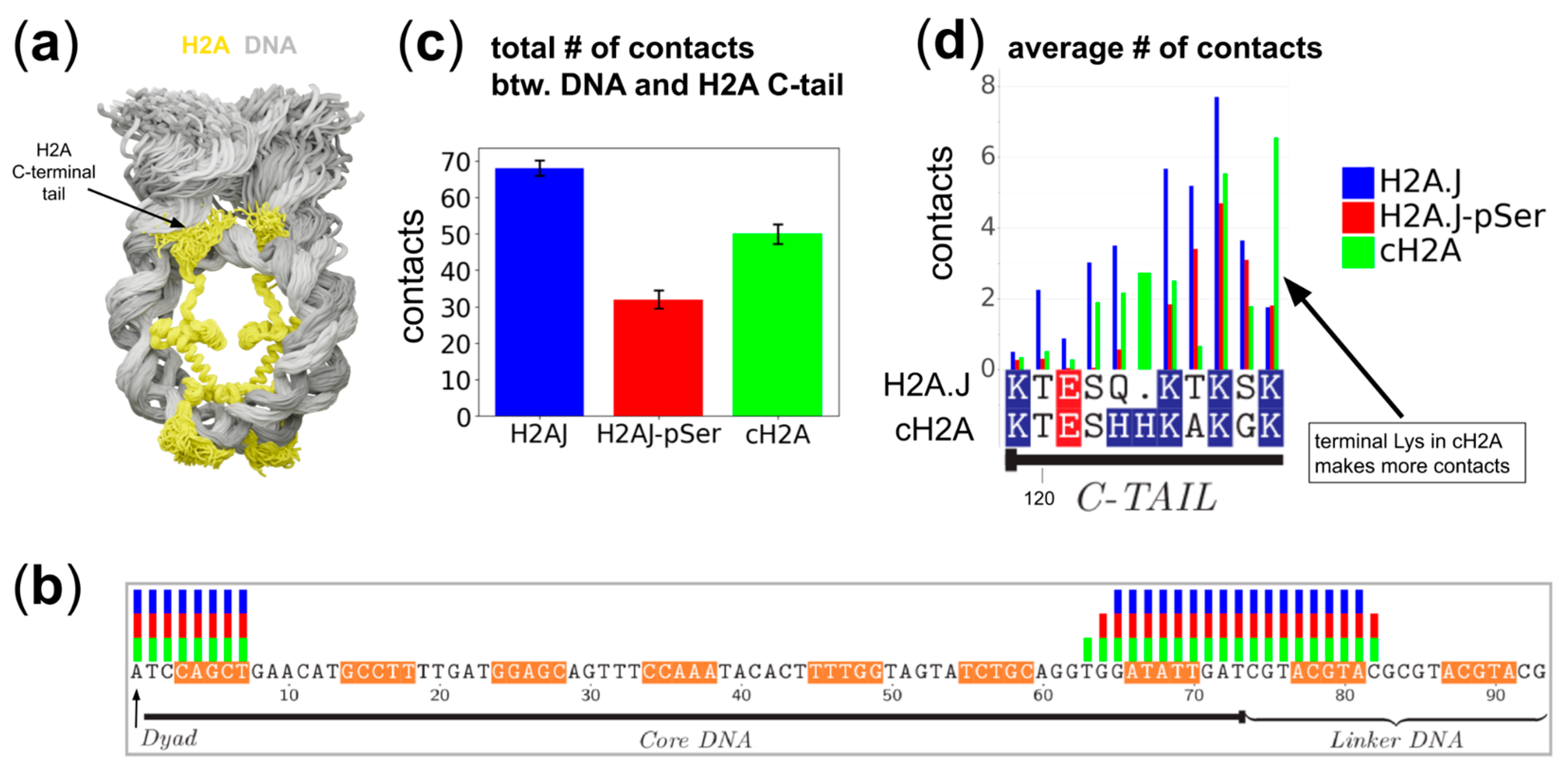
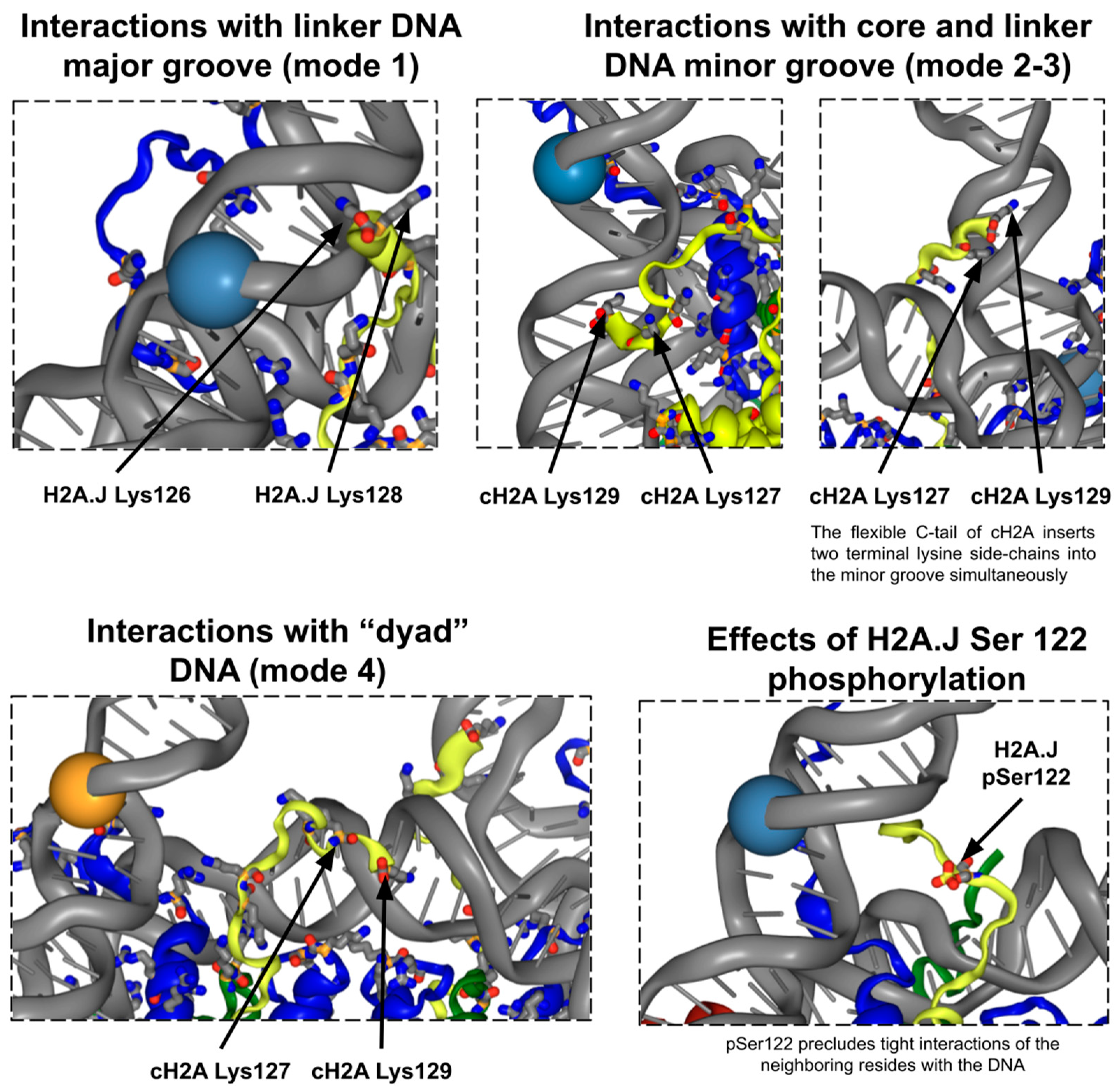
2.2. H2A C-Terminal Tail Interaction Patterns with the DNA Differ Between Histone Variants
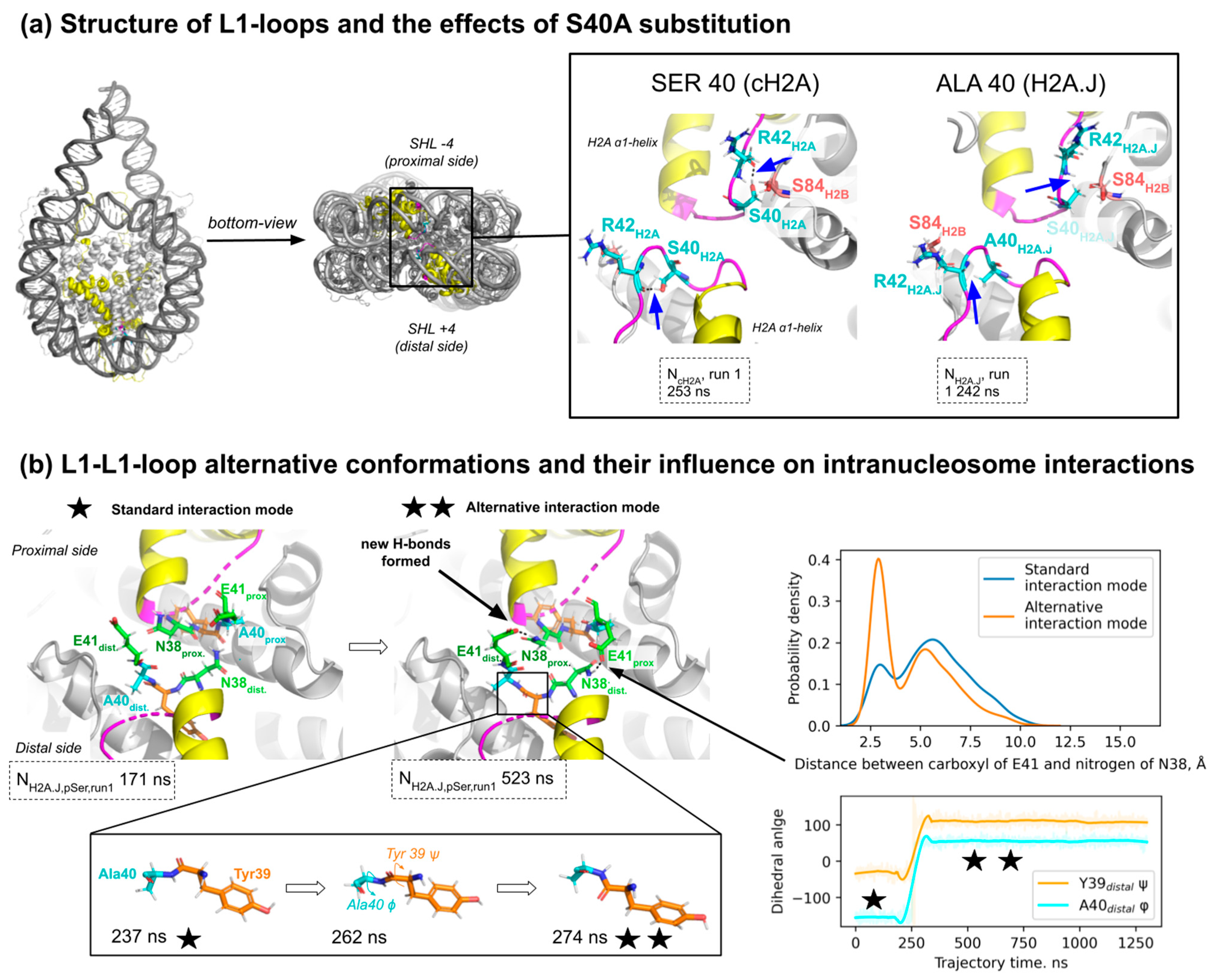
2.3. The Effect of Ser40Ala Substitution on L1-Loop Conformation and in Intra- and Inter-Dimer Interactions Within the Nucleosome
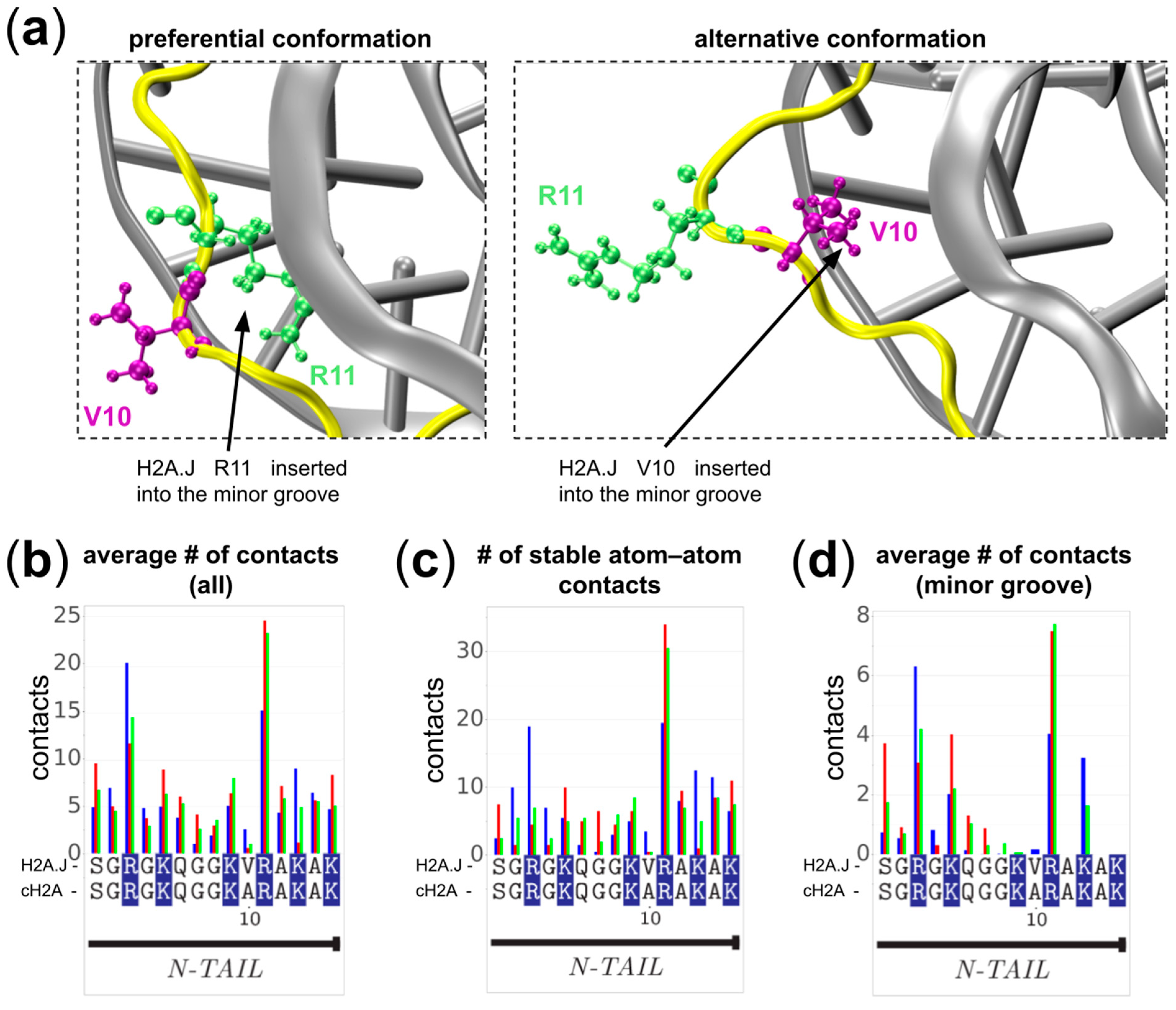
2.4. The Role of Ala10Val Substitution
3. Discussion
4. Materials and Methods
4.1. Building Nucleosome Systems for MD Simulations
4.2. Simulation Parameters
4.3. Trajectory Analysis
4.4. Force Field Modification
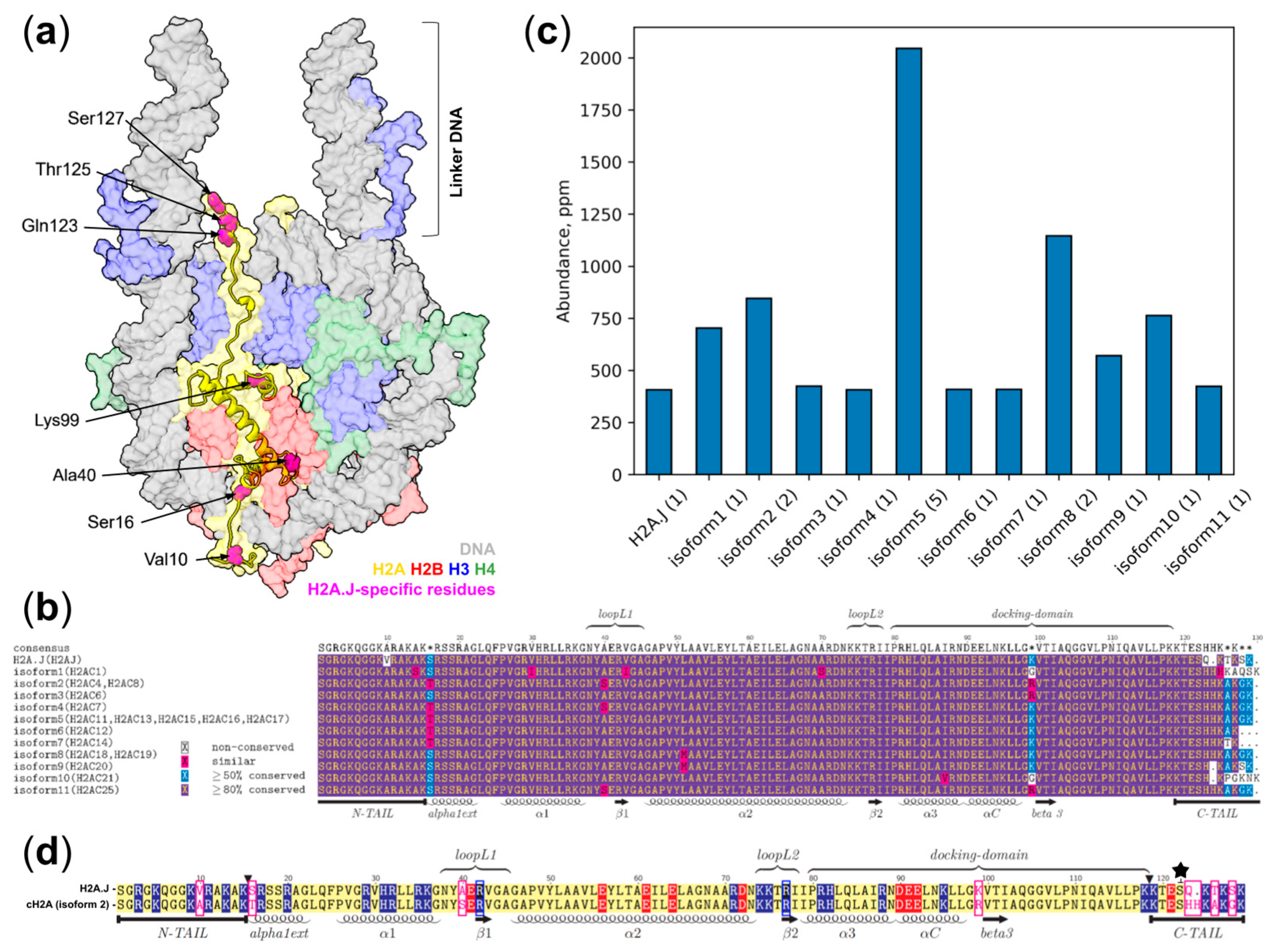
4.5. Histone Abundance Analysis Using Data from PaxDB
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, Z.; Li, S.; Subramaniam, S.; Shyy, J.Y.-J.; Chien, S. Epigenetic Regulation: A New Frontier for Biomedical Engineers. Annu. Rev. Biomed. Eng. 2017, 19, 195–219. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, R.D. Chromatin Structure: A Repeating Unit of Histones and DNA. Science 1974, 184, 868–871. [Google Scholar] [CrossRef] [PubMed]
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal Structure of the Nucleosome Core Particle at 2.8 A Resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Van Holde, K.E. Chromatin; Springer Series in Molecular Biology; Springer: Berlin/Heidelberg, Germany, 1989; ISBN 978-0-387-96694-6. [Google Scholar]
- Lyubitelev, A.V.; Nikitin, D.V.; Shaytan, A.K.; Studitsky, V.M.; Kirpichnikov, M.P. Structure and Functions of Linker Histones. Biochem. Mosc. 2016, 81, 213–223. [Google Scholar] [CrossRef]
- Armeev, G.A.; Gribkova, A.K.; Pospelova, I.; Komarova, G.A.; Shaytan, A.K. Linking Chromatin Composition and Structural Dynamics at the Nucleosome Level. Curr. Opin. Struct. Biol. 2019, 56, 46–55. [Google Scholar] [CrossRef]
- Henikoff, S.; Smith, M.M. Histone Variants and Epigenetics. Cold Spring Harb. Perspect. Biol. 2015, 7, a019364. [Google Scholar] [CrossRef]
- Talbert, P.B.; Henikoff, S. Histone Variants at a Glance. J. Cell Sci. 2021, 134, jcs244749. [Google Scholar] [CrossRef] [PubMed]
- Skvortsova, K.; Iovino, N.; Bogdanović, O. Functions and Mechanisms of Epigenetic Inheritance in Animals. Nat. Rev. Mol. Cell Biol. 2018, 19, 774–790. [Google Scholar] [CrossRef]
- Shaytan, A.K.; Landsman, D.; Panchenko, A.R. Nucleosome Adaptability Conferred by Sequence and Structural Variations in Histone H2A-H2B Dimers. Curr. Opin. Struct. Biol. 2015, 32, 48–57. [Google Scholar] [CrossRef]
