
Ferroptosis and Its Potential Role in the Physiopathology of Skeletal Muscle Atrophy


Abstract
:1. Introduction
2. Ferroptosis: A New Way of Cell Death
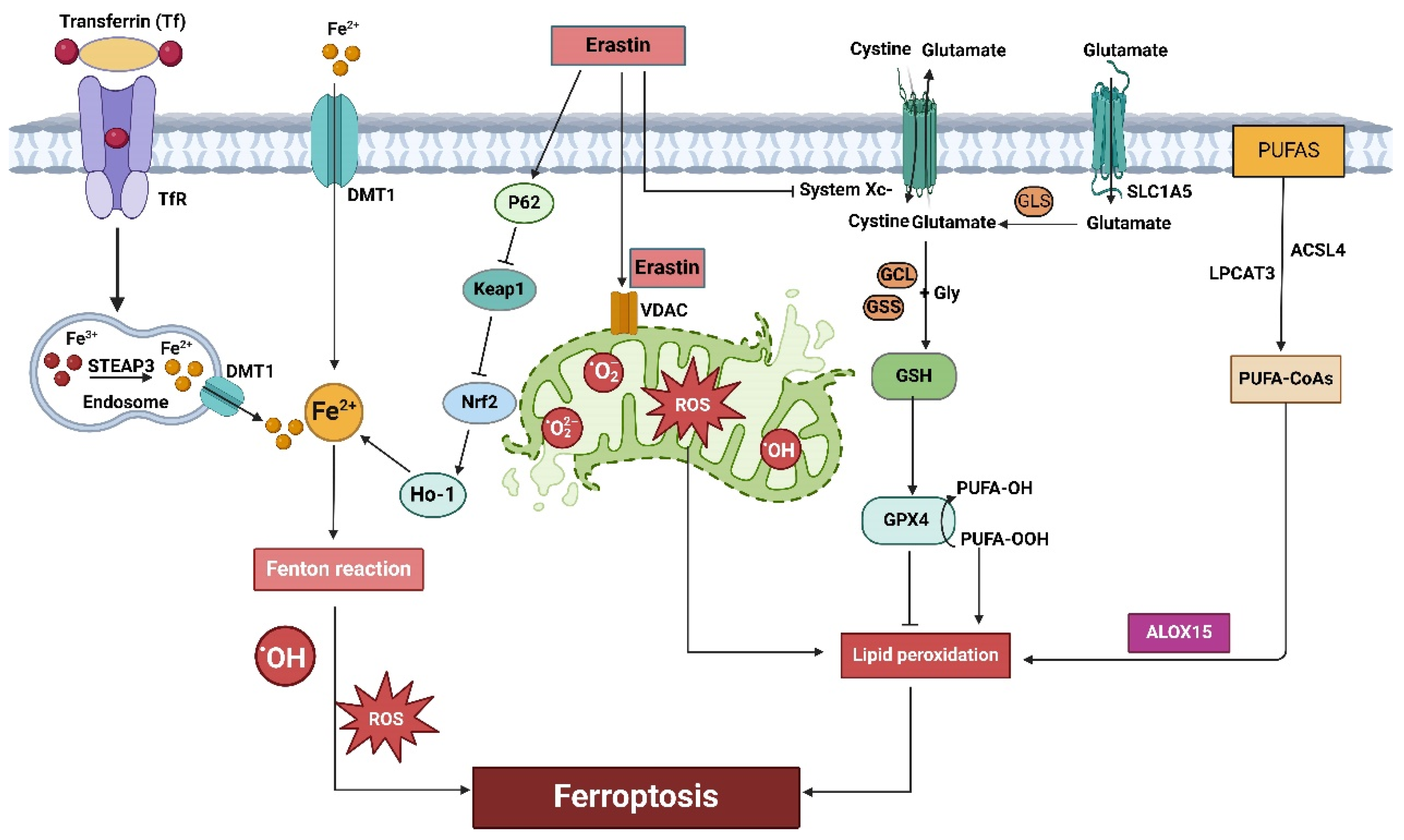
3. Mechanisms of Ferroptosis
3.1. The System Xc-GSH-GPX4 Pathway and Ferroptosis
3.2. Iron Metabolism and Ferroptosis
3.3. Lipid Metabolism and Ferroptosis
3.4. Amino Acids Metabolism and Ferroptosis
3.5. Mitochondrial Dysfunction and Ferroptosis
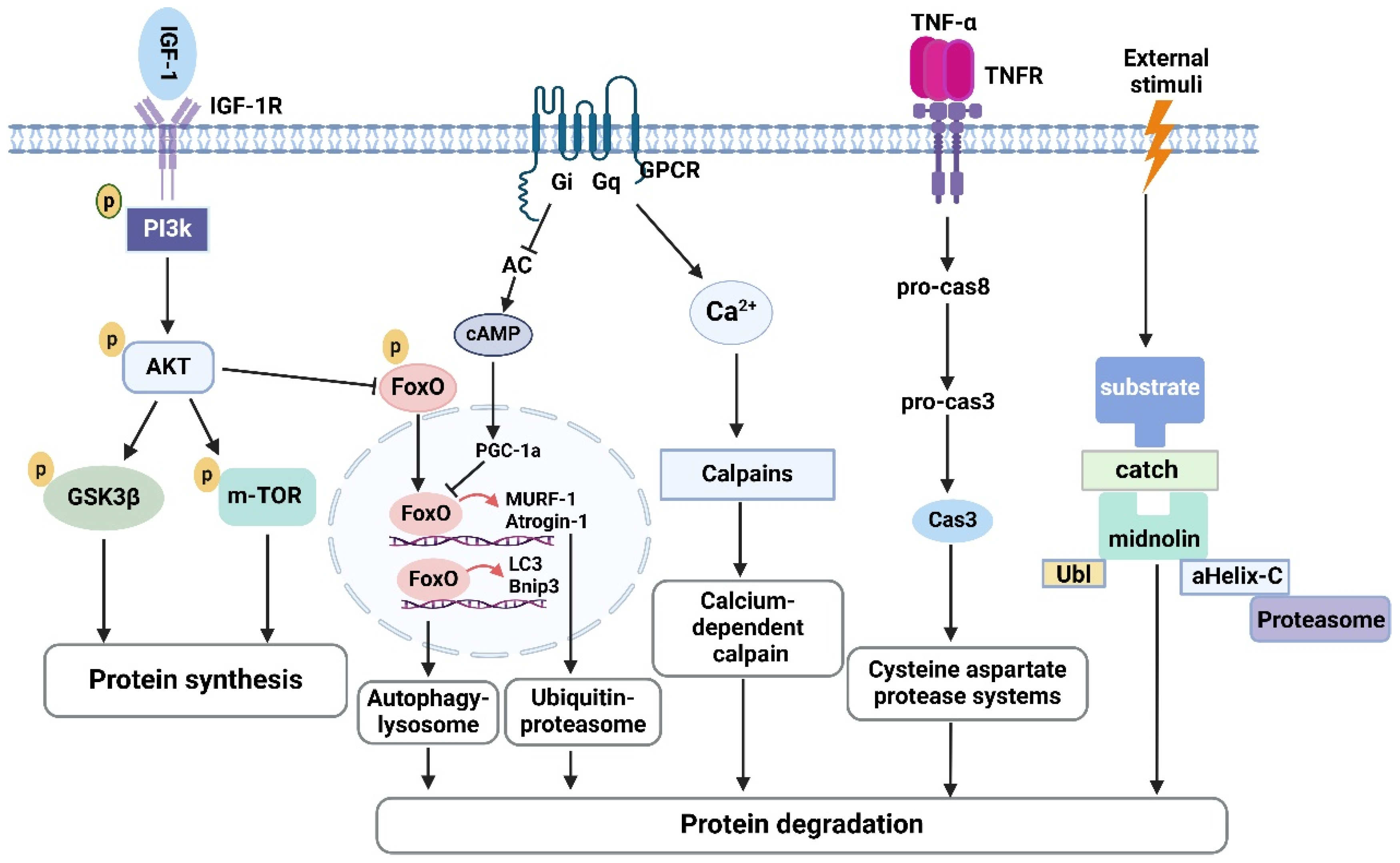
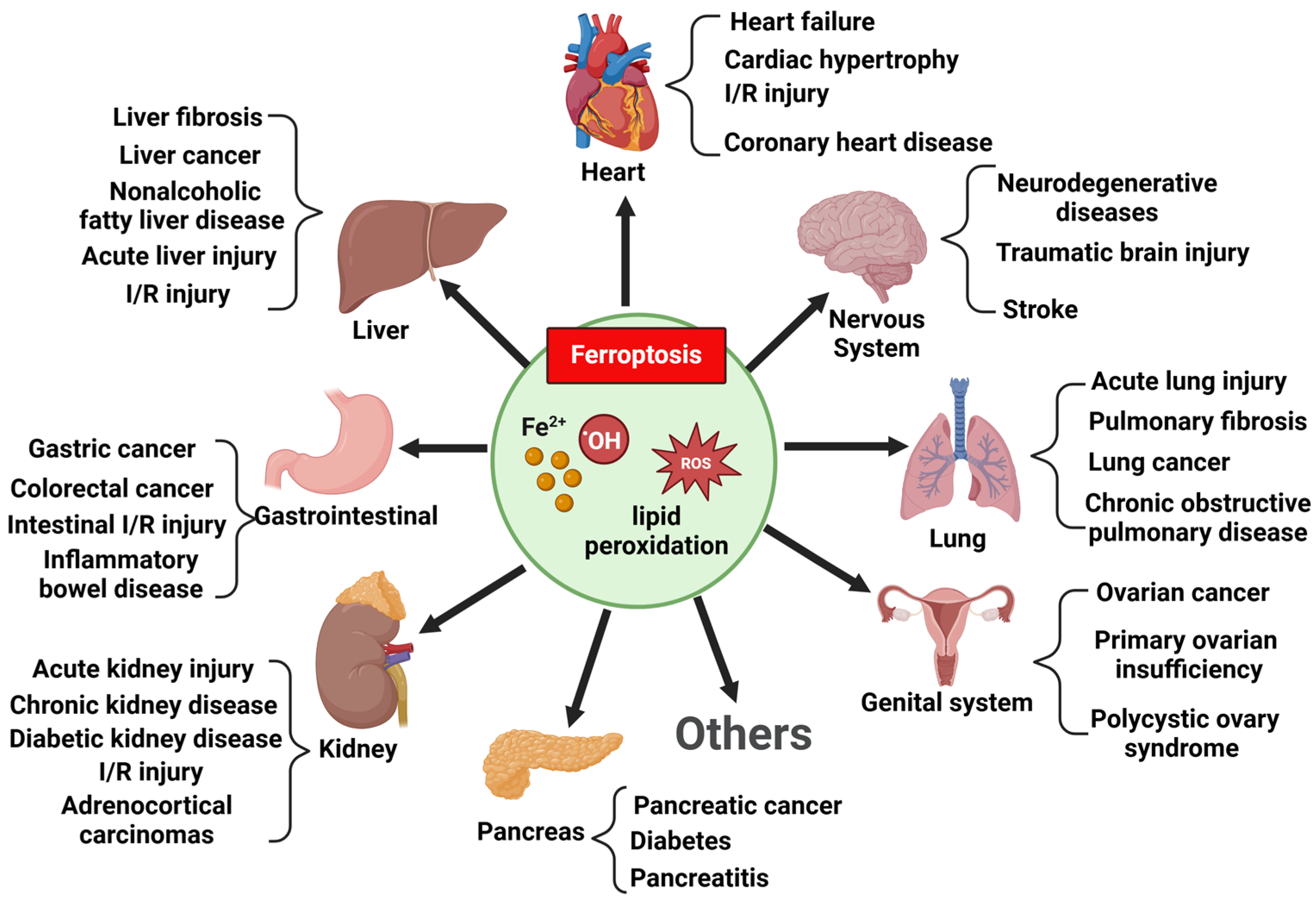
4. Ferroptosis and Skeletal Muscle Atrophy
4.1. Ferroptosis and Sarcopenia
4.2. Ferroptosis and CKD-Associated Muscle Atrophy
4.3. Ferroptosis and Sepsis-Induced Muscle Atrophy
4.4. Ferroptosis and Cisplatin-Induced Muscle Atrophy
4.5. Ferroptosis and Nerve Damage-Induced Muscle Atrophy
5. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Baskin, K.K.; Winders, B.R.; Olson, E.N. Muscle as a “mediator” of systemic metabolism. Cell Metab. 2015, 21, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Li, N.; Jia, W.; Wang, N.; Liang, M.; Yang, X.; Du, G. Skeletal muscle atrophy: From mechanisms to treatments. Pharmacol. Res. 2021, 172, 105807. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Xu, F.; Li, L.; Peng, C.; Sun, H.; Qiu, J.; Sun, J. Epigenetic control of skeletal muscle atrophy. Cell. Mol. Biol. Lett. 2024, 29, 99. [Google Scholar] [CrossRef] [PubMed]
- Furrer, R.; Handschin, C. Muscle Wasting Diseases: Novel Targets and Treatments. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 315–339. [Google Scholar] [CrossRef]
- Mercuri, E.; Sumner, C.J.; Muntoni, F.; Darras, B.T.; Finkel, R.S. Spinal muscular atrophy. Nat. Rev. Dis. Primers 2022, 8, 52. [Google Scholar] [CrossRef]
- Bilgic, S.N.; Domaniku, A.; Toledo, B.; Agca, S.; Weber, B.Z.C.; Arabaci, D.H.; Ozornek, Z.; Lause, P.; Thissen, J.P.; Loumaye, A.; et al. EDA2R-NIK signalling promotes muscle atrophy linked to cancer cachexia. Nature 2023, 617, 827–834. [Google Scholar] [CrossRef]
- Deane, C.S.; Piasecki, M.; Atherton, P.J. Skeletal muscle immobilisation-induced atrophy: Mechanistic insights from human studies. Clin. Sci. 2024, 138, 741–756. [Google Scholar] [CrossRef]
- Mishra, S.; Cosentino, C.; Tamta, A.K.; Khan, D.; Srinivasan, S.; Ravi, V.; Abbotto, E.; Arathi, B.P.; Kumar, S.; Jain, A.; et al. Sirtuin 6 inhibition protects against glucocorticoid-induced skeletal muscle atrophy by regulating IGF/PI3K/AKT signaling. Nat. Commun. 2022, 13, 5415. [Google Scholar] [CrossRef]
- Xiao, Q.; Sun, C.C.; Tang, C.F. Heme oxygenase-1: A potential therapeutic target for improving skeletal muscle atrophy. Exp. Gerontol. 2023, 184, 112335. [Google Scholar] [CrossRef]
- Gu, X.; Nardone, C.; Kamitaki, N.; Mao, A.; Elledge, S.J.; Greenberg, M.E. The midnolin-proteasome pathway catches proteins for ubiquitination-independent degradation. Science 2023, 381, eadh5021. [Google Scholar] [CrossRef]
- Pang, X.; Zhang, P.; Chen, X.; Liu, W. Ubiquitin-proteasome pathway in skeletal muscle atrophy. Front. Physiol. 2023, 14, 1289537. [Google Scholar] [CrossRef] [PubMed]
- Masiero, E.; Agatea, L.; Mammucari, C.; Blaauw, B.; Loro, E.; Komatsu, M.; Metzger, D.; Reggiani, C.; Schiaffino, S.; Sandri, M. Autophagy is required to maintain muscle mass. Cell Metab. 2009, 10, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Fu, T.; Guo, Q.; Zhou, D.; Sun, W.; Zhou, Z.; Chen, X.; Zhang, J.; Liu, L.; Xiao, L.; et al. Disuse-associated loss of the protease LONP1 in muscle impairs mitochondrial function and causes reduced skeletal muscle mass and strength. Nat. Commun. 2022, 13, 894. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.C.; Chiang, Y.F.; Huang, T.C.; Chen, H.Y.; Lin, P.H.; Ali, M.; Hsia, S.M. Capsaicin alleviates cisplatin-induced muscle loss and atrophy in vitro and in vivo. J. Cachexia Sarcopenia Muscle 2023, 14, 182–197. [Google Scholar] [CrossRef]
- Fang, W.Y.; Tseng, Y.T.; Lee, T.Y.; Fu, Y.C.; Chang, W.H.; Lo, W.W.; Lin, C.L.; Lo, Y.C. Triptolide prevents LPS-induced skeletal muscle atrophy via inhibiting NF-kappaB/TNF-alpha and regulating protein synthesis/degradation pathway. Br. J. Pharmacol. 2021, 178, 2998–3016. [Google Scholar] [CrossRef]
- Wang, H.H.; Sun, Y.N.; Qu, T.Q.; Sang, X.Q.; Zhou, L.M.; Li, Y.X.; Ren, F.Z. Nobiletin Prevents D-Galactose-Induced C2C12 Cell Aging by Improving Mitochondrial Function. Int. J. Mol. Sci. 2022, 23, 11963. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Zheng, J.; Conrad, M. The Metabolic Underpinnings of Ferroptosis. Cell Metab. 2020, 32, 920–937. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Zhou, Z.; Li, Q. The Role of Pyroptosis in the Pathogenesis of Kidney Diseases. Kidney Dis. 2023, 9, 443–458. [Google Scholar] [CrossRef]
- Yu, Y.; Yan, Y.; Niu, F.; Wang, Y.; Chen, X.; Su, G.; Liu, Y.; Zhao, X.; Qian, L.; Liu, P.; et al. Ferroptosis: A cell death connecting oxidative stress, inflammation and cardiovascular diseases. Cell Death Discov. 2021, 7, 193. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Broadening horizons: The role of ferroptosis in cancer. Nat. Rev. Clin. Oncol. 2021, 18, 280–296. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, J.; Kang, R.; Klionsky, D.J.; Tang, D. Ferroptosis: Machinery and regulation. Autophagy 2021, 17, 2054–2081. [Google Scholar] [CrossRef]
- Wang, Y.; Yan, S.; Liu, X.; Deng, F.; Wang, P.; Yang, L.; Hu, L.; Huang, K.; He, J. PRMT4 promotes ferroptosis to aggravate doxorubicin-induced cardiomyopathy via inhibition of the Nrf2/GPX4 pathway. Cell Death Differ. 2022, 29, 1982–1995. [Google Scholar] [CrossRef]
- Zhang, W.; Gong, M.; Zhang, W.; Mo, J.; Zhang, S.; Zhu, Z.; Wang, X.; Zhang, B.; Qian, W.; Wu, Z.; et al. Thiostrepton induces ferroptosis in pancreatic cancer cells through STAT3/GPX4 signalling. Cell Death Dis. 2022, 13, 630. [Google Scholar] [CrossRef]
- Chen, H.; Wang, C.; Liu, Z.; He, X.; Tang, W.; He, L.; Feng, Y.; Liu, D.; Yin, Y.; Li, T. Ferroptosis and Its Multifaceted Role in Cancer: Mechanisms and Therapeutic Approach. Antioxidants 2022, 11, 1504. [Google Scholar] [CrossRef]
- Zheng, Z.; Zhang, S.; Liu, X.; Wang, X.; Xue, C.; Wu, X.; Zhang, X.; Xu, X.; Liu, Z.; Yao, L.; et al. LRRK2 regulates ferroptosis through the system Xc-GSH-GPX4 pathway in the neuroinflammatory mechanism of Parkinson’s disease. J. Cell. Physiol. 2024, 239, e31250. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Cheff, D.M.; Huang, C.; Scholzen, K.C.; Gencheva, R.; Ronzetti, M.H.; Cheng, Q.; Hall, M.D.; Arner, E.S.J. The ferroptosis inducing compounds RSL3 and ML162 are not direct inhibitors of GPX4 but of TXNRD1. Redox Biol. 2023, 62, 102703. [Google Scholar] [CrossRef] [PubMed]
- Tuo, L.; Xiang, J.; Pan, X.; Gao, Q.; Zhang, G.; Yang, Y.; Liang, L.; Xia, J.; Wang, K.; Tang, N. PCK1 Downregulation Promotes TXNRD1 Expression and Hepatoma Cell Growth via the Nrf2/Keap1 Pathway. Front. Oncol. 2018, 8, 611. [Google Scholar] [CrossRef] [PubMed]
- Sabatier, P.; Beusch, C.M.; Gencheva, R.; Cheng, Q.; Zubarev, R.; Arner, E.S.J. Comprehensive chemical proteomics analyses reveal that the new TRi-1 and TRi-2 compounds are more specific thioredoxin reductase 1 inhibitors than auranofin. Redox Biol. 2021, 48, 102184. [Google Scholar] [CrossRef] [PubMed]
- Gonciarz, R.L.; Collisson, E.A.; Renslo, A.R. Ferrous Iron-Dependent Pharmacology. Trends Pharmacol. Sci. 2021, 42, 7–18. [Google Scholar] [CrossRef]
- Dev, S.; Babitt, J.L. Overview of iron metabolism in health and disease. Hemodial. Int. 2017, 21, S6–S20. [Google Scholar] [CrossRef]
- Zhang, K.; Fan, C.; Cai, D.; Zhang, Y.; Zuo, R.; Zhu, L.; Cao, Y.; Zhang, J.; Liu, C.; Chen, Y.; et al. Contribution of TGF-Beta-Mediated NLRP3-HMGB1 Activation to Tubulointerstitial Fibrosis in Rat with Angiotensin II-Induced Chronic Kidney Disease. Front. Cell Dev. Biol. 2020, 8, 1. [Google Scholar] [CrossRef]
- Fang, X.; Cai, Z.; Wang, H.; Han, D.; Cheng, Q.; Zhang, P.; Gao, F.; Yu, Y.; Song, Z.; Wu, Q.; et al. Loss of Cardiac Ferritin H Facilitates Cardiomyopathy via Slc7a11-Mediated Ferroptosis. Circ. Res. 2020, 127, 486–501. [Google Scholar] [CrossRef]
- Qin, X.; Zhang, J.; Wang, B.; Xu, G.; Yang, X.; Zou, Z.; Yu, C. Ferritinophagy is involved in the zinc oxide nanoparticles-induced ferroptosis of vascular endothelial cells. Autophagy 2021, 17, 4266–4285. [Google Scholar] [CrossRef]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J., 3rd; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef]
- Garcia-Casal, M.N.; Pasricha, S.R.; Martinez, R.X.; Lopez-Perez, L.; Pena-Rosas, J.P. Serum or plasma ferritin concentration as an index of iron deficiency and overload. Cochrane Database Syst. Rev. 2021, 5, CD011817. [Google Scholar] [CrossRef]