- Santoro, S.W.; Dulac, C. The Activity-Dependent Histone Variant H2BE Modulates the Life Span of Olfactory Neurons. eLife 2012, 1, e00070. [Google Scholar] [CrossRef]
- Espiritu, D.; Gribkova, A.K.; Gupta, S.; Shaytan, A.K.; Panchenko, A.R. Molecular Mechanisms of Oncogenesis through the Lens of Nucleosomes and Histones. J. Phys. Chem. B 2021, 125, 3963–3976. [Google Scholar] [CrossRef] [PubMed]
- Seal, R.L.; Denny, P.; Bruford, E.A.; Gribkova, A.K.; Landsman, D.; Marzluff, W.F.; McAndrews, M.; Panchenko, A.R.; Shaytan, A.K.; Talbert, P.B. A Standardized Nomenclature for Mammalian Histone Genes. Epigenetics Chromatin 2022, 15, 34. [Google Scholar] [CrossRef]
- Contrepois, K.; Coudereau, C.; Benayoun, B.A.; Schuler, N.; Roux, P.-F.; Bischof, O.; Courbeyrette, R.; Carvalho, C.; Thuret, J.-Y.; Ma, Z.; et al. Histone Variant H2A.J Accumulates in Senescent Cells and Promotes Inflammatory Gene Expression. Nat. Commun. 2017, 8, 14995. [Google Scholar] [CrossRef] [PubMed]
- Nishida, H.; Suzuki, T.; Tomaru, Y.; Hayashizaki, Y. A Novel Replication-Independent Histone H2a Gene in Mouse. BMC Genet. 2005, 6, 10. [Google Scholar] [CrossRef]
- Kim, S.-T.; Lim, D.-S.; Canman, C.E.; Kastan, M.B. Substrate Specificities and Identification of Putative Substrates of ATM Kinase Family Members. J. Biol. Chem. 1999, 274, 37538–37543. [Google Scholar] [CrossRef]
- Mangelinck, A.; Coudereau, C.; Courbeyrette, R.; Ouararhni, K.; Hamiche, A.; Redon, C.E.; Bonner, W.M.; Van Dijk, E.; Derbois, C.; Olaso, R.; et al. The H2A.J Histone Variant Contributes to Interferon-Stimulated Gene Expression in Senescence by Its Weak Interaction with H1 and the Derepression of Repeated DNA Sequences. bioRxiv 2020. [Google Scholar] [CrossRef]
- Tanaka, H.; Sato, S.; Koyama, M.; Kujirai, T.; Kurumizaka, H. Biochemical and Structural Analyses of the Nucleosome Containing Human Histone H2A.J. J. Biochem. 2020, 167, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Isermann, A.; Mann, C.; Rübe, C.E. Histone Variant H2A.J Marks Persistent DNA Damage and Triggers the Secretory Phenotype in Radiation-Induced Senescence. Int. J. Mol. Sci. 2020, 21, 9130. [Google Scholar] [CrossRef] [PubMed]
- Abd Al-razaq, M.A.; Freyter, B.M.; Isermann, A.; Tewary, G.; Mangelinck, A.; Mann, C.; Rübe, C.E. Role of Histone Variant H2A.J in Fine-Tuning Chromatin Organization for the Establishment of Ionizing Radiation-Induced Senescence. Cells 2023, 12, 916. [Google Scholar] [CrossRef]
- Lai, P.M.; Chan, K.M. Roles of Histone H2A Variants in Cancer Development, Prognosis, and Treatment. Int. J. Mol. Sci. 2024, 25, 3144. [Google Scholar] [CrossRef]
- Cornen, S.; Guille, A.; Adélaïde, J.; Addou-Klouche, L.; Finetti, P.; Saade, M.-R.; Manai, M.; Carbuccia, N.; Bekhouche, I.; Letessier, A.; et al. Candidate Luminal B Breast Cancer Genes Identified by Genome, Gene Expression and DNA Methylation Profiling. PLoS ONE 2014, 9, e81843. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Weremowicz, S.; Feng, B.; Gentleman, R.C.; Marks, J.R.; Gelman, R.; Brennan, C.; Polyak, K. Combined cDNA Array Comparative Genomic Hybridization and Serial Analysis of Gene Expression Analysis of Breast Tumor Progression. Cancer Res. 2006, 66, 4065–4078. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ghareeb, W.M.; Lu, X.; Huang, Y.; Huang, S.; Chi, P. Coexpression Network Analysis Linked H2AFJ to Chemoradiation Resistance in Colorectal Cancer. J. Cell. Biochem. 2019, 120, 10351–10362. [Google Scholar] [CrossRef]
- Liu, J.; Qiu, W.; Shen, X.; Sun, G. Bioinformatics Analysis Revealed Hub Genes and Pathways Involved in Sorafenib Resistance in Hepatocellular Carcinoma. Math. Biosci. Eng. 2019, 16, 6319–6334. [Google Scholar] [CrossRef]
- Lee, H.-H.; Lin, C.-H.; Lin, H.-Y.; Kuei, C.-H.; Zheng, J.-Q.; Wang, Y.-H.; Lu, L.-S.; Lee, F.-P.; Hu, C.-J.; Wu, D.; et al. Histone 2A Family Member J Drives Mesenchymal Transition and Temozolomide Resistance in Glioblastoma Multiforme. Cancers 2019, 12, 98. [Google Scholar] [CrossRef]
- Redon, C.E.; Schmal, Z.; Tewary, G.; Mangelinck, A.; Courbeyrette, R.; Thuret, J.-Y.; Aladjem, M.I.; Bonner, W.M.; Rübe, C.E.; Mann, C. Histone Variant H2A.J Is Enriched in Luminal Epithelial Gland Cells. Genes 2021, 12, 1665. [Google Scholar] [CrossRef]
- Kurumizaka, H.; Kujirai, T.; Takizawa, Y. Contributions of Histone Variants in Nucleosome Structure and Function. J. Mol. Biol. 2021, 433, 166678. [Google Scholar] [CrossRef]
- Armeev, G.A.; Gribkova, A.K.; Shaytan, A.K. NucleosomeDB—A Database of 3D Nucleosome Structures and Their Complexes with Comparative Analysis Toolkit. bioRxiv 2023. [Google Scholar] [CrossRef]