- Feng, H.; Schorpp, K.; Jin, J.; Yozwiak, C.E.; Hoffstrom, B.G.; Decker, A.M.; Rajbhandari, P.; Stokes, M.E.; Bender, H.G.; Csuka, J.M.; et al. Transferrin Receptor Is a Specific Ferroptosis Marker. Cell Rep. 2020, 30, 3411–3423.e7. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhang, H.; Cui, J.G.; Wang, J.X.; Chen, M.S.; Wang, H.R.; Li, X.N.; Li, J.L. Ferroptosis is critical for phthalates driving the blood-testis barrier dysfunction via targeting transferrin receptor. Redox Biol. 2023, 59, 102584. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Chen, S.; Pan, X.; Dai, X.; Pan, G.; Li, Z.; Mai, X.; Tian, Y.; Zhang, S.; Liu, B.; et al. Transferrin receptor 1 ablation in satellite cells impedes skeletal muscle regeneration through activation of ferroptosis. J. Cachexia Sarcopenia Muscle 2021, 12, 746–768. [Google Scholar] [CrossRef] [PubMed]
- Hiromatsu, M.; Toshida, K.; Itoh, S.; Harada, N.; Kohashi, K.; Oda, Y.; Yoshizumi, T. Transferrin Receptor is Associated with Sensitivity to Ferroptosis Inducers in Hepatocellular Carcinoma. Ann. Surg. Oncol. 2023, 30, 8675–8689. [Google Scholar] [CrossRef]
- Ma, H.; Huang, Y.; Tian, W.; Liu, J.; Yan, X.; Ma, L.; Lai, J. Endothelial transferrin receptor 1 contributes to thrombogenesis through cascade ferroptosis. Redox Biol. 2024, 70, 103041. [Google Scholar] [CrossRef]
- Wu, Y.; Jiao, H.; Yue, Y.; He, K.; Jin, Y.; Zhang, J.; Zhang, J.; Wei, Y.; Luo, H.; Hao, Z.; et al. Ubiquitin ligase E3 HUWE1/MULE targets transferrin receptor for degradation and suppresses ferroptosis in acute liver injury. Cell Death Differ. 2022, 29, 1705–1718. [Google Scholar] [CrossRef]
- Liu, P.; Feng, Y.; Li, H.; Chen, X.; Wang, G.; Xu, S.; Li, Y.; Zhao, L. Ferrostatin-1 alleviates lipopolysaccharide-induced acute lung injury via inhibiting ferroptosis. Cell Mol. Biol. Lett. 2020, 25, 10. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Liao, P.; Wang, W.; Wang, W.; Kryczek, I.; Li, X.; Bian, Y.; Sell, A.; Wei, S.; Grove, S.; Johnson, J.K.; et al. CD8(+) T cells and fatty acids orchestrate tumor ferroptosis and immunity via ACSL4. Cancer Cell 2022, 40, 365–378.e6. [Google Scholar] [CrossRef]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef]
- Cao, Z.; Liu, X.; Zhang, W.; Zhang, K.; Pan, L.; Zhu, M.; Qin, H.; Zou, C.; Wang, W.; Zhang, C.; et al. Biomimetic Macrophage Membrane-Camouflaged Nanoparticles Induce Ferroptosis by Promoting Mitochondrial Damage in Glioblastoma. ACS Nano 2023, 17, 23746–23760. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.; Liu, X.; Wang, X.; Shi, X.; Ma, L.; Zhang, Y.; Zhou, T.; Zhao, C.; Zhang, X.; Fan, B.; et al. Edaravone Modulates Neuronal GPX4/ACSL4/5-LOX to Promote Recovery After Spinal Cord Injury. Front. Cell Dev. Biol. 2022, 10, 849854. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.; Shchepinov, M.S.; Pratt, D.A. Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. ACS Cent. Sci. 2018, 4, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Henry, W.S.; Ricq, E.L.; Graham, E.T.; Phadnis, V.V.; Maretich, P.; Paradkar, S.; Boehnke, N.; Deik, A.A.; Reinhardt, F.; et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature 2020, 585, 603–608. [Google Scholar] [CrossRef]
- Tesfay, L.; Paul, B.T.; Konstorum, A.; Deng, Z.; Cox, A.O.; Lee, J.; Furdui, C.M.; Hegde, P.; Torti, F.M.; Torti, S.V. Stearoyl-CoA Desaturase 1 Protects Ovarian Cancer Cells from Ferroptotic Cell Death. Cancer Res. 2019, 79, 5355–5366. [Google Scholar] [CrossRef]
- Yang, Y.; Zhu, T.; Wang, X.; Xiong, F.; Hu, Z.; Qiao, X.; Yuan, X.; Wang, D. ACSL3 and ACSL4, Distinct Roles in Ferroptosis and Cancers. Cancers 2022, 14, 5896. [Google Scholar] [CrossRef]
- Magtanong, L.; Ko, P.J.; To, M.; Cao, J.Y.; Forcina, G.C.; Tarangelo, A.; Ward, C.C.; Cho, K.; Patti, G.J.; Nomura, D.K.; et al. Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem. Biol. 2019, 26, 420–432.e9. [Google Scholar] [CrossRef]
- Yoo, H.C.; Yu, Y.C.; Sung, Y.; Han, J.M. Glutamine reliance in cell metabolism. Exp. Mol. Med. 2020, 52, 1496–1516. [Google Scholar] [CrossRef]
- Yao, H.; Jiang, W.; Liao, X.; Wang, D.; Zhu, H. Regulatory mechanisms of amino acids in ferroptosis. Life Sci. 2024, 351, 122803. [Google Scholar] [CrossRef]
- Chen, Y.; Cui, Y.; Li, M.; Xia, M.; Xiang, Q.; Mao, Y.; Li, H.; Chen, J.; Zeng, W.; Zheng, X.; et al. A novel mechanism of ferroptosis inhibition-enhanced atherosclerotic plaque stability: YAP1 suppresses vascular smooth muscle cell ferroptosis through GLS1. FASEB J. 2024, 38, e23850. [Google Scholar] [CrossRef]
- Suzuki, S.; Venkatesh, D.; Kanda, H.; Nakayama, A.; Hosokawa, H.; Lee, E.; Miki, T.; Stockwell, B.R.; Yokote, K.; Tanaka, T.; et al. GLS2 Is a Tumor Suppressor and a Regulator of Ferroptosis in Hepatocellular Carcinoma. Cancer Res. 2022, 82, 3209–3222. [Google Scholar] [CrossRef] [PubMed]
- Roos, N.; Benz, R.; Brdiczka, D. Identification and characterization of the pore-forming protein in the outer membrane of rat liver mitochondria. Biochim. Biophys. Acta 1982, 686, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Yagoda, N.; von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Luo, M.; Zhang, K.; Zhang, J.; Gao, T.; Connell, D.O.; Yao, F.; Mu, C.; Cai, B.; Shang, Y.; et al. Nedd4 ubiquitylates VDAC2/3 to suppress erastin-induced ferroptosis in melanoma. Nat. Commun. 2020, 11, 433. [Google Scholar] [CrossRef] [PubMed]
- Niu, B.; Lei, X.; Xu, Q.; Ju, Y.; Xu, D.; Mao, L.; Li, J.; Zheng, Y.; Sun, N.; Zhang, X.; et al. Protecting mitochondria via inhibiting VDAC1 oligomerization alleviates ferroptosis in acetaminophen-induced acute liver injury. Cell Biol. Toxicol. 2022, 38, 505–530. [Google Scholar] [CrossRef]
- Mazure, N.M. VDAC in cancer. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 665–673. [Google Scholar] [CrossRef]
- Atkinson, A.; Winge, D.R. Metal acquisition and availability in the mitochondria. Chem. Rev. 2009, 109, 4708–4721. [Google Scholar] [CrossRef]
- Paradkar, P.N.; Zumbrennen, K.B.; Paw, B.H.; Ward, D.M.; Kaplan, J. Regulation of mitochondrial iron import through differential turnover of mitoferrin 1 and mitoferrin 2. Mol. Cell. Biol. 2009, 29, 1007–1016. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef]
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Porto Freitas, F.; Seibt, T.; et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 2018, 172, 409–422.e21. [Google Scholar] [CrossRef]
- Yang, R.; Gao, W.; Wang, Z.; Jian, H.; Peng, L.; Yu, X.; Xue, P.; Peng, W.; Li, K.; Zeng, P. Polyphyllin I induced ferroptosis to suppress the progression of hepatocellular carcinoma through activation of the mitochondrial dysfunction via Nrf2/HO-1/GPX4 axis. Phytomedicine 2024, 122, 155135. [Google Scholar] [CrossRef] [PubMed]
- Ye, T.; Yang, W.; Gao, T.; Yu, X.; Chen, T.; Yang, Y.; Guo, J.; Li, Q.; Li, H.; Yang, L. Trastuzumab-induced cardiomyopathy via ferroptosis-mediated mitochondrial dysfunction. Free Radic. Biol. Med. 2023, 206, 143–161. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Zandkarimi, F.; Zhang, Y.; Meena, J.K.; Kim, J.; Zhuang, L.; Tyagi, S.; Ma, L.; Westbrook, T.F.; Steinberg, G.R.; et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat. Cell Biol. 2020, 22, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Dong, X.; Du, W.; Shi, X.; Chen, K.; Zhang, W.; Gao, M. LKB1-AMPK axis negatively regulates ferroptosis by inhibiting fatty acid synthesis. Signal Transduct. Target. Ther. 2020, 5, 187. [Google Scholar] [CrossRef]
- Wei, S.; Qiu, T.; Yao, X.; Wang, N.; Jiang, L.; Jia, X.; Tao, Y.; Wang, Z.; Pei, P.; Zhang, J.; et al. Arsenic induces pancreatic dysfunction and ferroptosis via mitochondrial ROS-autophagy-lysosomal pathway. J. Hazard. Mater. 2020, 384, 121390. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, Q.; Chang, M.; Pan, Y.; Yahaya, B.H.; Liu, Y.; Lin, J. Chemotherapy impairs ovarian function through excessive ROS-induced ferroptosis. Cell Death Dis. 2023, 14, 340. [Google Scholar] [CrossRef]
- Liu, M.; Fan, Y.; Li, D.; Han, B.; Meng, Y.; Chen, F.; Liu, T.; Song, Z.; Han, Y.; Huang, L.; et al. Ferroptosis inducer erastin sensitizes NSCLC cells to celastrol through activation of the ROS-mitochondrial fission-mitophagy axis. Mol. Oncol. 2021, 15, 2084–2105. [Google Scholar] [CrossRef]
- Tai, P.; Chen, X.; Jia, G.; Chen, G.; Gong, L.; Cheng, Y.; Li, Z.; Wang, H.; Chen, A.; Zhang, G.; et al. WGX50 mitigates doxorubicin-induced cardiotoxicity through inhibition of mitochondrial ROS and ferroptosis. J. Transl. Med. 2023, 21, 823. [Google Scholar] [CrossRef]
- Wang, X.; Shen, T.; Lian, J.; Deng, K.; Qu, C.; Li, E.; Li, G.; Ren, Y.; Wang, Z.; Jiang, Z.; et al. Resveratrol reduces ROS-induced ferroptosis by activating SIRT3 and compensating the GSH/GPX4 pathway. Mol. Med. 2023, 29, 137. [Google Scholar] [CrossRef]
- Li, S.; Zheng, L.; Zhang, J.; Liu, X.; Wu, Z. Inhibition of ferroptosis by up-regulating Nrf2 delayed the progression of diabetic nephropathy. Free Radic. Biol. Med. 2021, 162, 435–449. [Google Scholar] [CrossRef]