- Horikoshi, N.; Sato, K.; Shimada, K.; Arimura, Y.; Osakabe, A.; Tachiwana, H.; Hayashi-Takanaka, Y.; Iwasaki, W.; Kagawa, W.; Harata, M.; et al. Structural Polymorphism in the L1 Loop Regions of Human H2A.Z.1 and H2A.Z.2. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 2431–2439. [Google Scholar] [CrossRef]
- Chakravarthy, S.; Gundimella, S.K.Y.; Caron, C.; Perche, P.-Y.; Pehrson, J.R.; Khochbin, S.; Luger, K. Structural Characterization of the Histone Variant macroH2A. Mol. Cell. Biol. 2005, 25, 7616–7624. [Google Scholar] [CrossRef]
- Bishop, T.C. Molecular Dynamics Simulations of a Nucleosome and Free DNA. J. Biomol. Struct. Dyn. 2005, 22, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Ruscio, J.Z.; Onufriev, A. A Computational Study of Nucleosomal DNA Flexibility. Biophys. J. 2006, 91, 4121–4132. [Google Scholar] [CrossRef] [PubMed]
- Fedulova, A.S.; Armeev, G.A.; Romanova, T.A.; Singh-Palchevskaia, L.; Kosarim, N.A.; Motorin, N.A.; Komarova, G.A.; Shaytan, A.K. Molecular Dynamics Simulations of Nucleosomes Are Coming of Age. WIREs Comput. Mol. Sci. 2024, 14, e1728. [Google Scholar] [CrossRef]
- Bowerman, S.; Wereszczynski, J. Effects of MacroH2A and H2A.Z on Nucleosome Dynamics as Elucidated by Molecular Dynamics Simulations. Biophys. J. 2016, 110, 327–337. [Google Scholar] [CrossRef]
- Kniazeva, A.S.; Armeev, G.A.; Shaytan, A.K. H2A-H2B Histone Dimer Plasticity and Its Functional Implications. Cells 2022, 11, 2837. [Google Scholar] [CrossRef] [PubMed]
- Armeev, G.A.; Kniazeva, A.S.; Komarova, G.A.; Kirpichnikov, M.P.; Shaytan, A.K. Histone Dynamics Mediate DNA Unwrapping and Sliding in Nucleosomes. Nat. Commun. 2021, 12, 2387. [Google Scholar] [CrossRef]
- Kohestani, H.; Wereszczynski, J. Effects of H2A.B Incorporation on Nucleosome Structures and Dynamics. Biophys. J. 2021, 120, 1498–1509. [Google Scholar] [CrossRef]
- Peng, J.; Yuan, C.; Hua, X.; Zhang, Z. Molecular Mechanism of Histone Variant H2A.B on Stability and Assembly of Nucleosome and Chromatin Structures. Epigenetics Chromatin 2020, 13, 28. [Google Scholar] [CrossRef]
- Li, S.; Wei, T.; Panchenko, A.R. Histone Variant H2A.Z Modulates Nucleosome Dynamics to Promote DNA Accessibility. Nat. Commun. 2023, 14, 769. [Google Scholar] [CrossRef]
- Peng, Y.; Li, S.; Onufriev, A.; Landsman, D.; Panchenko, A.R. Binding of Regulatory Proteins to Nucleosomes is Modulated by Dynamic Histone Tails. Nat. Commun. 2021, 12, 5280. [Google Scholar] [CrossRef]