- Yang, S.; Xie, Z.; Pei, T.; Zeng, Y.; Xiong, Q.; Wei, H.; Wang, Y.; Cheng, W. Salidroside attenuates neuronal ferroptosis by activating the Nrf2/HO1 signaling pathway in Abeta(1-42)-induced Alzheimer’s disease mice and glutamate-injured HT22 cells. Chin. Med. 2022, 17, 82. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Chen, S.; Guo, H.; Jiang, H.; Liu, H.; Fu, H.; Wang, D. Forsythoside A Mitigates Alzheimer’s-like Pathology by Inhibiting Ferroptosis-mediated Neuroinflammation via Nrf2/GPX4 Axis Activation. Int. J. Biol. Sci. 2022, 18, 2075–2090. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhu, Q.; Wang, Y.; Peng, J.; Shao, L.; Li, X. Irisin protects against sepsis-associated encephalopathy by suppressing ferroptosis via activation of the Nrf2/GPX4 signal axis. Free Radic. Biol. Med. 2022, 187, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.S.; Chen, J.C.; Lin, L.; Cheng, Y.Z.; Zhao, Y.; Zhang, Y.; Pan, X.D. Dendrobine rescues cognitive dysfunction in diabetic encephalopathy by inhibiting ferroptosis via activating Nrf2/GPX4 axis. Phytomedicine 2023, 119, 154993. [Google Scholar] [CrossRef]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef]
- Roh, J.L.; Kim, E.H.; Jang, H.; Shin, D. Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biol. 2017, 11, 254–262. [Google Scholar] [CrossRef]
- Fan, Z.; Wirth, A.K.; Chen, D.; Wruck, C.J.; Rauh, M.; Buchfelder, M.; Savaskan, N. Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis 2017, 6, e371. [Google Scholar] [CrossRef]
- Chen, L.; Hambright, W.S.; Na, R.; Ran, Q. Ablation of the Ferroptosis Inhibitor Glutathione Peroxidase 4 in Neurons Results in Rapid Motor Neuron Degeneration and Paralysis. J. Biol. Chem. 2015, 290, 28097–28106. [Google Scholar] [CrossRef]
- Huang, Y.; Wu, B.; Shen, D.; Chen, J.; Yu, Z.; Chen, C. Ferroptosis in a sarcopenia model of senescence accelerated mouse prone 8 (SAMP8). Int. J. Biol. Sci. 2021, 17, 151–162. [Google Scholar] [CrossRef]
- He, J.; He, Z.; Wang, H.; Zhang, C.; Pei, T.; Yan, S.; Yan, Y.; Wang, F.; Chen, Y.; Yuan, N.; et al. Caffeic acid alleviates skeletal muscle atrophy in 5/6 nephrectomy rats through the TLR4/MYD88/NF-kB pathway. Biomed. Pharmacother. 2024, 174, 116556. [Google Scholar] [CrossRef]
- Ni, S.H.; Zhang, X.J.; OuYang, X.L.; Ye, T.C.; Li, J.; Li, Y.; Sun, S.N.; Han, X.W.; Long, W.J.; Wang, L.J.; et al. Lobetyolin Alleviates Ferroptosis of Skeletal Muscle in 5/6 Nephrectomized Mice via Activation of Hedgehog-GLI1 Signaling. Phytomedicine 2023, 115, 154807. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Z.; Xu, H.C.; Zhou, H.X.; Zhang, C.K.; Li, B.M.; He, J.H.; Ni, P.S.; Yu, X.M.; Liu, Y.Q.; Li, F.H. Long-term detraining reverses the improvement of lifelong exercise on skeletal muscle ferroptosis and inflammation in aging rats: Fiber-type dependence of the Keap1/Nrf2 pathway. Biogerontology 2023, 24, 753–769. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Z.; Yu, Z.; Wang, M.; Zhou, R.; Chen, S.; Yu, X.; Li, F. Targeting STAT6 to mitigate sepsis-induced muscle atrophy and weakness: Modulation of mitochondrial dysfunction, ferroptosis, and CHI3L1-Mediated satellite cell loss. Biochem. Biophys. Rep. 2024, 37, 101608. [Google Scholar] [CrossRef] [PubMed]
- Ju, L.; Diao, J.; Zhang, J.; Dai, F.; Zhou, H.; Han, Z.; Hu, R.; Pei, T.; Wang, F.; He, Z.; et al. Shenshuai Yingyang Jiaonang ameliorates chronic kidney disease-associated muscle atrophy in rats by inhibiting ferroptosis mediated by the HIF-1alpha/SLC7A11 pathway. Heliyon 2024, 10, e29093. [Google Scholar] [CrossRef]
- Liu, X.; Xu, M.; Yu, Y.; Chen, Y.; Weng, X.; Zhu, L. PD-1 Alleviates Cisplatin-Induced Muscle Atrophy by Regulating Inflammation and Oxidative Stress. Antioxidants 2022, 11, 1839. [Google Scholar] [CrossRef]
- Wang, W.; Ren, W.; Zhu, L.; Hu, Y.; Ye, C. Identification of genes and key pathways underlying the pathophysiological association between sarcopenia and chronic obstructive pulmonary disease. Exp. Gerontol. 2024, 187, 112373. [Google Scholar] [CrossRef]
- Yang, B.; Pan, J.; Zhang, X.N.; Wang, H.; He, L.; Rong, X.; Li, X.; Peng, Y. NRF2 activation suppresses motor neuron ferroptosis induced by the SOD1(G93A) mutation and exerts neuroprotection in amyotrophic lateral sclerosis. Neurobiol. Dis. 2023, 184, 106210. [Google Scholar] [CrossRef]