- Bui, M.; Pitman, M.; Nuccio, A.; Roque, S.; Donlin-Asp, P.G.; Nita-Lazar, A.; Papoian, G.A.; Dalal, Y. Internal Modifications in the CENP-A Nucleosome Modulate Centromeric Dynamics. Epigenetics Chromatin 2017, 10, 17. [Google Scholar] [CrossRef] [PubMed]
- Lebedenko, O.O.; Salikov, V.A.; Izmailov, S.A.; Podkorytov, I.S.; Skrynnikov, N.R. Using NMR Diffusion Data to Validate MD Models of Disordered Proteins: Test Case of N-Terminal Tail of Histone H4. Biophys. J. 2024, 123, 80–100. [Google Scholar] [CrossRef] [PubMed]
- Winogradoff, D.; Aksimentiev, A. Molecular Mechanism of Spontaneous Nucleosome Unraveling. J. Mol. Biol. 2019, 431, 323–335. [Google Scholar] [CrossRef]
- Huertas, J.; Schöler, H.R.; Cojocaru, V. Histone Tails Cooperate to Control the Breathing of Genomic Nucleosomes. PLoS Comput. Biol. 2021, 17, e1009013. [Google Scholar] [CrossRef]
- Shaytan, A.K.; Xiao, H.; Armeev, G.A.; Wu, C.; Landsman, D.; Panchenko, A.R. Hydroxyl-Radical Footprinting Combined with Molecular Modeling Identifies Unique Features of DNA Conformation and Nucleosome Positioning. Nucleic Acids Res. 2017, 45, 9229–9243. [Google Scholar] [CrossRef]
- Sun, W.; Lebedenko, O.O.; Salguero, N.G.; Shannon, M.D.; Zandian, M.; Poirier, M.G.; Skrynnikov, N.R.; Jaroniec, C.P. Conformational and Interaction Landscape of Histone H4 Tails in Nucleosomes Probed by Paramagnetic NMR Spectroscopy. J. Am. Chem. Soc. 2023, 145, 25478–25485. [Google Scholar] [CrossRef] [PubMed]
- Ghoneim, M.; Fuchs, H.A.; Musselman, C.A. Histone Tail Conformations: A Fuzzy Affair with DNA. Trends Biochem. Sci. 2021, 46, 564–578. [Google Scholar] [CrossRef]
- Vogler, C.; Huber, C.; Waldmann, T.; Ettig, R.; Braun, L.; Izzo, A.; Daujat, S.; Chassignet, I.; Lopez-Contreras, A.J.; Fernandez-Capetillo, O.; et al. Histone H2A C-Terminus Regulates Chromatin Dynamics, Remodeling, and Histone H1 Binding. PLoS Genet. 2010, 6, e1001234. [Google Scholar] [CrossRef]
- Singhpalchevsk, L.; Shaytan, A.K. Diversity of H2A Histones and Their Effect on Nucleosome Structural Properties. Mosc. Univ. Biol. Sci. Bull. 2023, 78, 212–218. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA Double-Stranded Breaks Induce Histone H2AX Phosphorylation on Serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef]