- Wang, T.; Tomas, D.; Perera, N.D.; Cuic, B.; Luikinga, S.; Viden, A.; Barton, S.K.; McLean, C.A.; Samson, A.L.; Southon, A.; et al. Ferroptosis mediates selective motor neuron death in amyotrophic lateral sclerosis. Cell Death Differ. 2022, 29, 1187–1198. [Google Scholar] [CrossRef]
- Nogradi, B.; Nogradi-Halmi, D.; Erdelyi-Furka, B.; Kadar, Z.; Csont, T.; Gaspar, R. Mechanism of motoneuronal and pyramidal cell death in amyotrophic lateral sclerosis and its potential therapeutic modulation. Cell Death Discov. 2024, 10, 291. [Google Scholar] [CrossRef]
- Tu, L.F.; Zhang, T.Z.; Zhou, Y.F.; Zhou, Q.Q.; Gong, H.B.; Liang, L.; Hai, L.N.; You, N.X.; Su, Y.; Chen, Y.J.; et al. GPX4 deficiency-dependent phospholipid peroxidation drives motor deficits of ALS. J. Adv. Res. 2023, 43, 205–218. [Google Scholar] [CrossRef]
- Golko-Perez, S.; Amit, T.; Bar-Am, O.; Youdim, M.B.; Weinreb, O. A Novel Iron Chelator-Radical Scavenger Ameliorates Motor Dysfunction and Improves Life Span and Mitochondrial Biogenesis in SOD1(G93A) ALS Mice. Neurotox. Res. 2017, 31, 230–244. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Na, R.; Danae McLane, K.; Thompson, C.S.; Gao, J.; Wang, X.; Ran, Q. Overexpression of ferroptosis defense enzyme Gpx4 retards motor neuron disease of SOD1G93A mice. Sci. Rep. 2021, 11, 12890. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Jentoft, A.J.; Sayer, A.A. Sarcopenia. Lancet 2019, 393, 2636–2646. [Google Scholar] [CrossRef] [PubMed]
- Damluji, A.A.; Alfaraidhy, M.; AlHajri, N.; Rohant, N.N.; Kumar, M.; Al Malouf, C.; Bahrainy, S.; Ji Kwak, M.; Batchelor, W.B.; Forman, D.E.; et al. Sarcopenia and Cardiovascular Diseases. Circulation 2023, 147, 1534–1553. [Google Scholar] [CrossRef] [PubMed]
- Dent, E.; Morley, J.E.; Cruz-Jentoft, A.J.; Arai, H.; Kritchevsky, S.B.; Guralnik, J.; Bauer, J.M.; Pahor, M.; Clark, B.C.; Cesari, M.; et al. International Clinical Practice Guidelines for Sarcopenia (ICFSR): Screening, Diagnosis and Management. J. Nutr. Health Aging 2018, 22, 1148–1161. [Google Scholar] [CrossRef]
- Altun, M.; Edstrom, E.; Spooner, E.; Flores-Moralez, A.; Bergman, E.; Tollet-Egnell, P.; Norstedt, G.; Kessler, B.M.; Ulfhake, B. Iron load and redox stress in skeletal muscle of aged rats. Muscle Nerve 2007, 36, 223–233. [Google Scholar] [CrossRef]
- DeRuisseau, K.C.; Park, Y.M.; DeRuisseau, L.R.; Cowley, P.M.; Fazen, C.H.; Doyle, R.P. Aging-related changes in the iron status of skeletal muscle. Exp. Gerontol. 2013, 48, 1294–1302. [Google Scholar] [CrossRef]
- Aydemir, T.B.; Troche, C.; Kim, J.; Kim, M.H.; Teran, O.Y.; Leeuwenburgh, C.; Cousins, R.J. Aging amplifies multiple phenotypic defects in mice with zinc transporter Zip14 (Slc39a14) deletion. Exp. Gerontol. 2016, 85, 88–94. [Google Scholar] [CrossRef]
- Romagnani, P.; Remuzzi, G.; Glassock, R.; Levin, A.; Jager, K.J.; Tonelli, M.; Massy, Z.; Wanner, C.; Anders, H.J. Chronic kidney disease. Nat. Rev. Dis. Primers 2017, 3, 17088. [Google Scholar] [CrossRef]
- Wang, K.; Liu, Q.; Tang, M.; Qi, G.; Qiu, C.; Huang, Y.; Yu, W.; Wang, W.; Sun, H.; Ni, X.; et al. Chronic kidney disease-induced muscle atrophy: Molecular mechanisms and promising therapies. Biochem. Pharmacol. 2023, 208, 115407. [Google Scholar] [CrossRef]
- Jankowski, J.; Floege, J.; Fliser, D.; Bohm, M.; Marx, N. Cardiovascular Disease in Chronic Kidney Disease: Pathophysiological Insights and Therapeutic Options. Circulation 2021, 143, 1157–1172. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Anderson, E.M.; Thome, T.; Lu, G.; Salyers, Z.R.; Cort, T.A.; O’Malley, K.A.; Scali, S.T.; Ryan, T.E. Skeletal myopathy in CKD: A comparison of adenine-induced nephropathy and 5/6 nephrectomy models in mice. Am. J. Physiol. Ren. Physiol. 2021, 321, F106–F119. [Google Scholar] [CrossRef] [PubMed]
- Ku, E.; Del Vecchio, L.; Eckardt, K.U.; Haase, V.H.; Johansen, K.L.; Nangaku, M.; Tangri, N.; Waikar, S.S.; Wiecek, A.; Cheung, M.; et al. Novel anemia therapies in chronic kidney disease: Conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2023, 104, 655–680. [Google Scholar] [CrossRef] [PubMed]
- Reiss, A.B.; Miyawaki, N.; Moon, J.; Kasselman, L.J.; Voloshyna, I.; D’Avino, R., Jr.; De Leon, J. CKD, arterial calcification, atherosclerosis and bone health: Inter-relationships and controversies. Atherosclerosis 2018, 278, 49–59. [Google Scholar] [CrossRef]
- Wang, X.H.; Mitch, W.E. Mechanisms of muscle wasting in chronic kidney disease. Nat. Rev. Nephrol. 2014, 10, 504–516. [Google Scholar] [CrossRef]
- Massini, G.; Caldiroli, L.; Molinari, P.; Carminati, F.M.I.; Castellano, G.; Vettoretti, S. Nutritional Strategies to Prevent Muscle Loss and Sarcopenia in Chronic Kidney Disease: What Do We Currently Know? Nutrients 2023, 15, 3107. [Google Scholar] [CrossRef]
- Cecconi, M.; Evans, L.; Levy, M.; Rhodes, A. Sepsis and septic shock. Lancet 2018, 392, 75–87. [Google Scholar] [CrossRef]
- Nakanishi, N.; Oto, J.; Tsutsumi, R.; Akimoto, Y.; Nakano, Y.; Nishimura, M. Upper limb muscle atrophy associated with in-hospital mortality and physical function impairments in mechanically ventilated critically ill adults: A two-center prospective observational study. J. Intensive Care 2020, 8, 87. [Google Scholar] [CrossRef]
- Boulikas, T.; Vougiouka, M. Recent clinical trials using cisplatin, carboplatin and their combination chemotherapy drugs (review). Oncol. Rep. 2004, 11, 559–595. [Google Scholar] [CrossRef]
- Conte, E.; Bresciani, E.; Rizzi, L.; Cappellari, O.; De Luca, A.; Torsello, A.; Liantonio, A. Cisplatin-Induced Skeletal Muscle Dysfunction: Mechanisms and Counteracting Therapeutic Strategies. Int. J. Mol. Sci. 2020, 21, 1242. [Google Scholar] [CrossRef]
- Cooper, C.; Burden, S.T.; Cheng, H.; Molassiotis, A. Understanding and managing cancer-related weight loss and anorexia: Insights from a systematic review of qualitative research. J. Cachexia Sarcopenia Muscle 2015, 6, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Kasarskis, E.J.; Tandon, L.; Lovell, M.A.; Ehmann, W.D. Aluminum, calcium, and iron in the spinal cord of patients with sporadic amyotrophic lateral sclerosis using laser microprobe mass spectroscopy: A preliminary study. J. Neurol. Sci. 1995, 130, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zeng, X.; Bian, W.; Li, H.; Tegeleqi, B.; Gao, Z.; Liu, J. Exosomes from Muscle-Derived Stem Cells Repair Peripheral Nerve Injury by Inhibiting Ferroptosis via the Keap1-Nrf2-Ho-1 Axis. J. Cell Biochem. 2024, 125, e30614. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Morphologic Features | Biochemical Features | Regulatory Signal | Key Genes | Inhibitor | Inducer | |
|---|---|---|---|---|---|---|
| Ferroptosis | Small mitochondria with increased membrane density, outer membrane rupture, and cristae reduction in or vanishing | Iron accumulation, lipid peroxidation, and ROS excessive accumulation | Xc-/GPX4, E-cadherin-NF2-Hippo-YAP, AMPK and Hypoxia signaling, P53-SAT1-ALOX15, P62-Keap1-NRF2 pathway, FSP1-COQ10-NAD(P)H pathway, HSPB1-TRF1 | GPX4, ACSL4, NRF2, TfR1, FTH1, LPCAT3, SLC7A11, SLC39A14, NCOA4, FSP1, COX2, ACSL4, P53, HSPB1 | Fer-1, Liproxststatin-1, mesylate, SRS16-86, SRS11-9, Vitamin E, deferoxamine, 2,2′-pyridine Deferoxamine | Erastin, RSL3, ML162, FIN56 FINO2, Sorafenib, Sulfasalazine, (1S,3R)-RSL3, DPI7, DPI10, |
| Apoptosis | Cell shrinkage, membrane blebbing, nuclear fragmentation, chromatin condensation and margination, formation of apoptotic bodies, and disintegration of the cytoskeleton | DNA fragmentation | Growth factor, Nutrient deprivation, DNA damage, P53, Bcl-2, mitochondrion pathway and endoplasmic reticulum pathway, Fas ligand, TNF or TNF-related apoptosis-inducing ligand, Caspase | Cytochrome c, pro-caspase-9, pro-caspase-3, pro-caspase-7, BCL-2, BAX, BCL-X, APAF1 | Nerve growth factor, fibroblast growth factor 10, metformin, resveratrol, forsythiaside B, rehmannioside A, baicalein, anthocyanins, apsinini, apigenin, delphinidin, rosmarinic acid, IGF-1 | TGF-β, IL-10, IL-2, Dexamethasone, PKC-delta, Resveratrol, Curcumin, Yessotoxin, TNF family, Metal cadmium, HIF-1α |
| Autophagy | formation of double-membrane enclosed vesicles | Formation of double-membraned autolysosomes, including microautophagy chaperone-mediated autophagy, and macroautophagy | mTOR, AMPK, Beclin-1, P53 signaling, | Atg5/Atg7, LC3, Atg6/Beclin-1, p62/SQSTM1, Ulk-1 | Rubicon, chloroquine, VPS34 inhibitors, ULK1 inhibitors, Atg4B inhibitors, Lys05, quinacrine, VATG-027, VATG-032, hydroxychloroquine | Resveratrol, Spermidine, SMER28, Luteolin, Apigenin, Salidroside, ABT-737, GX15-070 (Obatoclax mesylate), Metformin, Rapamycin and rapalogs, Curcumin, Quercetin |