- Tsai, C.-H.; Chen, Y.-J.; Yu, C.-J.; Tzeng, S.-R.; Wu, I.-C.; Kuo, W.-H.; Lin, M.-C.; Chan, N.-L.; Wu, K.-J.; Teng, S.-C. SMYD3-Mediated H2A.Z.1 Methylation Promotes Cell Cycle and Cancer Proliferation. Cancer Res. 2016, 76, 6043–6053. [Google Scholar] [CrossRef] [PubMed]
- Shukla, M.S.; Syed, S.H.; Goutte-Gattat, D.; Richard, J.L.; Montel, F.; Hamiche, A.; Travers, A.; Faivre-Moskalenko, C.; Bednar, J.; Hayes, J.J.; et al. The Docking Domain of Histone H2A Is Required for H1 Binding and RSC-Mediated Nucleosome Remodeling. Nucleic Acids Res. 2011, 39, 2559–2570. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; De Falco, L.; Padavattan, S.; Rao, C.; Geifman-Shochat, S.; Liu, C.-F.; Davey, C.A. PARP1 Exhibits Enhanced Association and Catalytic Efficiency with γH2A.X-Nucleosome. Nat. Commun. 2019, 10, 5751. [Google Scholar] [CrossRef] [PubMed]
- Morrison, E.A.; Bowerman, S.; Sylvers, K.L.; Wereszczynski, J.; Musselman, C.A. The Conformation of the Histone H3 Tail Inhibits Association of the BPTF PHD Finger with the Nucleosome. eLife 2018, 7, e31481. [Google Scholar] [CrossRef]
- Pilotto, S.; Speranzini, V.; Tortorici, M.; Durand, D.; Fish, A.; Valente, S.; Forneris, F.; Mai, A.; Sixma, T.K.; Vachette, P.; et al. Interplay among Nucleosomal DNA, Histone Tails, and Corepressor CoREST Underlies LSD1-Mediated H3 Demethylation. Proc. Natl. Acad. Sci. USA 2015, 112, 2752–2757. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and Recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef]
- Kim, J.J.; Kingston, R.E. Context-Specific Polycomb Mechanisms in Development. Nat. Rev. Genet. 2022, 23, 680–695. [Google Scholar] [CrossRef]
- Casa, V.; Gabellini, D. A Repetitive Elements Perspective in Polycomb Epigenetics. Front. Genet. 2012, 3, 199. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Reddy, D.; Jani, V.; Gadewal, N.; Shah, S.; Reddy, R.; Bose, K.; Sonavane, U.; Joshi, R.; Gupta, S. Histone Isoform H2A1H Promotes Attainment of Distinct Physiological States by Altering Chromatin Dynamics. Epigenetics Chromatin 2017, 10, 48. [Google Scholar] [CrossRef]
- Millán-Zambrano, G.; Burton, A.; Bannister, A.J.; Schneider, R. Histone Post-Translational Modifications—Cause and Consequence of Genome Function. Nat. Rev. Genet. 2022, 23, 563–580. [Google Scholar] [CrossRef]
- Weirich, S.; Kusevic, D.; Schnee, P.; Reiter, J.; Pleiss, J.; Jeltsch, A. Discovery of NSD2 Non-Histone Substrates and Design of a Super-Substrate. Commun. Biol. 2024, 7, 707. [Google Scholar] [CrossRef] [PubMed]