| Necroptosis | Cytoplasmic and organelle swelling, plasma membrane rupture, pore formation on cell membranes, moderate chromatin condensation, loss of cellular and organelle integrity | Phosphorylation of MLKL by receptor RIPK1 and RIPK3, the assembly of necrosome | RIPK1/RIPK3/MLKL pathway, Fas/FasL, Toll-like receptors, TNF-R1, ROS, RIG-I and STING, PKC-MAPK-AP-1 related signaling, | RIPK1, RIPK3, FADD, MLKL, caspase-8, caspase-10 | Necrostatin-1, GSK2982772, GSK’840, GSK’843, GSK’872, dabrafenib, ponatinib, pazopanib, GSK’074 | Z-DNA-binding protein (ZBP1), Doxorubicin, Convallatoxin, Apurinic/apyrimidinic endonuclease 1, Cisplatin, Acetylshikonin, TNF-α, Alcohol, Tunicamycin |
| Pyroptosis | Cells swelling, cell membrane-forming pore, rupture, and bubbling of plasma membranes, nuclear condensation, and DNA damage | Gasdermin cleavage, gasdermin E dependent inflammasome formation, caspase-dependent, release of IL-1β and IL-18 | Canonical inflammasome pathway, non-canonical inflammasome pathway, apoptotic caspases-mediated pathway, granzyme-mediated pathway | GSDMD, caspase-1, caspase-3, caspase-4, caspase-5, caspase-11, IL-1β, IL-18, NLRP3 | MCC950, P2 × 7 inhibitor, silybin, dihydroquerceti, liraglutide, caspase-1 inhibitor, rosiglitazone, IL-β receptor antagonist | Triptolide, Paclitaxel, Cisplatin, Dibutyl phthalate, Copper-bacteriochlorin nanosheet, Cucurbitacin B, Simvastatin, Nobiletin, Arsenic, Metformin |
| Feature | Comment | References |
|---|---|---|
| Conditional ablation of Gpx4 in neurons of mice | Increased ferritin deposition, motor neuron degeneration rapid paralysis, and severe muscle atrophy in mice | [88] |
| Increased iron in the skeletal muscle of old SAMP8 mice | Increased lipid peroxidation and MDA content and Ptgs2 mRNA levels; decreased NADPH and GSH content | [89] |
| Elevated levels of iron, ACSL4 and ALOX15, decreased xCT and GPX4 expression | Increased ferroptosis in rats with chronic kidney disease-induced muscle atrophy | [90] |
| Increased ferroptosis markers and lipid peroxidation products in the skeletal muscle of 5/6 nephrectomized mice | Reduced GSH/GSSG ratio, decreased GSH content, increased MDA production | [91] |
| Decreased FTL, FPN, and GPX4 expression in the quadriceps femoris of old rats | Decreased levels of FTL, FPN, and GPX4 in the quadriceps muscle of old rats, which were significantly increased after lifelong aerobic training | [92] |
| Increased levels of ferroptosis marker (COX2, ACSL4, and FTH1) and decrease GPX4 expression | Increased ferroptosis in skeletal muscle of CLP mice, inhibition of STAT6 activity attenuates ferroptosis | [93] |
| Elevated levels of iron and MDA and decreased NADPH, GSH, and GPX4 expression | Increased ferroptosis in rats with chronic kidney disease-induced muscle atrophy | [94] |
| Increased expression levels of ferroptosis ferroptosis-related genes (ACSL4, Sat1, SLC39A14) | Ferroptosis-related genes, such as ACSL4, Sat1and SLC39A14 expression levels were significantly increased in the cisplatin-treated mice atrophic muscles | [95] |
| Ferroptosis-related signaling pathways were significantly enriched in sarcopenia patients with chronic obstructive pulmonary disease (COPD) | A cohort study uncovers ferroptosis as a potential common mechanism of COPD complicated by sarcopenia | [96] |
| Elevated levels of ROS, MDA and decreased GSH, SLC7A11, and GPX4 expression | Increased ferroptosis in skeletal muscle of ALS mice induced by the SOD1G93A mutation, NRF2 activation suppresses motor neuron ferroptosis | [97,98,99] |
| Decreased accumulation of 4HNE and iron and lipid peroxidation in ALS mice | Fe-1, VAR10303 and overexpression of GPX4 ameliorated motor neurons and prolonged lifespan in SOD1G93A mice | [100,101,102] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, C.-C.; Xiao, J.-L.; Sun, C.; Tang, C.-F. Ferroptosis and Its Potential Role in the Physiopathology of Skeletal Muscle Atrophy. Int. J. Mol. Sci. 2024, 25, 12463. https://doi.org/10.3390/ijms252212463
Sun C-C, Xiao J-L, Sun C, Tang C-F. Ferroptosis and Its Potential Role in the Physiopathology of Skeletal Muscle Atrophy. International Journal of Molecular Sciences. 2024; 25(22):12463. https://doi.org/10.3390/ijms252212463
Chicago/Turabian StyleSun, Chen-Chen, Jiang-Ling Xiao, Chen Sun, and Chang-Fa Tang. 2024. "Ferroptosis and Its Potential Role in the Physiopathology of Skeletal Muscle Atrophy" International Journal of Molecular Sciences 25, no. 22: 12463. https://doi.org/10.3390/ijms252212463
APA StyleSun, C.-C., Xiao, J.-L., Sun, C., & Tang, C.-F. (2024). Ferroptosis and Its Potential Role in the Physiopathology of Skeletal Muscle Atrophy. International Journal of Molecular Sciences, 25(22), 12463. https://doi.org/10.3390/ijms252212463