- Shi, X. Unveiling Structural and Dynamical Features of Chromatin Using NMR Spectroscopy. Magn. Reson. Lett. 2024, 200153. [Google Scholar] [CrossRef]
- Gansen, A.; Felekyan, S.; Kühnemuth, R.; Lehmann, K.; Tóth, K.; Seidel, C.A.M.; Langowski, J. High Precision FRET Studies Reveal Reversible Transitions in Nucleosomes between Microseconds and Minutes. Nat. Commun. 2018, 9, 4628. [Google Scholar] [CrossRef]
- Yoo, J.; Aksimentiev, A. New Tricks for Old Dogs: Improving the Accuracy of Biomolecular Force Fields by Pair-Specific Corrections to Non-Bonded Interactions. Phys. Chem. Chem. Phys. 2018, 20, 8432–8449. [Google Scholar] [CrossRef]
- Shabane, P.S.; Izadi, S.; Onufriev, A.V. General Purpose Water Model Can Improve Atomistic Simulations of Intrinsically Disordered Proteins. J. Chem. Theory Comput. 2019, 15, 2620–2634. [Google Scholar] [CrossRef] [PubMed]
- Wakamori, M.; Fujii, Y.; Suka, N.; Shirouzu, M.; Sakamoto, K.; Umehara, T.; Yokoyama, S. Intra- and Inter-Nucleosomal Interactions of the Histone H4 Tail Revealed with a Human Nucleosome Core Particle with Genetically-Incorporated H4 Tetra-Acetylation. Sci. Rep. 2015, 5, 17204. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Lu, X.-J.; Olson, W.K. 3DNA: A Software Package for the Analysis, Rebuilding and Visualization of Three-Dimensional Nucleic Acid Structures. Nucleic Acids Res. 2003, 31, 5108–5121. [Google Scholar] [CrossRef]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical p Ka Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Kosarim, N.; Armeev, G.A.; Kirpichnikov, M.P.; Shaytan, A.K. Analysis of Ion Atmosphere Around Nucleosomes Using Supercomputer MD Simulations. Supercomput. Front. Innov. 2022, 9, 56–67. [Google Scholar] [CrossRef]
- Shaytan, A.K.; Armeev, G.A.; Goncearenco, A.; Zhurkin, V.B.; Landsman, D.; Panchenko, A.R. Coupling between Histone Conformations and DNA Geometry in Nucleosomes on a Microsecond Timescale: Atomistic Insights into Nucleosome Functions. J. Mol. Biol. 2016, 428, 221–237. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Ivani, I.; Dans, P.D.; Noy, A.; Pérez, A.; Faustino, I.; Hospital, A.; Walther, J.; Andrio, P.; Goñi, R.; Balaceanu, A.; et al. Parmbsc1: A Refined Force Field for DNA Simulations. Nat. Methods 2016, 13, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Voevodin, V.V.; Antonov, A.S.; Nikitenko, D.A.; Shvets, P.A.; Sobolev, S.I.; Sidorov, I.Y.; Stefanov, K.S.; Voevodin, V.V.; Zhumatiy, S.A. Supercomputer Lomonosov-2: Large Scale, Deep Monitoring and Fine Analytics for the User Community. Supercomput. Front. Innov. 2019, 6, 4–11. [Google Scholar] [CrossRef]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A Toolkit for the Analysis of Molecular Dynamics Simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System, Version 3.0 Schrödinger, LLC. Available online: https://www.pymol.org/ (accessed on 6 November 2024).
- Warnecke, A.; Sandalova, T.; Achour, A.; Harris, R.A. PyTMs: A Useful PyMOL Plugin for Modeling Common Post-Translational Modifications. BMC Bioinform. 2014, 15, 370. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; O’Mara, M.L. PsiRESP: Calculating RESP Charges with Psi4. J. Open Source Softw. 2022, 7, 4100. [Google Scholar] [CrossRef]
- Sousa da Silva, A.W.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser interfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef]
- Huang, Q.; Szklarczyk, D.; Wang, M.; Simonovic, M.; von Mering, C. PaxDb 5.0: Curated Protein Quantification Data Suggests Adaptive Proteome Changes in Yeasts. Mol. Cell. Proteom. 2023, 22, 100640. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kosarim, N.A.; Fedulova, A.S.; Shariafetdinova, A.S.; Armeev, G.A.; Shaytan, A.K. Molecular Dynamics Simulations of Nucleosomes Containing Histone Variant H2A.J. Int. J. Mol. Sci. 2024, 25, 12136. https://doi.org/10.3390/ijms252212136
Kosarim NA, Fedulova AS, Shariafetdinova AS, Armeev GA, Shaytan AK. Molecular Dynamics Simulations of Nucleosomes Containing Histone Variant H2A.J. International Journal of Molecular Sciences. 2024; 25(22):12136. https://doi.org/10.3390/ijms252212136
Chicago/Turabian StyleKosarim, Nikita A., Anastasiia S. Fedulova, Aleksandra S. Shariafetdinova, Grigoriy A. Armeev, and Alexey K. Shaytan. 2024. "Molecular Dynamics Simulations of Nucleosomes Containing Histone Variant H2A.J" International Journal of Molecular Sciences 25, no. 22: 12136. https://doi.org/10.3390/ijms252212136
APA StyleKosarim, N. A., Fedulova, A. S., Shariafetdinova, A. S., Armeev, G. A., & Shaytan, A. K. (2024). Molecular Dynamics Simulations of Nucleosomes Containing Histone Variant H2A.J. International Journal of Molecular Sciences, 25(22), 12136. https://doi.org/10.3390/ijms252212136